 Nathan Karin
Nathan Karin- Department of Immunology, Faculty of Medicine, Technion—Israel Institute of Technology, Haifa, Israel
Myeloid-derived suppressor cells (MDSC) represent a heterogeneous population of immature myeloid cells. Under normal conditions, they differentiate into macrophages, dendritic cells, and granulocytes. Under pathological conditions, such as chronic inflammation, or cancer, they tend to maintain their immature state as immature myeloid cells that, within the tumor microenvironment, become suppressor cells and assist tumor escape from immune eradication. MDSC are comprised of two major subsets: monocytic MDSC (M-MDSC) and polymorphonuclear MDSC (PMN-MDSC). Monocytic myeloid cells give rise to monocytic cells, whereas PMN-MDSC share similarities with neutrophils. Based on their biological activities, a two-stage model that includes the mobilization of the periphery as myeloid cells and their activation within the tumor microenvironment converting them into suppressor cells was previously suggested by D. Gabrilovich. From the migratory viewpoint, we are suggesting a more complex setup. It starts with crosstalk between the tumor site and the hematopoietic stem and progenitor cells (HSPCs) at the bone marrow (BM) and secondary lymphatic organs, resulting in rapid myelopoiesis followed by mobilization to the blood. Although myelopoiesis is coordinated by several cytokines and transcription factors, mobilization is selectively directed by chemokine receptors and may differ between M-MDSC and PMN-MDSC. These myeloid cells may then undergo further expansion at these secondary lymphatic organs and then home to the tumor site. Finally, selective homing of T cell subsets has been associated with retention at the target organs directed by adhesion molecules or chemokine receptors. The possible relevance to myeloid cells is still speculative but is discussed.
Introduction
The tumor microenvironment (TME) is the environment around the tumor that includes sounding blood vessels; immune cells; fibroblasts; soluble mediators, such as cytokines, chemokines, and growth factors; and extracellular matrix (ECM). Among the immune cells that enable tumor escape from immune eradication are myeloid-derived suppressor cells (MDSC). These are a heterogeneous population of cells that consists of myeloid progenitor cells and immature myeloid cells (IMCs). Under nonpathological conditions, these IMCs differentiate into monocytic cells that later become macrophages, dendritic cells (DC), and mature granulocytes. However, under stress and during chronic inflammation, particularly cancer, they tend to response to “emergency signals” (1, 2), and as a result, their maturation into fully differentiated cells is inhibited while retaining their suppressive activity (3–7). Their mechanism of action includes secretion of Arginase 1 (encoded by ARG1) and inducible nitric oxide synthase (iNOS, also known as NOS2) as well as an increase in their production of nitric oxide (NO) and reactive oxygen species (ROS) [for a recent review, see (8)]. MDSC also express immune checkpoint inhibitors, among them PD-L1 and also PD-1 (9). Along with this, very recently it has been reported that targeted deletion of PD-1 from MDSC induces highly effective antitumor immunity (10). Altogether, these render MDSC immune suppressive, in particular of effector T cells, which enables tumor escape from immune eradication (3–7). It is, thus, believed that these cells play a major role in enabling tumors to escape their elimination or blockade, which could be beneficial for cancer immunotherapy (11).
Myeloid cells, as other bone marrow (BM)-derived cells, are generated from hematopoietic stem and progenitor cells (HSPCs) in a process termed myelopoiesis and then are mobilized from the BM to the blood. HSPCs also migrate from the BM to secondary lymph nodes and spleen (12). At these organs, the presence of myeloid cells has also been recorded [reviewed in (13)]. Recently, it has been reported that under “emergency” conditions occurring during stress, inflammation, and cancer diseases, the retinoic acid–related orphan receptor (RORC1/ROR/γ) orchestrates emergency myelopoiesis by suppressing negative (Socs3 and Bcl3) and promoting positive (C/EBPb) regulators of granulopoiesis as well as the key transcriptional mediators of myeloid progenitor commitment and differentiation to the monocytic/macrophage lineage (IRF8 and PU.1) (2). This may suggest that, under emergency conditions, myelopoiesis and rapid extension of myeloid cells may also take place at secondary lymphatic organs and spleen and, by so doing, allow massive accumulation of these cells at tumor sites (2) [(see also reviews in ((1) and (14)) (Figure 1)].
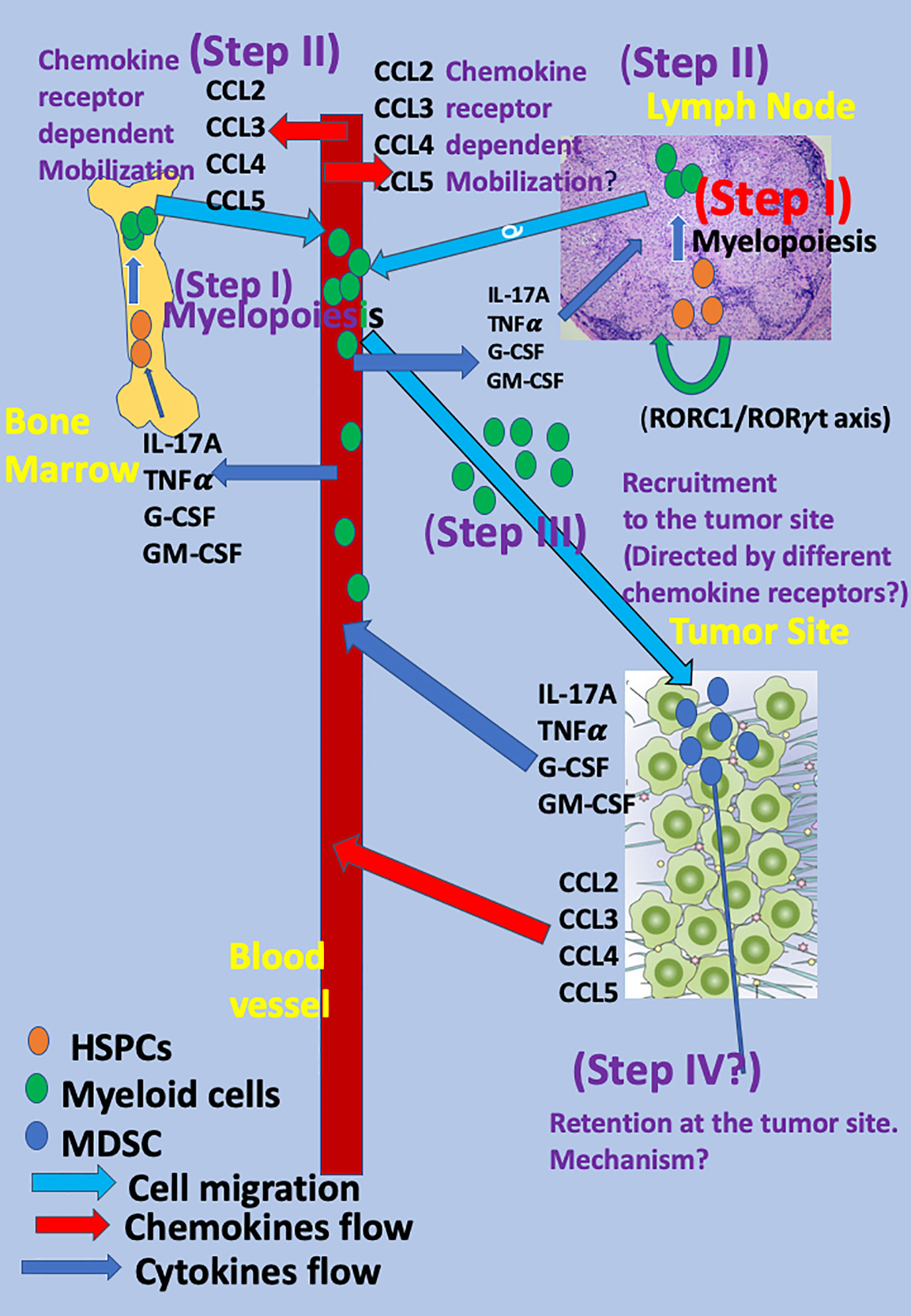
Figure 1 The mobilization and migration of myeloid cells to the tumor site as a multistep event The mobilization and migration of myeloid cells to the tumor site is a multistep event in which cytokines, chemokines, and transcription factors released from the tumor site reach the blood and, thereafter, the BM and LNs and direct the different steps in myeloid cell differentiation and migration. The first step (Step I) is rapid myelopoiesis of myeloid cells at the BM and secondary lymphatic organs (LNs and spleen) and is directed by several cytokines, among them interleukin-17A (IL-17A), G-CSF, GM-CSF, TNFα, and others. Recently, the key role of the retinoic acid–related orphan receptor (RORC1/ROR/γ) in directing myelopoiesis in LNs has been observed (2). The subsequent step (Step II) includes the mobilization of myeloid cells to the blood and is directed by specific chemokine receptors: CCR2 for monocytic myeloid cells (15) and CCR5 for the polymorphonuclear myeloid cells (16) via CCR2 key ligand CCL2 and the CCR5 key ligands: CCL3, CCL4, and CCL5 (Step II). Homing to the tumor site is likely to be directed by many chemokines and chemokine receptors and is likely to have low specificity (Step III). Step IV includes the retention of these cells at the tumor site and, thus far, has been mostly studied for T cells (17–20). For myeloid cells, it is still speculative.
Chemokines are a subgroup of chemotactic cytokines that are well associated with chemo-attraction of various leukocytes, either from the BM to the blood (mobilization); from the blood to sites of inflammation, autoimmune sites, tumor sites, etc.; and from tissues and blood to the lymph nodes (21–23). The current review focuses on elaborating a sequential multistep model for characterizing their myelopoiesis, mobilization, recruitment, retention, and biological function. In this model, the migratory properties of myeloid cells from BM (and perhaps also from secondary lymphatic organs) to the blood (mobilization), is likely to be directed by specific chemokine receptors (Figure 1). The model that we are suggesting does not contradict the two-stage model of Gabrilovich (11), but adds several steps that are associated with the migratory properties of these cells. For example, the first step in Dr. Gabrilovich’s model corresponds to activation of myelopoiesis, mobilization to the blood, and migration of myeloid cells to the tumor sites as suggested in our multistep model as different steps.
MDSC Subtypes
MDSC are comprised of two major subsets: monocytic MDSC (M-MDSC), and polymorphonuclear MDSC (PMN-MDSC). In human, M-MDSC are defined as CD11b+ CD14+ CD15−HLA-DRlow/− cells. Due to the low or absent HLA-DR expression, M-MDSC can be distinguished from monocytes. Human PMN-MDSC are characterized as CD11b+ CD14−CD15+ HLA-DR− or CD11b+CD14−CD66b+ (24).
In mice, M-MDSC are defined as CD11b+Ly6G−Ly6Chigh and share phenotypical and morphological characteristics with monocytes. PMN-MDSC are described as CD11b+Ly6GhighLy6Clow cells and resemble neutrophils (24).
M-MDSC and tumor-associated macrophages (TAMs) share many features (25). Thus, it is believed that, at the tumor site, M-MDSC may become TAMs. The question of whether PMN-MDSC may also become mature granulocytes is still an open question. There are two lines of evidence that support this hypothesis: 1. Tumor-associated neutrophils and G-MDSC represent similar functional states of cells originating from the same cell type and induced within a tumor host. 2. Neutrophils isolated from a normal tumor-free host substantially differ from tumor-associated neutrophils or G-MDSC obtained from a tumor-bearing host [reviewed in (26)].
Both types of MDSC express many chemokine receptors, among them CCR5 and CCR2 (27). Within the TME, a vast majority of MDSC are PM-MDSC (about 80%) even though they have a shorter lifetime (11). Both also operate via similar mechanisms of immunosuppression with a few differences: Arginase-1 and prostaglandin E2 (PGE2) are preferentially produced by PMN-MDSC, whereas NO is by M-MDSC [for a recent review, see (3)]. It is also believed that M-MDSC are more prominent than PMN-MDSC as they are thought to rapidly differentiate to TAMs at the tumor site (28–35), whereas PMN-MDSC play a major role in inducing peripheral T cell tolerance (3, 11).
The Two-Stage Model of Myeloid Cells Mobilization and Function
Myelopoiesis during acute infection, stress, or trauma results in rapid terminal differentiation of myeloid cells. By contrast, in cancer and chronic inflammation, myelopoiesis is associated with defective myeloid cell differentiation, which results in the accumulation and persistence of immature myeloid cells at cancer sites or chronic inflammatory sites. These cells then function as suppressor cells and are, therefore, referred to as MDSC (4, 6–8). Based on the above, D. Gabrilovich et al. suggest a two-stage model that is based on the biological function of myeloid cells during cancer and chronic inflammation (11). It includes the myelopoiesis of these cells in BM, their mobilization to the blood and secondary lymphatic organs as myeloid cells (stage I), and later their transition and maintenance as MDSC (stage II), which mostly takes place at the tumor site (11) or, respectively, sites of chronic inflammation (36).
In both type of diseases, the rapid myelopoiesis of myeloid cells at the BM is likely to be directed by several cytokines and transcription factors, among them interleukin-17A (IL-17A) ROR1C that induces IL-17A, G-CSF, GM-CSF, TNFα and others (2, 4, 6, 14, 37, 38), whereas maintenance of the suppressive function is driven by several components that affect the activities of MDSC at the tumor site, including interaction with other cells, particularly T cells cytokines, chemokines, and transcription factors, and the effect of microRNA released from exosomes (39–41).
The second stage includes two distinct yet partially overlapping types of signals. The first is associated with the expansion of the immature myeloid cells and inhibition of their terminal differentiation, and the second is their pathologic activation as suppressor cells (42). The first group of signals is mostly driven by tumor-derived growth factors as well as STAT3, IRF8, C/EBPb, Notch, adenosine receptors A2b signaling, and NLRP3 (43) and of microRNA released from exosomes (39–41). The second type of signal is mediated by factors produced mostly by the tumor stroma (proinflammatory cytokines, HMGB1) and includes the NF-kB pathway, STAT1, STAT6, prostaglandin E2 (PGE2), and cyclooxygenase 2 (COX2) as reviewed in (42).
It should be noted that the mechanisms controlling the suppressive activities may vary between PMN-MDSC and M-MDSC. The first are short-lived (44), and their activity may require close cell-to-cell contact with T cells (45), whereas M-MDSC are long-lived and are likely to give rise to TAMs that, under the TME milieu, suppress antitumor immune reactivity by different mechanisms (46).
Despite the clear definition between myeloid cells in the periphery and MDSC at the tumor site (11), it has been reported in cancer MDSC in spleen, and secondary lymphatic organs function as suppressor cells and execute far-reaching immune suppression by reducing expression of the L-selectin lymph node (LN) homing receptor on naive T and B cells, and impair T cell activation also by inhibiting the homing of naïve CD4+ and CD8+ T cells to LNs (47).
The Recruitment of MDSC at Tumor Sites as a Multistep Event Directed in Part by Chemokine–Chemokine Receptor Pathways
The generation of myeloid cells and their recruitment to the tumor site could be viewed as a multistep event, in which the cross-talk between the tumor site and myeloid cells play a major role. We are suggesting a four-step event that characterizes the homing of these cells (step I–IV) and an additional two steps that aim to focus on two complementary signaling events within the TME that enable the transformation of myeloid cells into suppressor cells and maintains their immature state as such (Figure 1) as follows:
Step I
The first step is myelopoiesis. It could occur in the BM and also possibly in the LNs and spleen as HSPCs also migrate from BM to LNs, spleen, and peripheral tissues (12) and undergo myelopoiesis there (2). Several key cytokines take a major role in this step, including IL-17A, granulocyte-colony stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), and TNFα. All these cytokines are largely produced at tumor sites, and their blood levels increase during cancer diseases (48–56). Concurrent myelopoiesis at the BM and secondary lymphatic organs may allow intensive accumulation of myeloid cells at the tumor site (1, 2, 14).
Step II
The subsequent step (step II) is the mobilization of myeloid cells that rapidly proliferated along myelopoiesis from the BM and possibly secondary lymphatic organs to the blood. It is not yet clear whether the mechanism by which these cells are mobilized from the BM to the blood differs from the one by which they are mobilized from the lymph nodes to the blood. Accumulating evidence votes for a pivotal role of chemokine–chemokine receptor interactions at this step (15, 16, 57). Several key chemokines are largely produced at tumor sites, and their blood level increases during cancer diseases, among them the CCR2 ligand CCL2 (58), and CCR5 ligands, in particular CCL5 (59, 60). These soluble mediators are likely to participate in the inter-talk between the tumor and leukocytes, either within the tumor site or at the periphery. The CCR2–CCL2 axis is highly relevant for monocytic and monocytic myeloid cells (15, 57), particularly in inflammation (15) and cancer (28–31, 33, 34). In 2003, Geissmann, Jung, and Littman reported different migratory properties for CX3CR1lowCCR2+Gr1+ and CX3CR1highCCR2-Gr1+ cells, showing that those that are CCR2+ preferentially home to inflammatory sites, whereas the others go to normal tissues (57). This links CCR2 to selective homing of monocytic myeloid cells to inflammatory sites (57). Three years later, Sebrina et al. demonstrated the pivotal role of CCR2 in directing the mobilization of Ly6Chigh monocytes from BM to the blood (15). This study shows that CCR2KO mice display fewer circulating Ly6Chigh monocytes and, after infection with listeria monocytogenes, accumulate activated monocytes in BM (15). This study also shows that the later chemotaxis of these cells to the inflammatory site is not necessarily CCR2-dependent and also occurs if using monocytic cells from CCR2 KO mice (15). Later studies further explore the pivotal role of CCR2 in directing the recruitment of CCR2+ monocytic cells to the tumor site to support its development and suppress antitumor immunity (28–31, 33, 34). More recently, Chang et al. observed in murine glioma that CCL2 produced by microglia recruited CCR2+Ly6C+ monocytic MDSCs (M-MDSCs) to the tumor site, which is absent in CCR2KO mice (61). Among the different ligands that bind CCR2, CCL2 has been thought to be the dominant chemokine. An additional chemokine that is likely to hold similar properties is CCL12 (62).
Less is known about the mobilization of polymorphonuclear myeloid cells from the BM (and perhaps lymphatic organs) to the blood. Our group found interest in exploring the role of CCR5 and its ligands in cancer. Individuals with a functional mutation in CCR5 (deletion of the N-terminal 32 nucleotides) display a high state of HIV resistance (63). Later, it was found that they also show low prevalence of cancer diseases, particularly cancer of the prostate (64). This motivated us to explore the underlying mechanism by which the absence of CCR5 confers cancer resistance. In so doing, we have used CCR5KO mice and an autologous model of prostate cancer in immunocompetent mice (16). In this study, we observed that 1. CCR5 ligands directly support tumor growth via CCR5, and thus, blockade of CCR5 ligands in a chimeric system in which CCR5KO mice bearing CCR5+ tumor cells, targeting CCR5 ligands restrains tumor growth (16). 2. CCR5 drives the accumulation of MDSC at the tumor site; thus, in CCR5KO mice, the relative number of GR1+ CD11b+ myeloid cells at the tumor site is very low, and tumor development is arrested. Reconstitution of these mice with GR1+ CD11b+ myeloid cells from WT mice (CCR5+) reconstituted tumor growth (16). Further investigation shows that, along with tumor development the level of CCR5 ligands that are largely expressed with the TME, increases in the blood. This leads to a rapid increase in the expression of CCR5 on myeloid cells at the BM to a rapid mobilization of CD11b+Ly6GhighLy6Clow myeloid cells that become PMN-MDSC at the tumor site (16). It has yet to be studied if limited accumulation of PMN-MDSC at the tumor site in CCR5KO mice exclusively results from reduction in their mobilization from the BM to the blood, and/or from secondary lymphatic organs to the blood, or also due to possible roles of CCR5 in directing the accumulation of these cells at the tumor site. In this study, we also observed that the CCR5–CCR5 ligands interaction also potentiates the suppressive activities of PMN-MDSC by increasing the expression of Arginase 1 and possibly other mediators that suppress effector T cell function (16). A recent manuscript used the technology of deleting the genomic locus incorporating the iCCRs of different chemokine receptors that have been associated with myelomonocytic cell population migration, including CCR1, CCR2, CCR3, and CCR5 to show that tissue-resident myelomonocytic cell populations are established even in their absence, whereas during inflammation, CCR2 holds a key role in their targeted recruitment (65). Dyer et al. have not explored their setup in a cancer disease model.
Step III
The third step (step III) includes the accumulation of MDSC at the tumor site and their retention there. In our opinion, this step is more complex and less understood than most leukocyte subtypes. The major obstacle is that myeloid cells express many different chemokine receptors and may, thus, respond to many different chemokines that are largely expressed at tumor sites. Then, what causes chemokine receptor specificity? Indeed, many studies show a significant role of different chemokines in myeloid cell recruitment to tumor sites (Table 1). Among them, 1. the CCL15-CCR1 signaling pathway (68, 69), 2. the CX3CL1 - CCL26 pathway for recruiting M-MDSC (70), 3. the CXCL5/CXCL2/CXCL1 chemokines were suggested to recruit PMN-MDSCs to tumor tissue via CXCR2 in murine spontaneous melanoma model (71), 4. CXCL13-CXCR5 signaling mediates the migration of MDSCs to tumor tissue (72). Moreover, in different cancer diseases, poor or good prognosis was associated with high or low levels of these chemokines (Table 2) [for a recent review, see (73)]. How could these observations take place with the predominate role of the CCR5-axis for directing PMN-MDSC recruitment and the CCR2-axis for M-MDSC selective recruitment at tumor sites? We are suggesting, within the three-step model described above, the highly selective step that determines receptor specificity is the mobilization from the BM and perhaps from the secondary LNs to the blood and that this step serves as a bottleneck for selectivity in myeloid cell migration.
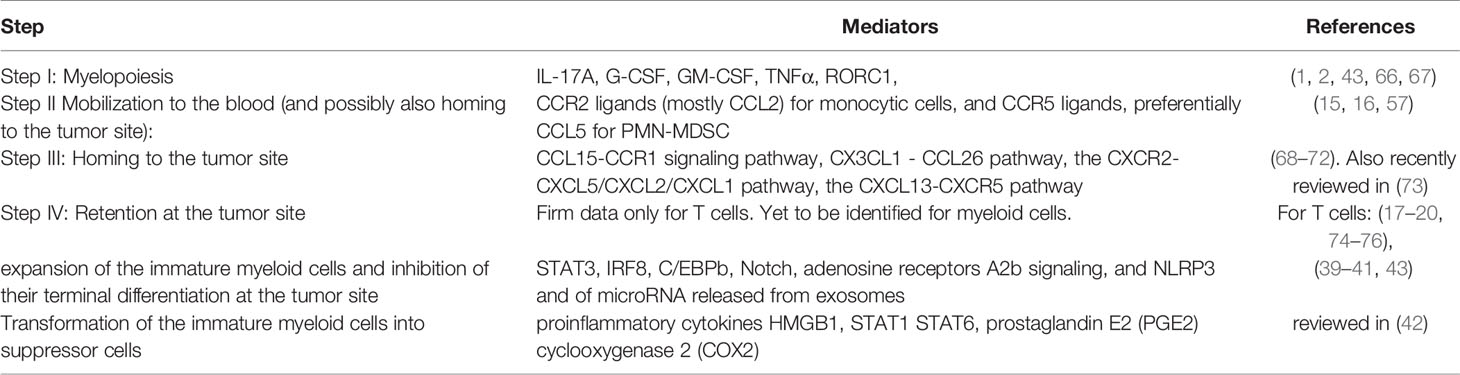
Table 1 The role of chemokines, cytokines, and other mediators in directing the different steps in myeloid cell migration and function.

Table 2 Key chemokines associated with myeloid cell homing and cancer prognosis.
Step IV
Retention at the tumor site: This step is still speculative and has been mostly studied for T cells thus far. It suggests that, tentatively, myeloid cells could be recruited to tumor sites by many different chemokine receptors, but their retention there is more specific and may involve a limited number of chemokine receptors and/or adhesion molecules (74). This option has been explored thus far only for T cell migration. Key examples are the retention of CD103+ memory T cells to tissues where they become tissue resident memory T cells due to the interaction of CD103 (an αE integrin) that binds a β7 counter integrin (17–20), the L-selectin serving as a homing receptor for naïve T cells (75), and the role of the α4β1 integrin in the retention of CD4+ T cells in the inflamed brain (76). The relevance of this concept to other leukocyte subtypes is yet to be studied.
Post-Translational Modification (PTM) of Chemokines and Selective Migration of PMN-MDSC
An important mechanism of fine-tuning chemokine activity is PTM of chemokines and their receptors. One of the mechanisms that may show high relevance to CCR5-dependent selective migration of PMN-MDSC is PTM by CD26 [for a recent relevant review, see (115)]. CD26 is a cell-bound enzyme ubiquitously expressed on blood cells, especially on activated T cells, fibroblasts, and epithelial cells. Two of the three CCR5 ligands, CCL4, and CCL5 are truncated by CD26, which may selectively reduce CCL4/CCL5 activity on T cells but to a lesser degree extend PMN-MDSC.
Clinical Implications in Cancer Diseases: Could Redundancy in Chemokines Be Overcome via Monotherapy?
Thus far, many clinical trials in humans in which single chemokines or their receptors were targeted for therapy of inflammatory autoimmunity or cancer failed. Two major potential reasons could be taken into account: redundancy, that is, different chemokines with similar properties bind the same chemokine receptor, and overcompensation by production of increased levels of targeted chemokine. Two possible approaches to partially overcome this obstacle is by designing a compound that would target all ligands of a single receptor or prefer a receptor blockade over targeting a single chemokine. We have taken the first approach and generated a chimeric CCR5 soluble receptor study (116) that could effectively inhibit cancer of the prostate in C57Bl/6 mice (16). Then, together with Viktor Umansky and his team, this study was further extended to a transgenic model of melanoma showing that indeed the CCR5-CCR5 ligand axis directs the accumulation of PMN-MDSC at the tumor site and that CCR5-Ig also effectively inhibited the development and progression of this disease (117). Others used blocking mAbs to CCR5 or even one of its three ligands, CCL5, to inhibit metastasis and improve the survival of tumor-bearing mice (118, 119) and also enhance anti-PD1 efficacy in gastric cancer (120). As for humans, Halama et al. recently showed success in blocking colorectal cancer using a CCR5 small molecule inhibitor that was previously developed for therapy of HIV (121). If successful, we think that extension of this therapeutic approach as a monotherapy or part of a combined therapy could be further considered.
A Future View of the Classical Two-Stage Model in Light of Modern Technologies
The traditional classification of myeloid cells in the periphery and MDSC at the tumor site have recently been revised using several modern technologies, aiming at categorizing single cells based on their gene signature (single-cell RNAseq) and expression of cell surface receptors and some intracellular proteins (mass cytometry, CyTOF). The basic hypothesis is that, beyond the variety between human and mouse in the classification of these cells (24), in each species, the gene signature and cell surface protein expression may vary depending on the organ from which cells are isolated (BM, spleen, blood, LNs, tumor site) and may also differ between tumor types (122–126). These studies are still in early development but may pave a new direction in scientific research and its translational implications.
Can the Multistep Model Explain the Paradox of Redundancy in Chemokine–Chemokine Receptor Interactions and Selective Migration?
MDSC express many chemokine receptors and may, therefore, potentially migrate in response to each of them (Table 1). The multistep model suggests that, among the four different steps (myelopoiesis, mobilization to the blood, recruitment, and retention) the step of mobilization to the blood is likely to be the more highly selective stage. In this event, CCL2 signals via CCR2 to allow the effective mobilization of monocytic cells, including monocytic myeloid cells (15, 57), whereas CCR5 via its ligands, mostly CCL5, is likely to direct the mobilization of PMN myeloid cells (16). The last has mostly been studied in our laboratory and has to be further confirmed by others. It is conceivable that the CCR2 and CCR5 axes are also involved, together with other axes in step III of homing to the tumor site (Table 1). This may explain why CCR2 and perhaps CCR5 are indeed key drivers in the migratory cascade of myeloid cells.
Among these four steps, not much is known for the last one (retention) for myeloid cells. For T cells, its selectivity and specificity are mostly directed by selective adhesion receptors (18–20, 75, 76). We do not exclude the possibility that a key adhesion molecule or a key chemokine receptor may direct this stage, making it a highly selective step as well.
Conclusions
Based on their biological function, the development of MDSC includes two major stages: the first starts with myelopoiesis in the BM and lymphatic organs and the second upon their entry to the tumor site where they acquire suppressive capabilities and retain their amateur state of development. Nevertheless, based on their migratory properties, their generation and migration to the tumor site could be described as a more detailed multistep event in which their mobilization to the blood seems to be chemokine-receptor dependent and also determines the selectivity of their migration. We have uncovered a key role of the CCR5 axis in directing the mobilization of PMN-MDSC and suggest CCR5 blocking as a potential way for monotherapy or part of combined therapy for cancer diseases.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This study was supported by the Ministry of Science & Technology of the state of Israel (MOST) and the German Cancer Research Center (DKFZ) (MOST- DKFZ grant # 3-16001 and grant # GR-2471) Israel Cancer Research fund (ICRF) (PG-19-105 and PG-171961), Israel Cancer Association (ICA 2020-1209), Collek Research fund, and Israel Science Foundation (ISF, 283/19).
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Consonni FM, Porta C, Marino A, Pandolfo C, Mola S, Bleve A, et al. Myeloid-Derived Suppressor Cells: Ductile Targets in Disease. Front Immunol (2019) 10:949. doi: 10.3389/fimmu.2019.00949
2. Strauss L, Sangaletti S, Consonni FM, Szebeni G, Morlacchi S, Totaro MG, et al. RORC1 Regulates Tumor-Promoting “Emergency” Granulo-Monocytopoiesis. Cancer Cell (2015) 28:253–69. doi: 10.1016/j.ccell.2015.07.006
3. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol (2018) 19:108–19. doi: 10.1038/s41590-017-0022-x
4. Tcyganov E, Mastio J, Chen E, Gabrilovich DI. Plasticity of myeloid-derived suppressor cells in cancer. Curr Opin Immunol (2018) 51:76–82. doi: 10.1016/j.coi.2018.03.009
5. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
6. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol (2016) 37:208–20. doi: 10.1016/j.it.2016.01.004
7. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9:162–74. doi: 10.1038/nri2506
8. Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, et al. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front Immunol (2018) 9:398. doi: 10.3389/fimmu.2018.00398
9. Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, et al. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front Immunol (2018) 9:1310. doi: 10.3389/fimmu.2018.01310
10. Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol (2020) 5:eaa1863. doi: 10.1126/sciimmunol.aay1863
11. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12:253–68. doi: 10.1038/nri3175
12. Massberg S, Schaerli P, Knezevic-Maramica I, Kollnberger M, Tubo N, Moseman EA, et al. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell (2007) 131:994–1008. doi: 10.1016/j.cell.2007.09.047
13. Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol (2009) 182:4499–506. doi: 10.4049/jimmunol.0802740
14. Gabrilovich D, Nefedova Y. ROR1C Regulates Differentiation of Myeloid-Derived Suppressor Cells. Cancer Cell (2015) 28:147–9. doi: 10.1016/j.ccell.2015.07.007
15. Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol (2006) 7:311–7. doi: 10.1038/ni1309
16. Hawila E, Razon H, Wildbaum G, Blattner C, Sapir Y, Shaked Y, et al. CCR5 Directs the Mobilization of CD11b(+)Gr1(+)Ly6C(low) Polymorphonuclear Myeloid Cells from the Bone Marrow to the Blood to Support Tumor Development. Cell Rep (2017) 21:2212–22. doi: 10.1016/j.celrep.2017.10.104
17. Edwards J, Wilmott JS, Madore J, Gide TN, Quek C, Tasker A, et al. CD103(+) Tumor-Resident CD8(+) T Cells Are Associated with Improved Survival in Immunotherapy-Naive Melanoma Patients and Expand Significantly During Anti-PD-1 Treatment. Clin Cancer Res (2018) 24:3036–45. doi: 10.1158/1078-0432.CCR-17-2257
18. Corgnac S, Boutet M, Kfoury M, Naltet C, Mami-Chouaib F. The Emerging Role of CD8(+) Tissue Resident Memory T (TRM) Cells in Antitumor Immunity: A Unique Functional Contribution of the CD103 Integrin. Front Immunol (2018) 9:1904. doi: 10.3389/fimmu.2018.01904
19. Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol (2013) 14:1294–301. doi: 10.1038/ni.2744
20. Annacker O, Coombes JL, Malmstrom V, Uhlig HH, Bourne T, Johansson-Lindbom B, et al. Essential role for CD103 in the T cell-mediated regulation of experimental colitis. J Exp Med (2005) 202:1051–61. doi: 10.1084/jem.20040662
21. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol (2014) 32:659–702. doi: 10.1146/annurev-immunol-032713-120145
22. Luster AD. Chemokines–chemotactic cytokines that mediate inflammation. N Engl J Med (1998) 338:436–45. doi: 10.1056/NEJM199802123380706
23. Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med (2006) 354:610–21. doi: 10.1056/NEJMra052723
24. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun (2016) 7:12150. doi: 10.1038/ncomms12150
25. Mantovani A, Sica A, Allavena P, Garlanda C, Locati M. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum Immunol (2009) 70:325–30. doi: 10.1016/j.humimm.2009.02.008
26. Brandau S, Moses K, Lang S. The kinship of neutrophils and granulocytic myeloid-derived suppressor cells in cancer: cousins, siblings or twins? Semin Cancer Biol (2013) 23:171–82. doi: 10.1016/j.semcancer.2013.02.007
27. Ben-Meir K, Twaik N, Baniyash M. Plasticity and biological diversity of myeloid derived suppressor cells. Curr Opin Immunol (2018) 51:154–61. doi: 10.1016/j.coi.2018.03.015
28. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
29. Frankenberger C, Rabe D, Bainer R, Sankarasharma D, Chada K, Krausz T, et al. Metastasis Suppressors Regulate the Tumor Microenvironment by Blocking Recruitment of Prometastatic Tumor-Associated Macrophages. Cancer Res (2015) 75:4063–73. doi: 10.1158/0008-5472.CAN-14-3394
30. Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res (2006) 66:605–12. doi: 10.1158/0008-5472.CAN-05-4005
31. Bolat F, Kayaselcuk F, Nursal TZ, Yagmurdur MC, Bal N, Demirhan B, et al. expression, and tumor-associated macrophages in breast tumors: correlations with prognostic parameters. J Exp Clin Cancer Res (2006) 25:365–72.
32. Saji H, Koike M, Yamori T, Saji S, Seiki M, Matsushima K, et al. Significant correlation of monocyte chemoattractant protein-1 expression with neovascularization and progression of breast carcinoma. Cancer (2001) 92:1085–91. doi: 10.1002/1097-0142(20010901)92:5<1085::AID-CNCR1424>3.0.CO;2-K
33. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature (2011) 475:222–5. doi: 10.1038/nature10138
34. Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res (2006) 66:11238–46. doi: 10.1158/0008-5472.CAN-06-1278
35. Izhak L, Wildbaum G, Jung S, Stein A, Shaked Y, Karin N. Dissecting the autocrine and paracrine roles of the CCR2-CCL2 axis in tumor survival and angiogenesis. PloS One (2012) 7:e28305. doi: 10.1371/journal.pone.0028305
36. Meyer M, Hensbergen PJ, van der Raaij-Helmer EM, Brandacher G, Margreiter R, Heufler C, et al. Cross reactivity of three T cell attracting murine chemokines stimulating the CXC chemokine receptor CXCR3 and their induction in cultured cells and during allograft rejection. Eur J Immunol (2001) 31:2521–7. doi: 10.1002/1521-4141(200108)31:8<2521::AID-IMMU2521>3.0.CO;2-Q
37. Porta C, Consonni FM, Morlacchi S, Sangaletti S, Bleve A, Totaro MG, et al. Tumor-Derived Prostaglandin E2 Promotes p50 NF-kappaB-Dependent Differentiation of Monocytic MDSCs. Cancer Res (2020) 80:2874–88. doi: 10.1158/0008-5472.CAN-19-2843
38. Travelli C, Consonni FM, Sangaletti S, Storto M, Morlacchi S, Grolla AA, et al. Nicotinamide Phosphoribosyltransferase Acts as a Metabolic Gate for Mobilization of Myeloid-Derived Suppressor Cells. Cancer Res (2019) 79:1938–51. doi: 10.1158/0008-5472.CAN-18-1544
39. Su Y, Qiu Y, Qiu Z, Qu P. MicroRNA networks regulate the differentiation, expansion and suppression function of myeloid-derived suppressor cells in tumor microenvironment. J Cancer (2019) 10:4350–6. doi: 10.7150/jca.35205
40. Guo X, Qiu W, Wang J, Liu Q, Qian M, Wang S, et al. Glioma exosomes mediate the expansion and function of myeloid-derived suppressor cells through microRNA-29a/Hbp1 and microRNA-92a/Prkar1a pathways. Int J Cancer (2019) 144:3111–26. doi: 10.1002/ijc.32052
41. Li L, Zhang J, Diao W, Wang D, Wei Y, Zhang CY, et al. MicroRNA-155 and MicroRNA-21 promote the expansion of functional myeloid-derived suppressor cells. J Immunol (2014) 192:1034–43. doi: 10.4049/jimmunol.1301309
42. Gabrilovich DI. Myeloid-Derived Suppressor Cells. Cancer Immunol Res (2017) 5:3–8. doi: 10.1158/2326-6066.CIR-16-0297
43. Condamine T, Mastio J, Gabrilovich DI. Transcriptional regulation of myeloid-derived suppressor cells. J Leukoc Biol (2015) 98:913–22. doi: 10.1189/jlb.4RI0515-204R
44. Condamine T, Kumar V, Ramachandran IR, Youn JI, Celis E, Finnberg N, et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J Clin Invest (2014) 124:2626–39. doi: 10.1172/JCI74056
45. Nagaraj S, Schrum AG, Cho HI, Celis E, Gabrilovich DI. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol (2010) 184:3106–16. doi: 10.4049/jimmunol.0902661
46. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med (2010) 207:2439–53. doi: 10.1084/jem.20100587
47. Ku AW, Muhitch JB, Powers CA, Diehl M, Kim M, Fisher DT, et al. Tumor-induced MDSC act via remote control to inhibit L-selectin-dependent adaptive immunity in lymph nodes. Elife (2016) 5:e1375. doi: 10.7554/eLife.17375
48. Wang Z, Zhou Q, Zeng H, Zhang H, Liu Z, Huang Q, et al. Tumor-infiltrating IL-17A(+) cells determine favorable prognosis and adjuvant chemotherapeutic response in muscle-invasive bladder cancer. Oncoimmunology (2020) 9:1747332. doi: 10.1080/2162402X.2020.1747332
49. Frasci G, Comella P, Carreca I, DeCataldis G, Muci D, Brunetti C, et al. Weekly dose-dense cisplatin-epirubicin-paclitaxel administration with granulocyte colony-stimulating factor support does not substantially improve prognosis in extensive disease small-cell lung cancer. A SICOG Phase II Study Oncol (2005) 68:223–9. doi: 10.1159/000086778
50. Brockstein B, Haraf DJ, Stenson K, Sulzen L, Witt ME, Weichselbaum RW, et al. A phase I-II study of concomitant chemoradiotherapy with paclitaxel (one-hour infusion), 5-fluorouracil and hydroxyurea with granulocyte colony stimulating factor support for patients with poor prognosis head and neck cancer. Ann Oncol (2000) 11:721–8. doi: 10.1023/A:1008324131519
51. Brockstein B, Haraf DJ, Stenson K, Fasanmade A, Stupp R, Glisson B, et al. Phase I study of concomitant chemoradiotherapy with paclitaxel, fluorouracil, and hydroxyurea with granulocyte colony-stimulating factor support for patients with poor-prognosis cancer of the head and neck. J Clin Oncol (1998) 16:735–44. doi: 10.1200/JCO.1998.16.2.735
52. Zhang N, Wang Q, Tian Y, Xiong S, Li G, Xu L. Expressions of IL-17 and TNF-alpha in patients with Hashimoto’s disease combined with thyroid cancer before and after surgery and their relationship with prognosis. Clin Transl Oncol (2020) 22:1280–7. doi: 10.1007/s12094-019-02253-1
53. Gao Y, Wang Y, Wang X, Wang Y, Zhang X, Sun X. TNF-like ligand 1A is associated with progression and prognosis of human gastric cancer. Onco Targets Ther (2019) 12:7715–23. doi: 10.2147/OTT.S210939
54. Mikami S, Mizuno R, Kosaka T, Saya H, Oya M, Okada Y. Expression of TNF-alpha and CD44 is implicated in poor prognosis, cancer cell invasion, metastasis and resistance to the sunitinib treatment in clear cell renal cell carcinomas. Int J Cancer (2015) 136:1504–14. doi: 10.1002/ijc.29137
55. Tahara T, Shibata T, Nakamura M, Yamashita H, Yoshioka D, Okubo M, et al. Effect of IL-1beta and TNF-alpha polymorphisms on the prognosis and survival of gastric cancer patients. Clin Exp Med (2011) 11:211–7. doi: 10.1007/s10238-010-0129-y
56. Takagi K, Tomita K, Fukushima Y, Chikamoto A, Kanou J, Honda T, et al. Endogenous TNF inducibility and prognosis of colorectal cancer. Anticancer Res (1998) 18:4141–6.
57. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity (2003) 19:71–82. doi: 10.1016/S1074-7613(03)00174-2
58. Zhang J, Yan Y, Cui X, Zhang J, Yang Y, Li H, et al. CCL2 expression correlates with Snail expression and affects the prognosis of patients with gastric cancer. Pathol Res Pract (2017) 213:217–21. doi: 10.1016/j.prp.2016.12.013
59. Shan J, Chouchane A, Mokrab Y, Saad M, Boujassoum S, Sayaman RW, et al. Genetic Variation in CCL5 Signaling Genes and Triple Negative Breast Cancer: Susceptibility and Prognosis Implications. Front Oncol (2019) 9:1328. doi: 10.3389/fonc.2019.01328
60. Wang T, Wei Y, Tian L, Song H, Ma Y, Yao Q, et al. C-C motif chemokine ligand 5 (CCL5) levels in gastric cancer patient sera predict occult peritoneal metastasis and a poorer prognosis. Int J Surg (2016) 32:136–42. doi: 10.1016/j.ijsu.2016.07.008
61. Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res (2016) 76:5671–82. doi: 10.1158/0008-5472.CAN-16-0144
62. Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye-Ogala R, Arisa S, et al. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin Cancer Res (2017) 23:5187–201. doi: 10.1158/1078-0432.CCR-17-0741
63. Wang B, Palasanthiran P, Zeigler J, Cunningham A, Saksena NK. CCR5-delta 32 gene deletion in HIV-1 infected patients. Lancet (1997) 350:742. doi: 10.1016/S0140-6736(05)63554-4
64. Balistreri CR, Carruba G, Calabro M, Campisi I, Di Carlo D, Lio D, et al. CCR5 proinflammatory allele in prostate cancer risk: a pilot study in patients and centenarians from Sicily. Ann N Y Acad Sci (2009) 1155:289–92. doi: 10.1111/j.1749-6632.2008.03691.x
65. Dyer DP, Medina-Ruiz L, Bartolini R, Schuette F, Hughes CE, Pallas K, et al. Chemokine Receptor Redundancy and Specificity Are Context Dependent. Immunity (2019) 50:378–89.e5. doi: 10.1016/j.immuni.2019.01.009
66. Al Sayed MF, Amrein MA, Buhrer ED, Huguenin AL, Radpour R, Riether C, et al. T-cell-Secreted TNFalpha Induces Emergency Myelopoiesis and Myeloid-Derived Suppressor Cell Differentiation in Cancer. Cancer Res (2019) 79:346–59. doi: 10.1158/0008-5472.CAN-17-3026
67. Liu B, Tan W, Barsoum A, Gu X, Chen K, Huang W, et al. IL-17 is a potent synergistic factor with GM-CSF in mice in stimulating myelopoiesis, dendritic cell expansion, proliferation, and functional enhancement. Exp Hematol (2010) 38:877–84.e1. doi: 10.1016/j.exphem.2010.06.004
68. Inamoto S, Itatani Y, Yamamoto T, Minamiguchi S, Hirai H, Iwamoto M, et al. Loss of SMAD4 Promotes Colorectal Cancer Progression by Accumulation of Myeloid-Derived Suppressor Cells through the CCL15-CCR1 Chemokine Axis. Clin Cancer Res (2016) 22:492–501. doi: 10.1158/1078-0432.CCR-15-0726
69. Li Y, Wu J, Zhang P. CCL15/CCR1 axis is involved in hepatocellular carcinoma cells migration and invasion. Tumour Biol (2016) 37:4501–7. doi: 10.1007/s13277-015-4287-0
70. Okuma A, Hanyu A, Watanabe S, Hara E. p16(Ink4a) and p21(Cip1/Waf1) promote tumour growth by enhancing myeloid-derived suppressor cells chemotaxis. Nat Commun (2017) 8:2050. doi: 10.1038/s41467-017-02281-x
71. Toh B, Wang X, Keeble J, Sim WJ, Khoo K, Wong WC, et al. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PloS Biol (2011) 9:e1001162. doi: 10.1371/journal.pbio.1001162
72. Ding Y, Shen J, Zhang G, Chen X, Wu J, Chen W. CD40 controls CXCR5-induced recruitment of myeloid-derived suppressor cells to gastric cancer. Oncotarget (2015) 6:38901–11. doi: 10.18632/oncotarget.5644
73. Li BH, Garstka MA, Li ZF. Chemokines and their receptors promoting the recruitment of myeloid-derived suppressor cells into the tumor. Mol Immunol (2020) 117:201–15. doi: 10.1016/j.molimm.2019.11.014
74. Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol (2005) 6:1182–90. doi: 10.1038/ni1275
75. Lasky LA, Singer MS, Dowbenko D, Imai Y, Henzel WJ, Grimley C, et al. An endothelial ligand for L-selectin is a novel mucin-like molecule. Cell (1992) 69:927–38. doi: 10.1016/0092-8674(92)90612-G
76. Schlager C, Korner H, Krueger M, Vidoli S, Haberl M, Mielke D, et al. Effector T-cell trafficking between the leptomeninges and the cerebrospinal fluid. Nature (2016) 530:349–53. doi: 10.1038/nature16939
77. Feng L, Qi Q, Wang P, Chen H, Chen Z, Meng Z, et al. Serum level of CCL2 predicts outcome of patients with pancreatic cancer. Acta Gastroenterol Belg (2020) 83:295–9.
78. Eckstein M, Epple E, Jung R, Weigelt K, Lieb V, Sikic D, et al. CCL2 Expression in Tumor Cells and Tumor-Infiltrating Immune Cells Shows Divergent Prognostic Potential for Bladder Cancer Patients Depending on Lymph Node Stage. Cancers (Basel) (2020) 12:1253. doi: 10.3390/cancers12051253
79. Heiskala M, Leidenius M, Joensuu K, Heikkila P. High expression of CCL2 in tumor cells and abundant infiltration with CD14 positive macrophages predict early relapse in breast cancer. Virchows Arch (2019) 474:3–12. doi: 10.1007/s00428-018-2461-7
80. Yang Y, Zhai C, Chang Y, Zhou L, Shi T, Tan C, et al. High expression of chemokine CCL2 is associated with recurrence after surgery in clear-cell renal cell carcinoma. Urol Oncol (2016) 34:238.e19–26. doi: 10.1016/j.urolonc.2015.11.026
81. Tsaur I, Noack A, Makarevic J, Oppermann E, Waaga-Gasser AM, Gasser M, et al. CCL2 Chemokine as a Potential Biomarker for Prostate Cancer: A Pilot Study. Cancer Res Treat (2015) 47:306–12. doi: 10.4143/crt.2014.015
82. Fader AN, Rasool N, Vaziri SA, Kozuki T, Faber PW, Elson P, et al. CCL2 expression in primary ovarian carcinoma is correlated with chemotherapy response and survival outcomes. Anticancer Res (2010) 30:4791–8.
83. Zijlmans HJ, Fleuren GJ, Baelde HJ, Eilers PH, Kenter GG, Gorter A. The absence of CCL2 expression in cervical carcinoma is associated with increased survival and loss of heterozygosity at 17q11.2. J Pathol (2006) 208:507–17. doi: 10.1002/path.1918
84. Fujimoto Y, Inoue N, Morimoto K, Watanabe T, Hirota S, Imamura M, et al. Significant association between high serum CCL5 levels and better disease-free survival of patients with early breast cancer. Cancer Sci (2020) 111:209–18. doi: 10.1111/cas.14234
85. Suenaga M, Stintzing S, Cao S, Zhang W, Yang D, Ning Y, et al. Role of CCL5 and CCR5 gene polymorphisms in epidermal growth factor receptor signalling blockade in metastatic colorectal cancer: analysis of the FIRE-3 trial. Eur J Cancer (2019) 107:100–14. doi: 10.1016/j.ejca.2018.11.019
86. Kranjc MK, Novak M, Pestell RG, Lah TT. Cytokine CCL5 and receptor CCR5 axis in glioblastoma multiforme. Radiol Oncol (2019) 53:397–406. doi: 10.2478/raon-2019-0057
87. Derossi DR, Amarante MK, Guembarovski RL, de Oliveira CEC, Suzuki KM, Watanabe MAE, et al. CCL5 protein level: influence on breast cancer staging and lymph nodes commitment. Mol Biol Rep (2019) 46:6165–70. doi: 10.1007/s11033-019-05051-8
88. Sun K, Gong C, Peng H, Fang H, Zhou J, Li J, et al. High CCL5 expression is associated with osteosarcoma metastasis and poor prognosis of patients with osteosarcoma. Mol Med Rep (2017) 16:6953–7. doi: 10.3892/mmr.2017.7458
89. Zhang Q, Qin J, Zhong L, Gong L, Zhang B, Zhang Y, et al. CCL5-Mediated Th2 Immune Polarization Promotes Metastasis in Luminal Breast Cancer. Cancer Res (2015) 75:4312–21. doi: 10.1158/0008-5472.CAN-14-3590
90. Sadeghi M, Lahdou I, Oweira H, Daniel V, Terness P, Schmidt J, et al. Serum levels of chemokines CCL4 and CCL5 in cirrhotic patients indicate the presence of hepatocellular carcinoma. Br J Cancer (2015) 113:756–62. doi: 10.1038/bjc.2015.227
91. Yaal-Hahoshen N, Shina S, Leider-Trejo L, Barnea I, Shabtai EL, Azenshtein E, et al. The chemokine CCL5 as a potential prognostic factor predicting disease progression in stage II breast cancer patients. Clin Cancer Res (2006) 12:4474–80. doi: 10.1158/1078-0432.CCR-06-0074
92. Yin X, Han S, Song C, Zou H, Wei Z, Xu W, et al. Metformin enhances gefitinib efficacy by interfering with interactions between tumor-associated macrophages and head and neck squamous cell carcinoma cells. Cell Oncol (Dordr) (2019) 42:459–75. doi: 10.1007/s13402-019-00446-y
93. Liang Q, Tang C, Tang M, Zhang Q, Gao Y, Ge Z. TRIM47 is up-regulated in colorectal cancer, promoting ubiquitination and degradation of SMAD4. J Exp Clin Cancer Res (2019) 38:159. doi: 10.1186/s13046-019-1143-x
94. Yamamoto T, Kawada K, Itatani Y, Inamoto S, Okamura R, Iwamoto M, et al. Loss of SMAD4 Promotes Lung Metastasis of Colorectal Cancer by Accumulation of CCR1+ Tumor-Associated Neutrophils through CCL15-CCR1 Axis. Clin Cancer Res (2017) 23:833–44. doi: 10.1158/1078-0432.CCR-16-0520
95. Raja UM, Gopal G, Shirley S, Ramakrishnan AS, Rajkumar T. Immunohistochemical expression and localization of cytokines/chemokines/growth factors in gastric cancer. Cytokine (2017) 89:82–90. doi: 10.1016/j.cyto.2016.08.032
96. Li Y, Yu HP, Zhang P. CCL15 overexpression predicts poor prognosis for hepatocellular carcinoma. Hepatol Int (2016) 10:488–92. doi: 10.1007/s12072-015-9683-4
97. Bodelon C, Polley MY, Kemp TJ, Pesatori AC, McShane LM, Caporaso NE, et al. Circulating levels of immune and inflammatory markers and long versus short survival in early-stage lung cancer. Ann Oncol (2013) 24:2073–9. doi: 10.1093/annonc/mdt175
98. Lan Q, Lai W, Zeng Y, Liu L, Li S, Jin S, et al. CCL26 Participates in the PRL-3-Induced Promotion of Colorectal Cancer Invasion by Stimulating Tumor-Associated Macrophage Infiltration. Mol Cancer Ther (2018) 17:276–89. doi: 10.1158/1535-7163.MCT-17-0507
99. Zhang R, Liu Q, Peng J, Wang M, Li T, Liu J, et al. CXCL5 overexpression predicts a poor prognosis in pancreatic ductal adenocarcinoma and is correlated with immune cell infiltration. J Cancer (2020) 11:2371–81. doi: 10.7150/jca.40517
100. Alafate W, Li X, Zuo J, Zhang H, Xiang J, Wu W, et al. Elevation of CXCL1 indicates poor prognosis and radioresistance by inducing mesenchymal transition in glioblastoma. CNS Neurosci Ther (2020) 26:475–85. doi: 10.1111/cns.13297
101. Yu S, Yi M, Xu L, Qin S, Li A, Wu K. CXCL1 as an Unfavorable Prognosis Factor Negatively Regulated by DACH1 in Non-small Cell Lung Cancer. Front Oncol (2019) 9:1515. doi: 10.3389/fonc.2019.01515
102. Yamamoto Y, Kuroda K, Sera T, Sugimoto A, Kushiyama S, Nishimura S, et al. The Clinicopathological Significance of the CXCR2 Ligands, CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7, and CXCL8 in Gastric Cancer. Anticancer Res (2019) 39:6645–52. doi: 10.21873/anticanres.13879
103. Lu Y, Dong B, Xu F, Xu Y, Pan J, Song J, et al. CXCL1-LCN2 paracrine axis promotes progression of prostate cancer via the Src activation and epithelial-mesenchymal transition. Cell Commun Signal (2019) 17:118. doi: 10.1186/s12964-019-0434-3
104. Yildirim K, Colak E, Aktimur R, Gun S, Taskin MH, Nigdelioglu A, et al. Clinical Value of CXCL5 for Determining of Colorectal Cancer. Asian Pac J Cancer Prev (2018) 19:2481–4.
105. Zhao J, Ou B, Han D, Wang P, Zong Y, Zhu C, et al. Tumor-derived CXCL5 promotes human colorectal cancer metastasis through activation of the ERK/Elk-1/Snail and AKT/GSK3beta/beta-catenin pathways. Mol Cancer (2017) 16:70. doi: 10.1186/s12943-017-0629-4
106. Zhu X, Qiao Y, Liu W, Wang W, Shen H, Lu Y, et al. CXCL5 is a potential diagnostic and prognostic marker for bladder cancer patients. Tumour Biol (2016) 37:4569–77. doi: 10.1007/s13277-015-4275-4
107. le Rolle AF, Chiu TK, Fara M, Shia J, Zeng Z, Weiser MR, et al. The prognostic significance of CXCL1 hypersecretion by human colorectal cancer epithelia and myofibroblasts. J Transl Med (2015) 13:199. doi: 10.1186/s12967-015-0555-4
108. Zheng Z, Cai Y, Chen H, Chen Z, Zhu D, Zhong Q, et al. CXCL13/CXCR5 Axis Predicts Poor Prognosis and Promotes Progression Through PI3K/AKT/mTOR Pathway in Clear Cell Renal Cell Carcinoma. Front Oncol (2018) 8:682. doi: 10.3389/fonc.2018.00682
109. Wei Y, Lin C, Li H, Xu Z, Wang J, Li R, et al. CXCL13 expression is prognostic and predictive for postoperative adjuvant chemotherapy benefit in patients with gastric cancer. Cancer immunology immunotherapy CII (2018) 67:261–9. doi: 10.1007/s00262-017-2083-y
110. Kim SJ, Ryu KJ, Hong M, Ko YH, Kim WS. The serum CXCL13 level is associated with the Glasgow Prognostic Score in extranodal NK/T-cell lymphoma patients. J Hematol Oncol (2015) 8:49. doi: 10.1186/s13045-015-0200-y
111. Duan Z, Gao J, Zhang L, Liang H, Huang X, Xu Q, et al. Phenotype and function of CXCR5+CD45RA-CD4+ T cells were altered in HBV-related hepatocellular carcinoma and elevated serum CXCL13 predicted better prognosis. Oncotarget (2015) 6:44239–53. doi: 10.18632/oncotarget.6235
112. Chen L, Huang Z, Yao G, Lyu X, Li J, Hu X, et al. The expression of CXCL13 and its relation to unfavorable clinical characteristics in young breast cancer. J Transl Med (2015) 13:168. doi: 10.1186/s12967-015-0521-1
113. Hussain SK, Zhu W, Chang SC, Breen EC, Vendrame E, Magpantay L, et al. Serum levels of the chemokine CXCL13, genetic variation in CXCL13 and its receptor CXCR5, and HIV-associated non-hodgkin B-cell lymphoma risk. Cancer Epidemiol Biomarkers Prev (2013) 22:295–307. doi: 10.1158/1055-9965.EPI-12-1122
114. Panse J, Friedrichs K, Marx A, Hildebrandt Y, Luetkens T, Barrels K, et al. Chemokine CXCL13 is overexpressed in the tumour tissue and in the peripheral blood of breast cancer patients. Br J Cancer (2008) 99:930–8. doi: 10.1038/sj.bjc.6604621
115. Vanheule V, Metzemaekers M, Janssens R, Struyf S, Proost P. How post-translational modifications influence the biological activity of chemokines. Cytokine (2018) 109:29–51. doi: 10.1016/j.cyto.2018.02.026
116. Sapir Y, Vitenshtein A, Barsheshet Y, Zohar Y, Wildbaum G, Karin N. A fusion protein encoding the second extracellular domain of CCR5 arrests chemokine-induced cosignaling and effectively suppresses ongoing experimental autoimmune encephalomyelitis. J Immunol (2010) 185:2589–99. doi: 10.4049/jimmunol.1000666
117. Blattner C, Fleming V, Weber R, Himmelhan B, Altevogt P, Gebhardt C, et al. CCR5(+) Myeloid-Derived Suppressor Cells Are Enriched and Activated in Melanoma Lesions. Cancer Res (2018) 78:157–67. doi: 10.1158/0008-5472.CAN-17-0348
118. Tang Q, Jiang J, Liu J. CCR5 Blockade Suppresses Melanoma Development Through Inhibition of IL-6-Stat3 Pathway via Upregulation of SOCS3. Inflammation (2015) 38:2049–56. doi: 10.1007/s10753-015-0186-1
119. Zhang Y, Lv D, Kim HJ, Kurt RA, Bu W, Li Y, et al. A novel role of hematopoietic CCL5 in promoting triple-negative mammary tumor progression by regulating generation of myeloid-derived suppressor cells. Cell Res (2013) 23:394–408. doi: 10.1038/cr.2012.178
120. Yang L, Wang B, Qin J, Zhou H, Majumdar APN, Peng F. Blockade of CCR5-mediated myeloid derived suppressor cell accumulation enhances anti-PD1 efficacy in gastric cancer. Immunopharmacol Immunotoxicol (2018) 40:91–7. doi: 10.1080/08923973.2017.1417997
121. Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez-Carmona M, et al. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell (2016) 29:587–601. doi: 10.1016/j.ccell.2016.03.005
122. D’Aveni M, Notarantonio AB, Bertrand A, Boulange L, Pochon C, Rubio MT. Myeloid-Derived Suppressor Cells in the Context of Allogeneic Hematopoietic Stem Cell Transplantation. Front Immunol (2020) 11:989. doi: 10.3389/fimmu.2020.00989
123. Alshetaiwi H, Pervolarakis N, McIntyre LL, Ma D, Nguyen Q, Rath JA, et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci Immunol (2020) 5:eaay6017. doi: 10.1126/sciimmunol.aay6017
124. Metzger P, Kirchleitner SV, Kluge M, Koenig LM, Horth C, Rambuscheck CA, et al. Immunostimulatory RNA leads to functional reprogramming of myeloid-derived suppressor cells in pancreatic cancer. J Immunother Cancer (2019) 7:288. doi: 10.1186/s40425-019-0778-7
125. Roussel M, Ferrell Jr PB, Greenplate AR, Lhomme F, Le Gallou S, Diggins KE, et al. Mass cytometry deep phenotyping of human mononuclear phagocytes and myeloid-derived suppressor cells from human blood and bone marrow. J Leukoc Biol (2017) 102:437–47. doi: 10.1189/jlb.5MA1116-457R
Keywords: CCR5, CCR2 chemokines, cancer, myeloid derived suppressor cells, chemokine
Citation: Karin N (2020) The Development and Homing of Myeloid-Derived Suppressor Cells: From a Two-Stage Model to a Multistep Narrative. Front. Immunol. 11:557586. doi: 10.3389/fimmu.2020.557586
Received: 30 April 2020; Accepted: 03 September 2020;
Published: 26 October 2020.
Edited by:
Paolo Serafini, University of Miami, United StatesReviewed by:
Stefano Ugel, University of Verona, ItalyYulia Nefedova, Wistar Institute, United States
Copyright © 2020 Karin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nathan Karin, nkarin10@gmail.com