 Guang Ji1,2
Guang Ji1,2 Ning Wang1,2
Ning Wang1,2 Xu Han1,2
Xu Han1,2 Yaye Wang1,2Jinru Zhang1,2Yue Wu1,2Hongran Wu1,2Shaojuan Ma1,2Xueqin Song1,2*
Yaye Wang1,2Jinru Zhang1,2Yue Wu1,2Hongran Wu1,2Shaojuan Ma1,2Xueqin Song1,2*- 1Department of Neurology, Second Hospital of Hebei Medical University, Shijiazhuang, China
- 2Neurological Laboratory of Hebei Province, Shijiazhuang, China
DNAJB6 was identified as the causative gene of limb-girdle muscular dystrophy type 1D. In recent years, the phenotypic and molecular spectrum of DNAJB6-myopathy has been expanded, and several mutations of DNAJB6 have been identified in Europe, North America, and Asia. Interestingly, almost all identified mutations in previous reports were point mutations, and most of them were clustered in exon 5, which encodes the G/F domain of DNAJB6. The so-far unique splice site mutation eliminating the entire G/F domain was reported to cause a severe, early-onset phenotype. Here, we report a juvenile-onset Chinese patient who presented with proximal–distal myopathy as well as esotropia and facial weakness. Muscle pathology showed rimmed vacuolation and myofibrillar disarrangement. A novel splice-site mutation NM_058246:c.236-1_240delGGTGGA of the DNAJB6 gene was identified by targeted exome sequencing, which results in a severe defect of the G/F domain. This rare mutation type expands the molecular spectrum of DNAJB6-myopathy and further underlines the importance of the G/F region.
Introduction
The DNAJB6 gene encodes the DNAJB6 protein, which is a member of the heat shock protein (HSP) 40 family of co-chaperones, interacting with the chaperone of HSP70 through their J domains (Kampinga and Craig, 2010). There are two isoforms of DNAJB6 distinguished by their C-terminal parts: DNAJB6a and DNAJB6b (Sarparanta et al., 2020). Both isoforms of DNAJB6 harbor the J domain at the N-terminus, the glycine/phenylalanine-rich (G/F) region, and a serine/threonine-rich (S/T) region that recognizes and binds to the substrates. DNAJB6 is ubiquitously expressed and is of great importance in neurodegenerative diseases by inhibiting the aggregation of misfolded proteins, such as α-synuclein, TDP-43, polyglutamine-containing huntingtin, Aβ42, etc. (Chuang et al., 2002; Sarparanta et al., 2012; Mansson et al., 2014; Stein et al., 2014). DNAJB6 was identified as the pathogenic gene of limb-girdle muscular dystrophy type 1D (LGMD1D) in 2012 (Harms et al., 2012; Sarparanta et al., 2012), which was characterized by an autosomal dominant–inherited, progressive proximal muscular weakness. Pathologically, all reported muscular disorders associated with DNAJB6 mutations have exhibited common histological features characterized by sarcomeric protein aggregation, autophagic vacuolization, and myofibrillar degeneration, so the DNAJB6 myopathy was also classified as myofibrillar myopathy for pathological classification.
When identified, DNAJB6 myopathy was originally known as a late-onset, slowly progressive disease, and most patients can keep walking independently even at old age. However, with the discovery of novel mutations, some mutations were found to cause an earlier, more severe, or distal-onset disease (Harms et al., 2012; Nam et al., 2015; Palmio et al., 2015; Ruggieri et al., 2015; Monies et al., 2016; Tsai et al., 2017; Kim et al., 2018). In recent years, several DNAJB6 mutations were identified in different regions, and the patients were mainly reported in Europe (Palmio et al., 2015; Ruggieri et al., 2015; Jonson et al., 2018) and North America (Suarez-Cedeno et al., 2014; Ruggieri et al., 2015; Nallamilli et al., 2018). In Asia, mutations causing DNAJB6 myopathy have been reported in Saudi Arabia (Monies et al., 2016; Bohlega et al., 2018), Korea (Nam et al., 2015; Kim et al., 2018), and Japan (Sato et al., 2013). For people of Han Chinese origin, a heterozygous mutation (p.Pro96Leu) in the DNAJB6 gene was first identified in a Taiwanese family (Tsai et al., 2017). Interestingly, regardless of regions and ethnic groups, the mutations reported before were clustered in exon 5, which encodes the G/F domain of DNAJB6, except for only two mutational sites in the DNAJB6 J domain identified as the causative gene of the disease (Palmio et al., 2020). Furthermore, almost all identified mutations in previous reports were point mutations, and the so-far unique splice site mutation eliminating the entire G/F domain which caused a severe, early-onset phenotype was reported in 2015 (Ruggieri et al., 2015). Here, we report a juvenile-onset Chinese patient who presented with a proximal–distal phenotype associated with a novel alternative splice-site mutation which leads to complete deletion or partial deletion of exon 5. This rare mutational type expands the molecular spectrum of DNAJB6-myopathy which further underlines the importance of the G/F region.
Case Description
Clinical Data
A 17-year-old female complained of fatigue for 2 years, especially in the lower limbs. She experienced weakness when squatting and climbing stairs, without disturbance when walking or performing daily activities involving the upper limbs. In the recent 3 months, the symptoms progressed, and she felt easy to trip and needed help in climbing upstairs. The patient had visited a primary hospital and performed an MRI of the brain and knee joints examination. The brain MRI was normal, but the knee joints MRI showed a small amount of effusion in the right knee cavity and atrophy of the right thigh and calf muscles; however, a definite diagnosis was not made, and the patient did not receive any therapeutic intervention (Table 1). Her parents and a 14-year-old brother showed no similar symptoms. The patient achieved normal developmental milestones and had no special past medical history but esotropia since childhood.

TABLE 1. The timeline with relevant data from the episode of care.
Neurological examination showed weakness of the upper and lower limbs, with both proximal and distal involved and the left side slightly more severe than the right. Atrophy of bilateral thenar and hypothenar could be observed. It also revealed bilateral esotropia and facial weakness. Ptosis, diplopia, and bulbar features were absent. Laboratory tests showed normal levels of serum CK. Electromyography showed myogenic damage with normal nerve conduction velocity. Electrocardiogram, cardiac ultrasonography, and lung function were normal. MRI of the muscles in the lower limbs showed fatty infiltration and muscle atrophy of the lower limb muscles, most prominent involvement in posterior muscles (Figure 1).
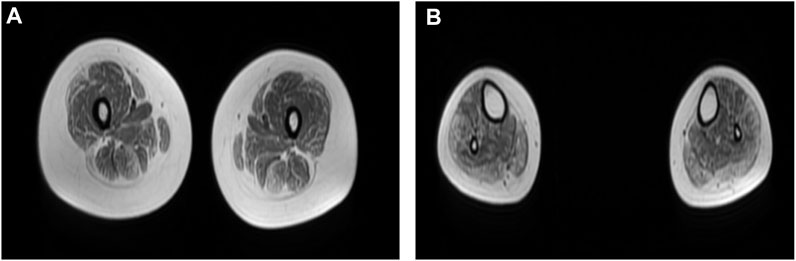
FIGURE 1. (A,B) Muscle MRI of lower limbs showed fatty infiltration and muscle atrophy of proximal and distal lower limb muscles, with the most prominent involvement in posterior muscles. (A) Thigh level. The biceps femoris (long head), semitendinosus, vastus lateralis, gracilis, and sartorius muscle are mildly to moderately affected. The rectus femoris is the best spared muscle. (B) Calf level. The gastrocnemius (medial head and lateral head), peroneus (fibularis) brevis, and peroneus (fibularis) longus muscle are moderately affected.
Pathological Findings
After informed consent, a muscle biopsy was performed on the left biceps brachii muscle of the patient. Histochemical staining of hematoxylin-eosin (HE) and modified Gomori trichrome (MGT) were processed according to traditional methods on frozen muscle sections. For immunohistochemical analysis, the following primary antibodies were applied to the muscle sections: desmin (ab8976, Abcam), p62 (P0067, Sigma-Aldrich), TDP-43 (12892-1-AP, Proteintech), LC3B (L7543, Sigma-Aldrich), and LAMP2 (#49067S, Cell Signaling Technology). Images were acquired using a light microscope (Olympus BX51, United States). For electron microscopy, 80 nm ultrathin sections were made, and images were acquired on a transmission electron microscope (TEM, JEM 1230, JEOL).
On light microscopy, HE and MGT staining (Figure 2A,B) revealed mildly proliferated connective tissue and adipose tissue in the perimysium and endomysium and fiber size variation with atrophic fibers. Rimmed vacuoles were found in a small portion of fibers. The eosinophilic cytoplasmic material deposition was also found in some fibers. On immunohistochemistry, the rimmed vacuoles showed positive reactivity for TDP-43, p62, and LAMP2 (Figure 2C–E), whereas components of LC3 reactivity were absent or less abundant in the rimmed vacuolar regions (Figure 2F), indicating impaired autophagic flux and defect degradation. Desmin reactivity showed moderate or strong expression in rimmed vacuolated fibers (Figure 2G). On electron microscopy, myelin bodies were found (Figure 2H), and myofibrillar structure was destroyed and replaced by loop debris materials in some regions (Figure 2I).
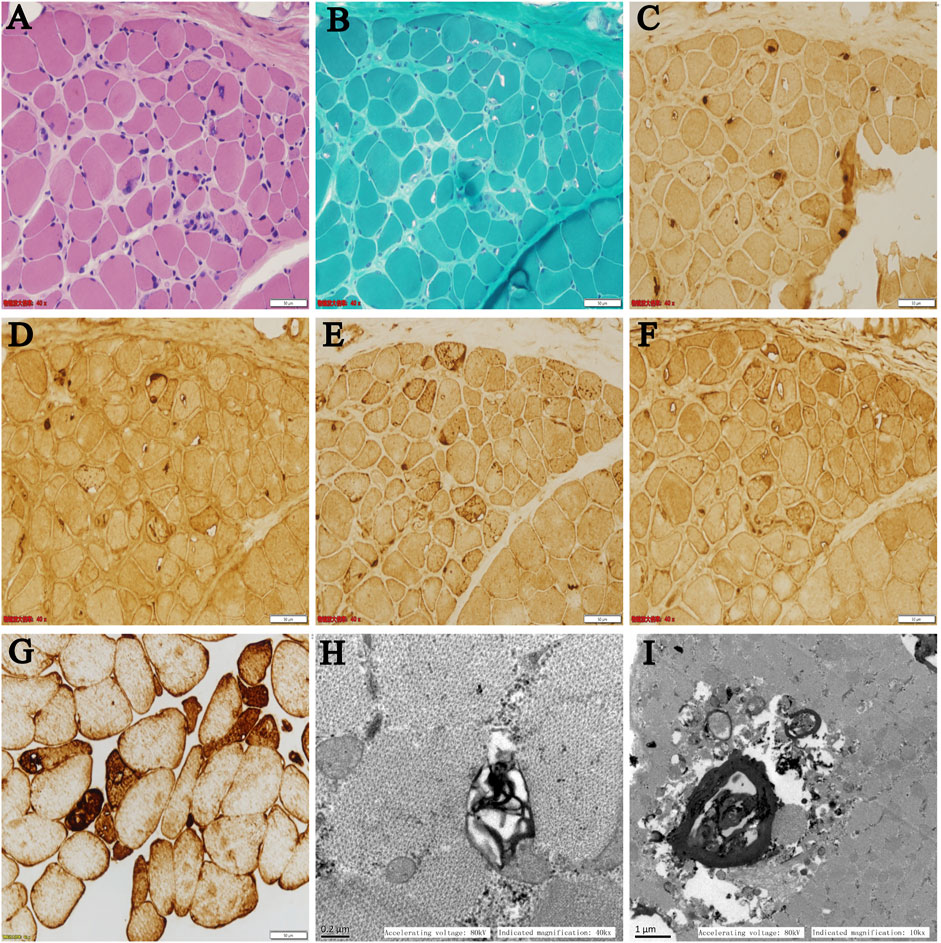
FIGURE 2. (A–F) Serial sections from the patient’s left biceps brachii muscle on light microscopy: (A) HE and (B) MGT staining revealed mildly proliferated connective tissue and fiber size variation with atrophic fibers. Rimmed vacuoles were found in a portion of the fibers. Immunohistochemistry showed positive reactivity for (C) TDP-43, (D) p62, and (E) LAMP2 in the rimmed vacuoles. (F) LC3 reactivity was absent or less abundant in the rimmed vacuolar regions. (G) Desmin reactivity showed moderate or strong expression in rimmed vacuolated fibers. (H,I) Ultrastructural changes on electron microscopy: (H) myelin bodies and (I) destroyed myofibrillar structure replaced by loop debris materials were found on transverse sections.
Targeted Exome Sequencing, Transcript Sequencing, and Molecular Modeling
Informed consent was signed by the patient’s parent for genetic analysis, and the peripheral blood of the patient and her parents was collected. Clinical exome sequencing analysis was accomplished by MyGenostics Inc., Beijing, China. A heterozygous mutation NM_058246:c.236-1_240delGGTGGA of the DNAJB6 gene was identified within exon 5 of DNAJB6 by genetic testing. The mutational site was confirmed by Sanger sequencing, and this site was normal in her parents (Figure 3A–C).
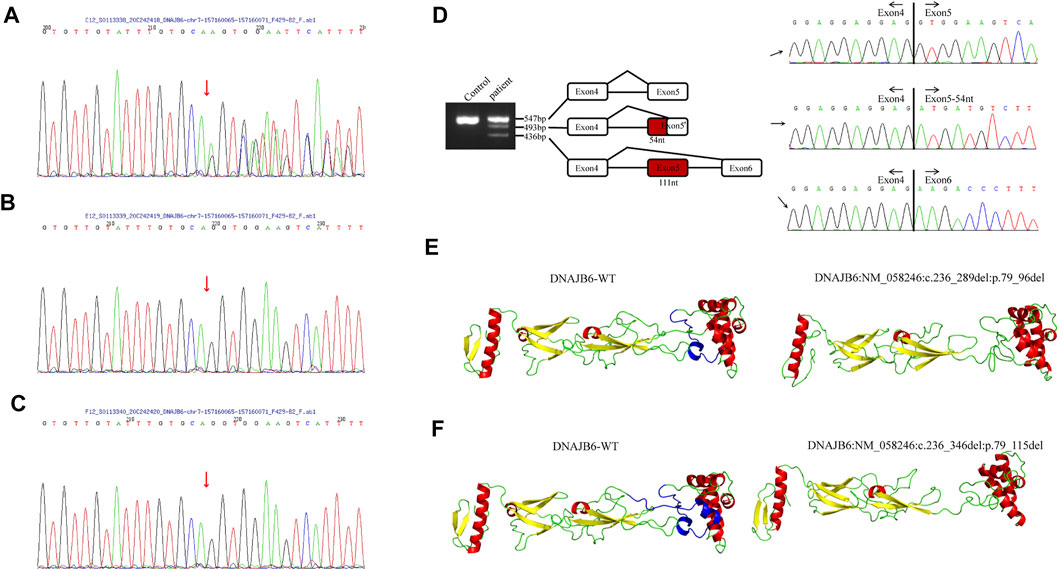
FIGURE 3. (A–C) The results of Sanger sequencing of the patient and her parents. (A) A heterozygous mutation c.236-1_240delGGTGGA (p.G79Efs*4) of the DNAJB6 gene was identified, and the mutational site of the patient was confirmed by Sanger sequencing. Sanger verification of her father (B) and mother (C) showed that this site was normal. (D). DNAJB6 transcript sequencing: Gel electrophoresis of the patient showed three different size bands, namely, 547, 493, and 436 bp. The 547 bp band is of the same size as the control one and is a wild-type transcript. By sequencing, the 493 bp band refers to 54-nucleotide deletion at the beginning of exon 5, and the 436 bp band refers to the whole 111-nucleotide delete of exon5, which encodes the G/F domain. (E,F) I-TASSER software was used to develop a suitable model to simulate the effect of the mutation region; the 3D protein modeling predicted that p.79_96del mutation would result in the loss of an α-helix (E) and p.79_115del mutation would result in the loss of two α-helices (F).
To verify the effect of variation on mRNA splicing, total RNA from the patient’s muscle tissues was extracted, and reverse transcription was performed. The RT-PCR products were separated by electrophoresis analysis and identified by TA cloning. Gel electrophoresis of the patient showed three different size bands, namely, 547, 493, and 436 bp (Figure 3D). The 547 bp band is of the same size as the control one and is a wild-type transcript. By sequencing, the 493 bp band refers to 54-nucleotide deletion at the beginning of exon 5, and the 436 bp band refers to the whole 111-nucleotide deletion of exon 5, which encodes the G/F domain (Figure 3D).
The I-TASSER software was used to develop a suitable model to simulate the effect of the mutation region (Zhang, 2008; Yang et al., 2015). The 3D protein modeling predicted that p.79_96del mutation would result in the loss of an α-helix (Figure 3E) and p.79_115del mutation would result in the loss of two α-helices (Figure 3F).
Discussion
Clinical Manifestation
DNAJB6 deficiency has been found to be associated with LGMD1D, a muscular disorder clinically characterized by slowly progressive proximal, limb-girdle muscle weakness. In recent years, however, mutations in the DNAJB6 gene have been reported to cause a wide spectrum of phenotypes based on the age of the onset, the severity of involvement, and the affected group of muscles. While most of the mutations have been reported to lead to a limb-girdle phenotype, some mutations were found to cause a distal myopathy or proximo-distal phenotype (Ruggieri et al., 2016; Sarparanta et al., 2020). The patient in this case also manifested a proximal–distal phenotype. Besides limb weakness, involvement of respiratory and bulbar muscles has been reported in some severe cases (Ruggieri et al., 2016), and facial weakness was also common in DNAJB6 myopathy (Ruggieri et al., 2015). Extraocular muscles have been spared in reported cases until now. Interestingly, esotropia was found in our patient, but the possibility that it was rather caused by extraocular muscles involvement in DNAJB6 myopathy or just a complicated disease remains unclear. Further MRI of the extraocular muscles of this patient was not performed, which may help distinguish the two situations.
Pathological Changes
On the pathological level, mutations in the DNAJB6 gene caused similar histological changes, including myofibrillar degeneration, protein accumulation, and autophagic vacuolation. Early changes in the pathology of DNAJB6 myopathy contain central myofibrillar lesions and Z-disc streaming followed by myofibrillar disintegration, and autophagic vacuoles appeared at later stages (Sandell et al., 2016). On light microscopy, DNAJB6 myopathy showed dystrophic or myopathic changes accompanied by rimmed vacuolation and myofibrillar aggregation in muscle pathology. This case showed definite rimmed vacuolar pathology; besides, eosinophilic cytoplasmic material deposition and the positive reactivity of desmin were also observed, which suggest myofibrillar aggregation. A previous study showed that the rimmed vacuoles were reactive for markers of impaired autophagy and defect degradation, such as LC3, p62, TDP-43, ubiquitin, and SMI-31 but do not stain for LAMP2, a marker of autophagosome–lysosome fusion (Sandell et al., 2016). However, the rimmed vacuoles in our patient were reactive for TDP-43, p62, and LAMP2, whereas the autophagosome marker LC3 reactivity was absent or less abundant, which was different from the previous study, suggesting that a variation in autophagy markers may appear in different patients or at different stages of the disease. On ultrastructure, these myofibrillar inclusions contained an abnormal accumulation of several proteins, such as myotilin, αB-crystallin, and desmin, and showed extensive myofibrillar disorganization with a mass of the Z-disk material. Pronounced larger pleomorphic myofibrillar aggregates were found in the later process of rare cases as major changes in rimmed vacuolar pathology (Sandell et al., 2016). Myelin bodies and myofibrillar disorganization were found in this case; however, large loop debris materials have not been defined, which may be formed by myofibrillar aggregation.
The Spectrum of Mutations
Until recently, 18 pathogenic mutations have been identified in DNAJB6 (Sarparanta et al., 2020). Interestingly, almost all identified mutations were clustered within exon 5 of the gene encoding a short stretch of amino acids in the G/F region, and only two pathogenic mutations (p.Ala50Val and p.Glu54Ala) in the J domain of DNAJB6 have been reported to cause distal or proximo-distal myopathies until now (Palmio et al., 2020), highlighting the G/F region as a mutational hot spot. Besides the patient in our report, another splice site mutation (c.346+5G>A) causing exon 5 skipping that eliminates the entire G/F domain has been reported, which further underlined the importance of the G/F region (Ruggieri et al., 2015). Currently, it is believed that the DNAJB6 G/F spiral folds play a critical role in the contact with the J domain (Ruggieri et al., 2015). By protein modeling, we predicted that the mutation in our patient would result in the loss of one or two α-helices of DNAJB6, showing evidence that the mutation in this patient may also act through disturbing these contacts, which is consistent with the previously reported DNAJB6 mutations (Ruggieri et al., 2015). However, the direct effect of the G/F domain defect on DNAJB6 myopathy is not clear. In addition, compared with the patient with a mutation that eliminates the entire G/F domain reported before, our patient developed symptoms later and presented with a proximal–distal phenotype without severe symptoms such as respiratory and bulbar involvement. This may be due to the alternative splicing only causing a small amount of abnormal mRNA, and the DNAJB6 protein, at least a part of it, still functions.
In addition, it has been known that LGMD1D was autosomal-dominantly inherited myopathy caused by gain-of-deleterious-function mutations in the DNAJB6 gene, and DNAJB6b was known as the pathogenic isoform. Interestingly, however, a homozygous mutation (p.Val232Gly fs*7) that localized to exon 9 of the DNAJB6 gene was reported to cause an autosomal-recessively inherited late-onset very recently (Qian et al., 2021). The novel mutation exclusively caused loss of the “a” region in DNAJB6a and resulted in a recessive toxic effect. These new findings uncovered the pathogenic role of DNAJB6a deficiency and indicated that many unclear issues remained to be studied on DNAJB6.
In summary, DNAJB6 myopathy is a rare muscular disorder with varying clinical phenotypes such as the age of the onset, the severity of disease, and the muscles involved. Although this disease is mainly characterized by slowly progressive proximal, limb-girdle muscle weakness, a distal myopathy or proximo-distal phenotype should also be noticed, especially when the rimmed vacuoles and myofibrillar aggregation were found in muscle pathology. Gene sequencing may help differentiate this from other muscular diseases.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Second Hospital of Hebei Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardians/next of kin.
Author Contributions
GJ designed the work and wrote the paper; NW, XH, YW, JZ, and YW collected clinical data; HW and SM performed pathological staining; XS designed the work and performed advisory supervision.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the cooperation of the patient and her parents. We are grateful to Shanghai We-Health Biomedical Technology Co. Ltd. and Beijing MyGenostics Inc. for their assistance in gene analysis techniques.
References
Bohlega, S. A., Alfawaz, S., Abou-Al-Shaar, H., Al-Hindi, H. N., Murad, H. N., Bohlega, M. S., et al. (2018). LGMD1D Myopathy with Cytoplasmic and Nuclear Inclusions in a Saudi Family Due to DNAJB6 Mutation. Acta Myol. 37 (3), 221–226.
Chuang, J.-Z., Zhou, H., Zhu, M., Li, S.-H., Li, X.-J., and Sung, C.-H. (2002). Characterization of a Brain-Enriched Chaperone, MRJ, that Inhibits Huntingtin Aggregation and Toxicity Independently. J. Biol. Chem. 277 (22), 19831–19838. doi:10.1074/jbc.m109613200
Harms, M. B., Sommerville, R. B., Allred, P., Bell, S., Ma, D., Cooper, P., et al. (2012). Exome Sequencing Reveals DNAJB6 Mutations in Dominantly-Inherited Myopathy. Ann. Neurol. 71 (3), 407–416. doi:10.1002/ana.22683
Jonson, P. H., Palmio, J., Johari, M., Penttilä, S., Evilä, A., Nelson, I., et al. (2018). Novel Mutations in DNAJB6 Cause LGMD1D and Distal Myopathy in French Families. Eur. J. Neurol. 25 (5), 790–794. doi:10.1111/ene.13598
Kampinga, H. H., and Craig, E. A. (2010). The HSP70 Chaperone Machinery: J Proteins as Drivers of Functional Specificity. Nat. Rev. Mol. Cell Biol. 11 (8), 579–592. doi:10.1038/nrm2941
Kim, K., Park, H. J., Lee, J. H., Hong, J., Ahn, S.-W., and Choi, Y.-C. (2018). Two Korean Families with Limb-Girdle Muscular Dystrophy Type 1D Associated with DNAJB6 Mutations. Yonsei Med. J. 59 (5), 698–701. doi:10.3349/ymj.2018.59.5.698
Månsson, C., Arosio, P., Hussein, R., Kampinga, H. H., Hashem, R. M., Boelens, W. C., et al. (2014). Interaction of the Molecular Chaperone DNAJB6 with Growing Amyloid-Beta 42 (Aβ42) Aggregates Leads to Sub-Stoichiometric Inhibition of Amyloid Formation. J. Biol. Chem. 289 (45), 31066–31076. doi:10.1074/jbc.m114.595124
Monies, D., Alhindi, H. N., Almuhaizea, M. A., Abouelhoda, M., Alazami, A. M., Goljan, E., et al. (2016). A First-Line Diagnostic Assay for Limb-Girdle Muscular Dystrophy and Other Myopathies. Hum. Genomics 10 (1), 32. doi:10.1186/s40246-016-0089-8
Nallamilli, B. R. R., Chakravorty, S., Kesari, A., Tanner, A., Ankala, A., Schneider, T., et al. (2018). Genetic Landscape and Novel Disease Mechanisms from a largeLGMDcohort of 4656 Patients. Ann. Clin. Transl. Neurol. 5 (12), 1574–1587. doi:10.1002/acn3.649
Nam, T.-S., Li, W., Heo, S.-H., Lee, K.-H., Cho, A., Shin, J.-H., et al. (2015). A Novel Mutation in DNAJB6, p.(Phe91Leu), in Childhood-Onset LGMD1D with a Severe Phenotypein Childhood-Onset LGMD1D with a Severe Phenotype. Neuromuscul. Disord. 25 (11), 843–851. doi:10.1016/j.nmd.2015.08.002
Palmio, J., Jonson, P. H., Evilä, A., Auranen, M., Straub, V., Bushby, K., et al. (2015). Novel Mutations in DNAJB6 Gene Cause a Very Severe Early-Onset Limb-Girdle Muscular Dystrophy 1D Disease. Neuromuscul. Disord. 25 (11), 835–842. doi:10.1016/j.nmd.2015.07.014
Palmio, J., Jonson, P. H., Inoue, M., Sarparanta, J., Bengoechea, R., Savarese, M., et al. (2020). Mutations in the J Domain of DNAJB6 Cause Dominant Distal Myopathy. Neuromuscul. Disord. 30 (1), 38–46. doi:10.1016/j.nmd.2019.11.005
Qian, F.-Y., Guo, Y.-D., Zu, J., Zhang, J.-H., Zheng, Y.-M., Abdoulaye, I. A., et al. (2021). A Novel Recessive Mutation Affecting DNAJB6a Causes Myofibrillar Myopathy. Acta Neuropathol. Commun. 9 (1), 23. doi:10.1186/s40478-020-01046-w
Ruggieri, A., Brancati, F., Zanotti, S., Maggi, L., Pasanisi, M. B., Saredi, S., et al. (2015). Complete Loss of the DNAJB6 G/F Domain and Novel Missense Mutations Cause Distal-Onset DNAJB6 Myopathy. Acta Neuropathol. Commun. 3, 44. doi:10.1186/s40478-015-0224-0
Ruggieri, A., Saredi, S., Zanotti, S., Pasanisi, M. B., Maggi, L., and Mora, M. (2016). DNAJB6 Myopathies: Focused Review on an Emerging and Expanding Group of Myopathies. Front. Mol. Biosci. 3, 63. doi:10.3389/fmolb.2016.00063
Sandell, S., Huovinen, S., Palmio, J., Raheem, O., Lindfors, M., Zhao, F., et al. (2016). Diagnostically Important Muscle Pathology in DNAJB6 Mutated LGMD1D. Acta Neuropathol. Commun. 4, 9. doi:10.1186/s40478-016-0276-9
Sarparanta, J., Jonson, P. H., Kawan, S., and Udd, B. (2020). Neuromuscular Diseases Due to Chaperone Mutations: A Review and Some New Results. Int. J. Mol. Sci. 21 (4), 1409. doi:10.3390/ijms21041409
Sarparanta, J., Jonson, P. H., Golzio, C., Sandell, S., Luque, H., Screen, M., et al. (2012). Mutations Affecting the Cytoplasmic Functions of the Co-chaperone DNAJB6 Cause Limb-Girdle Muscular Dystrophy. Nat. Genet. 44, 450–455. doi:10.1038/ng.1103
Sato, T., Hayashi, Y. K., Oya, Y., Kondo, T., Sugie, K., Kaneda, D., et al. (2013). DNAJB6 Myopathy in an Asian Cohort and Cytoplasmic/nuclear Inclusions. Neuromuscul. Disord. 23 (3), 269–276. doi:10.1016/j.nmd.2012.12.010
Stein, K. C., Bengoechea, R., Harms, M. B., Weihl, C. C., and True, H. L. (2014). Myopathy-causing Mutations in an HSP40 Chaperone Disrupt Processing of Specific Client Conformers. J. Biol. Chem. 289 (30), 21120–21130. doi:10.1074/jbc.m114.572461
Suarez-Cedeno, G., Winder, T., and Milone, M. (2014). DNAJB6 Myopathy: a Vacuolar Myopathy with Childhood Onset. Muscle Nerve 49 (4), 607–610. doi:10.1002/mus.24106
Tsai, P.-C., Tsai, Y.-S., Soong, B.-W., Huang, Y.-H., Wu, H.-T., Chen, Y.-H., et al. (2017). A novelDNAJB6mutation Causes Dominantly Inherited Distal-Onset Myopathy and Compromises DNAJB6 Function. Clin. Genet. 92 (2), 150–157. doi:10.1111/cge.13001
Yang, J., Yan, R., Roy, A., Xu, D., Poisson, J., and Zhang, Y. (2015). The I-TASSER Suite: Protein Structure and Function Prediction. Nat. Methods 12 (1), 7–8. doi:10.1038/nmeth.3213
Keywords: DNAJB6, LGMD1D, G/F domain, splice-site mutation, de novo, rimmed vacuoles
Citation: Ji G, Wang N, Han X, Wang Y, Zhang J, Wu Y, Wu H, Ma S and Song X (2022) Case Report: A Novel Splice-Site Mutation in DNAJB6 Associated With Juvenile-Onset Proximal–Distal Myopathy in a Chinese Patient. Front. Genet. 13:925926. doi: 10.3389/fgene.2022.925926
Received: 22 April 2022; Accepted: 13 May 2022;
Published: 23 June 2022.
Edited by:
Matthew E. R. Butchbach, Nemours Children’s Health Delaware, United StatesReviewed by:
Giovanna Cenacchi, University of Bologna, ItalyAlessandra Ruggieri, IRCCS Carlo Besta Neurological Institute Foundation, Italy
Copyright © 2022 Ji, Wang, Han, Wang, Zhang, Wu, Wu, Ma and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xueqin Song, sxq5679@163.com