 Artem Sharkov
Artem Sharkov Peter Sparber
Peter Sparber Anna Stepanova
Anna Stepanova Denis Pyankov
Denis Pyankov Sergei Korostelev1
Sergei Korostelev1 Mikhail Skoblov
Mikhail Skoblov- 1Genomed Ltd., Moscow, Russia
- 2Veltischev Research and Clinical Institute for Pediatrics of the Pirogov Russian National Research Medical University, Moscow, Russia
- 3Research Centre for Medical Genetics, Moscow, Russia
Febrile-associated epileptic encephalopathy is a large genetically heterogeneous group that is associated with pathogenic variants in SCN1A, PCDH19, SCN2A, SCN8A, and other genes. The disease onset ranges from neonatal or early-onset epileptic encephalopathy to late-onset epilepsy after 18 months. Some etiology-specific epileptic encephalopathies have target therapy which can serve as a clue for the correct genetic diagnosis. We present genetic, clinical, electroencephalographic, and behavioral features of a 4-year-old girl with epileptic encephalopathy related to a de novo intronic variant in the SCN2A gene. Initial NGS analysis revealed a frameshift variant in the KDM6A gene and a previously reported missense variant in SCN1A. Due to lack of typical clinical signs of Kabuki syndrome, we performed X-chromosome inactivation that revealed nearly complete skewed inactivation. Segregation analysis showed that the SCN1A variant was inherited from a healthy father. The proband had resistance to multiple antiseizure medications but responded well to sodium channel inhibitor Carbamazepine. Reanalysis of NGS data by a neurogeneticist revealed a previously uncharacterized heterozygous variant c.1035–7A>G in the SCN2A gene. Minigene assay showed that the c.1035–7A>G variant activates a cryptic intronic acceptor site which leads to 6-nucleotide extension of exon 9 (NP_066287.2:p.(Gly345_Gln346insTyrSer). SCN2A encephalopathy is a recognizable severe phenotype. Its electro-clinical and treatment response features can serve as a hallmark. In such a patient, reanalysis of genetic data is strongly recommended in case of negative or conflicting results of DNA analysis.
Introduction
Febrile seizures (FS) occur in about 2–3% of children from 3 months to 5 years of age with a phenotype outcome that varies from simple febrile seizures to Dravet-like syndrome and epileptic encephalopathy (Sharkov, 2020). Atypical features of febrile seizures such as focal semiology, prolonged episodes (>15 min), or multiple seizure types within the same fever episode are of increased risk of developing epilepsy. After the third febrile seizure, the risk of additional episodes is approaching 50% and the risk of epilepsy formation is up to 15.8% (Bertelsen et al., 2016). Recent studies showed the contribution of genetic causes in the development of febrile-associated epilepsy including inborn errors of metabolism (mitochondrial, peroxisomal, lysosomal diseases, organic acidurias, aminoacidopathies, glycosylation, and urea cycle disorders), monogenic early-onset epileptic encephalopathies, and epilepsy phenotype caused by copy number variations (CNVs) such as the Cornelia de Lange, Seckel, and Rubinstein–Taybi syndromes (Dadali et al., 2016; Wang et al., 2017; Deng et al., 2018).
Febrile onset of epilepsy or seizure exacerbation during febrile/afebrile illness is observed among patients with pathogenic variants in genes that are associated with epileptic and developmental encephalopathies (DEEs) including PRRT2, STX1B, and PCDH19, and genes encoding voltage-gated sodium channels such as SCN1A, SCN2A, SCN3A, and SCN8A are known as the cause for fever-associated seizures or epilepsy (Deng et al., 2018; Yokoi et al., 2018; Shibata et al., 2021).
However, the SCN1A gene is the most common cause of febrile-associated epilepsy among early-onset DEE (Verbeek et al., 2013; Symonds et al., 2019). SCN1A-related epilepsy is often represented by Dravet syndrome (DS) which typically presents in the first year of life in a normal child with prolonged, afebrile and febrile (triggered by mild fever or hot bath), focal (usually hemiclonic), and generalized tonic–clonic seizures. The first seizure occurs before 12 months of age in over 90% of cases, usually between 4 and 8 months (Gataullina and Dulac, 2017). Yet in rare cases, a later onset of seizures was described in patients up to 18–20 months of age (Wirrell et al., 2017).
The onset of myoclonic seizures is typically by the age of two in most cases. Non-convulsive status, focal seizures with impaired awareness, and atypical absence seizures generally occur after 2 years. Between 1 and 5 years of age, in the “worsening stage” of DS, motor seizures become more frequent but shorter, although their severity is still linked to mild hyperthermia.
Patients with DS have normal development prior to seizure onset, and neurological abnormalities typically become evident in 3–4 years of age and include crouched gait, hypotonia, incoordination, and impaired dexterity. Seizures are typically exacerbated with the use of sodium channel blocking drugs such as carbamazepine, oxcarbazepine, phenytoin, and lamotrigine (Gataullina and Dulac, 2017; Wirrell et al., 2017).
Here, we report a patient with epileptic and developmental symptoms similar to those of SCN1A-related encephalopathy but with an unusually good response to sodium channel blockers and his diagnostic odyssey.
Results
Case Presentation
A five-year-old girl, the first child of unrelated healthy parents, developed focal epilepsy at 18 months of age following a normal pregnancy and delivery. There was no family history of any neurodevelopmental disorders or epilepsy.
Autistic features and mild speech delay were recognized after 12 months and included poor eye contact, stereotypic movements, poor communication skills, and delay in the formation of joint attention. She spoke two to three simple words at the age of two.
Her psychomotor development had been considered normal until epilepsy onset which was characterized by bilateral tonic-clonic seizures, lasting 1 min, recurring several times within 2 days, and then increased in frequency to five seizures per day. An electroencephalogram (EEG) showed regional interictal spikes in the right central-frontal area (Figure 1A).

FIGURE 1. EEG longitudinal bipolar montage. Sweep: 30 mm/s; sensitivity: 150 mV/mm; bandpass: 1–70 Hz. Awake EEG (A) showing slow background activity with normal posterior dominant rhythm. Ictal EEG recording (B) showing myoclonic jerks with diffuse discharge and bursts of slow waves. 1.5T axial (C) and sagittal (D) T1 MRI indicated a mild diffuse brain atrophy, bilateral ventriculomegaly (blue arrows), and thinning of the corpus callosum (green arrows). Facial features (E) including a pronounced double curve of the upper lip (Cupid’s bow) and long palpebral fissures. Face2Gene analysis (F) showing low overlap with Kabuki syndrome.
Several antiseizure medications (ASMs) were used in different combinations:
• Levetiracetam (LEV) and carbamazepine (CBZ) stopped seizures for 6 months and improved the cognitive skills of the patient. After LEV was removed rare seizures appeared again.
• Topiramate (TPM)—led to cognition deterioration.
• Phenobarbital (PB)—led to severe weakness, drowsiness.
• CBZ with LEV and lamotrigine (LMT) decreased seizure frequency.
• Ethosuximide (ETX)—had no effect.
• Сlonazepam (CLZ)—had no effect and proved to be poorly tolerated.
• Valproate (VPA)—aggravated myoclonic seizures.
• Hormonal treatment (ACTH)—aggravated the seizures and led to agitation.
At a follow-up at the age of 2 years and 2 months, the proband displayed severe developmental delay and an increasing number of seizures up to multiple daily. She had almost a complete skills regression as she was not able to sit or walk independently and was non-verbal. The proband had both febrile provoked (triggered by mild fever) and daily afebrile generalized tonic, tonic-clonic, myoclonic, and myoclonic-atonic seizures, with rare status epilepticus. At the last clinical examination, the proband was 5 years old and had autistic features, suffered from severe cognitive impairment, and was unable to speak. She has mild dysmorphic features such as long palpebral fissures and tented upper lip but without eyelid ectropion, prominent fingertip pads, or major visceral anomalies or dysfunction.
Neurological examination showed muscle hypotonia with high tendon reflexes, discoordination, and ataxia. She had focal tonic (versive) seizures that persisted with secondary generalization that affected both sides alternately, with a frequency of one to two times a week. Fever occasionally aggravated seizure frequency up to one episode a day. There was no association of the seizures with sleep.
Slow background activity with normal posterior dominant rhythm, bifrontal and generalized discharges (sometimes followed by electrodecrement), low index during awakeness, and low-average index in the sleep appeared on EEG at 4 years of age (Figure 1B). Ictal EEG recorded short generalized tonic seizures and myoclonic jerks with diffuse discharge and burst of slow waves.
Brain MRI indicated mild diffuse brain atrophy, bilateral ventriculomegaly, and thinning of the corpus callosum. (Figures 1C,D).
Genetic Analysis
Due to the bilateral febrile provoked nature of the seizures, a monogenic etiology was suspected and an NGS-based custom gene panel of 2088 genes associated with epilepsy was performed. An in-house software pipeline was used designed to detect single-nucleotide variants (SNVs). The initial analysis revealed a heterozygous undescribed frameshift variant in the KDM6A gene—NM_021140.4:c.2831dupA (p.(Tyr944*)). The variant was classified as pathogenic according to the ACMG guidelines (Richards et al., 2015). Loss of function (LoF) variants in the KDM6A gene are associated with Kabuki syndrome (KS) 2 where haploinsufficiency is the main molecular mechanism (Lederer et al., 2012). The second reported variant was a heterozygous previously described missense variant in the SCN1A gene—NM_001165963.4:c.379C>G (p.(His127Asp)) (Zuberi et al., 2011; Cetica et al., 2017; Lindy et al., 2018; Brunklaus et al., 2020; Minardi et al., 2020). The variant affected a conserved amino acid residue where two other missense variants were reported as pathogenic in the literature (Xu et al., 2015; Djemie et al., 2016). The c.379C>G variant was predicted to be deleterious by several bioinformatic predictors including MutationTaster, PrimateAI, SIFT, and BadMut (Sim et al., 2012; Schwarz et al., 2014; Korvigo et al., 2018; Sundaram et al., 2018). According to the gnomAD database, the variant was observed 20 times in a heterozygous state with an allele frequency of 0.00007092. According to the ACMG guidelines, the variant was classified as a variant of uncertain significance (VUS).
Segregation analysis confirmed the de novo status for the variant in the KDM6A gene while the variant in the SCN1A gene was inherited from a healthy father with no clinical history of epilepsy (Supplementary Figure S1).
X-Chromosome Inactivation
Due to the fact that KS type 2 is an X-linked dominant syndrome, several studies linked the varying expressivity in females with skewed X-chromosome inactivation (XCI) (Lederer et al., 2014; Kim and Lee, 2017). To explore the possibility of a skewed XCI pattern in our patient we performed methyl sensitive quantitative fluorescent PCR (QF-PCR) of the polymorphic repeat (CAG)n in exon one of the AR gene. We observed almost completely skewed inactivation in the patient with an XCI ratio of 98:2 (Supplementary Figure S2).
Reanalysis of NGS Data
Reanalysis of the NGS data by neurogeneticists revealed a novel heterozygous intronic variant in the SCN2A gene - NG_008143.1(NM_021007.3):c.1035–7A>G which was classified as VUS. The variant was not found in the gnomAD database and it was predicted to not affect splicing by DANN (Quang et al., 2015). The nucleotide position appeared to be not strongly conserved. Moreover, another nucleotide variant located in the same position (c.1035–7A>C) was found 42 times in a heterozygous state in gnomAD with an allele frequency of 0.0001671. On the other hand, other splicing predictors suggested a potential impact on splicing including ADA (0.99924302), HSF 3.1 (+61,22%), and SpliceAI (acceptor gain: Δ score 0.91) (Desmet et al., 2009; Jaganathan et al., 2019). Sanger sequencing was performed in the family and confirmed the de novo status of the c.1035–7A>G variant in the SCN2A gene (Supplementary Figure S1).
Minigene Splicing Assay
For functional characterization of the c.1035–7A>G variant and due to the fact that SCN2A gene expression is brain restricted, a splicing minigene assay was performed. Exon 9, intron 9, and exon 10 with the adjusted intronic regions of the SCN2A gene were amplified from the proband genomic DNA and cloned into the pSpl3-Flu2 splicing vector (Sparber et al., 2020). Wild-type (WT) plasmid and a plasmid carrying c.1035–7A>G variant (MUT) were separately transfected to HEK293T cells and 48 h post-transfection total RNA was extracted and RT-PCR was performed. RT-PCR using plasmid-specific primers showed a single product in both WT and MUT constructions, however, with a slightly larger MUT fragment seen in PAGE (Figure 2B). Sanger sequencing revealed normal splicing pattern in the WT construction with the inclusion of both exons 9 and 10. In the MUT construction splicing alteration was noted. The c.1035–7A>G variant activated a cryptic intronic acceptor site with elongation of intron 8 by 6-nucleotides—p.(Gly345_Gln346insTyrSer) (Figure 2C). This insertion of two amino acids affects the extracellular domain of NaV1.2 between S5 and S6 in repeat one where several pathogenic missense variants were previously described in patients with SCN2A-related epilepsy (Shi et al., 2009; Wang et al., 2012; Wolff et al., 2017; Fernandez-Marmiesse et al., 2019). Based on the results of the minigene assay the c.1035–7A>G was classified as likely pathogenic (PM2, PM4, PP3, and PS2).
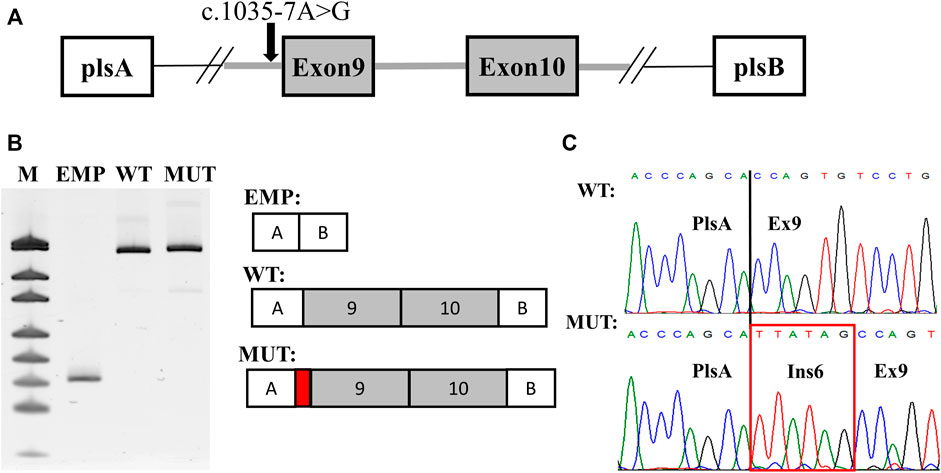
FIGURE 2. Results of the splicing minigene assay. (A) scheme of the minigene plasmid. Red arrow indicates the c.1035–7A > G variant. (B) plasmid-specific RT-PCR products in PAGE with urea. WT—wild-type isoform. MUT—mutated isoform. EMP—PCR product from an empty plasmid used as a control. M—pUC19 DNA molecular weight ladder. (C) Sanger sequencing of the detected isoforms. The 6-nucleotide extension of intron eight is highlighted by a red rectangle.
Discussion
Patients with KS have a pathogenic or likely pathogenic variant in KMT2D or KDM6A genes and distinctive facial features. The most commonly occurring features of KS are long palpebral fissures, eyelid ectropion, and fingertip pads. A large majority of people with KS are mildly or moderately intellectually disabled from birth, although some patients may have severe intellectual disability (ID). In a minority of patients the intelligence quotient (IQ) may be within the normal range as reported in (Matsumoto and Niikawa, 2003).
Boniel et al. demonstrated that patients with KS generally have low IQ levels and upon testing with the CARS, ADOS, and ADI-R scales (used in autism spectrum disorders), they tend to fulfill the criteria for autism. The prevalence of epilepsy in patients with KS has varied between 5 and 16–36% according to different sources. Seizure types most commonly include focal seizures, bilateral tonic–clonic seizures, and myoclonus. There are no reports of patients with KS and epileptic encephalopathies (Boniel et al., 2021).
Although, the segregation analysis confirmed the de novo status of the c.2831dupA variant in the KDM6A gene and the variant is predicted to lead to LoF which is in agreement with the molecular mechanism of KS we believe that the diagnosis could not be confirmed based solely on genetic data. The absence of typical clinical signs of KS and signs of epileptic encephalopathy in the proband raise doubts concerning its causality. The patient had only mild dysmorphic facial features (Figure 1E), had normal motor and near-normal developmental milestones until seizures onset, and had none of the supportive symptoms according to the international consensus diagnostic criteria of KS (Adam et al., 2019).
This incomplete penetrance in our patient could be explained by the almost complete skewed XCI, which is in agreement with previously reported clinically unaffected female carriers of a pathogenic variant in KDM6A (Lederer et al., 2014; Khodaeian et al., 2021). Several studies reported that even though the KDM6A gene escapes inactivation the expression level from the inactivated X-chromosome is significantly lower (Xu et al., 2008; Lederer et al., 2012). In this scenario, in our patient, there are near-normal levels of WT KDM6A protein which can rescue the phenotype. However, we could not fully exclude that the frame shifting variant in KDM6A, even with such skewed XCI may affect and modify the epilepsy phenotype in our patient.
The second reported variant p. His127Asp in the SCN1A gene was previously described in patients with DS and generalized epilepsy with the febrile seizures plus (GEFS+) phenotype (Zuberi et al., 2011; Cetica et al., 2017; Lindy et al., 2018; Brunklaus et al., 2020; Minardi et al., 2020). Nowadays, DS can be diagnosed among patients who met at least four of the five inclusion criteria: 1) normal or near-normal cognitive and motor development before seizure onset; 2) ≥2 febrile or afebrile seizures before 1 year of age; 3) seizure semiology consisting of myoclonic, hemiclonic, or generalized tonic-clonic seizures; 4) ≥2 seizures lasting longer than 10 min; and 5) failure to respond to first-line antiepileptic drug therapy with continued seizures after 2 years of age (Wu et al., 2015).
An additional feature of SCN1A-DS is an exacerbation of seizures during the use of sodium channel blocking drugs. Our patient had an unusually good response to carbamazepine. Also, we did not observe a non-convulsive status and atypical absence which generally occur after 2 years. Moreover, the proband had severe muscle hypotonia with increased tendon reflexes which is also rarely observed in SCN1A-related epilepsy.
Based on the fact that the variant in the SCN1A gene was observed 20 times in a heterozygous state in the gnomAD database, the fact that the proband father was an unaffected carrier and that the clinical picture was unusual for SCN1A-related epilepsy a different genetic etiology was suspected.
The favorable response to sodium channel inhibitors was suggestive for the SCN2A gene where in the previous analysis the intronic variant was missed. SCN2A encodes the pore-forming protein type 2 alpha subunit Nav1.2 of neuronal voltage-gated sodium channels. NaV1.2 is widely expressed throughout the human central nervous system, predominately in excitatory, glutamatergic neurons. It is located in the initial segment of the axons and is involved in the initiation and propagation of action potentials in a range of neuron classes (Boiko et al., 2003; Sanders et al., 2018).
SCN2A pathogenic variants have been identified as a prominent cause of a wide range of conditions, from benign neonatal or infantile seizures to neurodevelopmental disorders, including ASD, ID, and infantile-onset seizures of varying severity (Syrbe et al., 2016).
Also, few patients with unusual courses of diseases such as schizophrenia (Carroll et al., 2016) and recurrent ataxia (Leach et al., 2016) were reported.
SCN2A-epilepsy was divided into two groups depending on seizure onset: early infantile epilepsies (<3 months) and later onset (≥3 months), and described favorable effects to sodium channel blockers. Even in the late-onset epilepsy group, such medications were less effective and in some cases, induced seizure worsening (Wolff et al., 2017). Some patients with SCN2A-epilepsy have seizure exacerbation with intercurrent febrile or afebrile illnesses, including Dravet-like syndrome phenotype (Shi et al., 2009).
Reanalysis of the NGS data by neurogeneticists revealed a novel heterozygous intronic variant in the SCN2A gene: c.1035–7A>G. Following analysis of the variant has shown his potentially causative role and explained the course of the disease. Overall, based on the atypical clinical picture, lack of cosegregation with the previously reported variant in the SCN1A, and the result of the functional analysis which are concordant with the molecular pathogenesis of SCN2A-related DEE we believe that the intronic variant is causative in the reported patient.
Reanalysis of NGS data is an important tool in cases with no causative variants (Liu et al., 2019). An additional investigation by a novel specialist could dramatically increase the diagnostic yield, not only by finding causative variants in novel, previously undescribed genes, but also by finding variants that were filtered out during the first interpretation. In such cases, deep phenotyping and detailed clinical examination are of great importance.
A major challenge in DEE is establishing the pathogenicity of novel nucleotide variants and performing robust genotype-phenotype correlations. Considering the genetic heterogeneity of DEE, functional analysis is an important tool that helps in understanding the molecular consequences of the genetic variant and is crucial for proper genetic counseling. Unlike low-throughput, time-consuming approaches for investigation of the molecular defect on the protein level, for some variants, functional analysis on the RNA level can be used as an effective alternative (Wai et al., 2020). In such cases, the major limitation is the expression level of the gene of interest in an available biological sample. However, even for genes with tissue-specific expression patterns such as SCN2A for a subset of variants that are predicted to affect splicing functional analysis using the minigene assay can overcome this limitation.
Splicing variants are more and more recognized as a major cause of Mendelian disorders. Some estimate that up to 50–60% of all pathogenic variants in monogenic diseases could in fact be splicing variants (Wai et al., 2020). Abnormal splicing is more often associated with LoF, due to frameshift and nonsense-mediated decay activation. Yet, the exact molecular mechanism of splicing variants is very challenging to predict, given the complexity of the splicing machinery. In such cases, functional analysis using different approaches such as the minigene assay is crucial for a proper understanding of the molecular pathogenesis. In our patient, the splicing change leads to the insertion of two amino acids and does not alter the reading frame. This observation fits with the molecular mechanism of SCN2A-related DEE, where LoF variants are often found in patients with intellectual disability and GoF variants are linked to epileptic phenotype (Wolff et al., 2019).
Here, we report a diagnostic odyssey of a patient with an atypical Dravet-like phenotype, but with a good response to sodium channel inhibitors. Reanalysis of genetic data and functional analysis confirmed the causative role of the undescribed intronic variant in the SCN2A gene c.1035–7A>G. This report highlights the importance of a phenotype-driven strategy in DEE diagnosis and broadens the mutational spectrum of SCN2A epileptic disorders.
Materials and Methods
Subjects: The proband was clinically examined in the Veltischev Research and Clinical Institute for Pediatrics of the Pirogov Russian National Research Medical University, Russia. Genetic analysis was performed in Genomed Ltd., Russia. Functional analysis was performed in the Research Centre for Medical Genetics., Russia. We reviewed medical files, EEG tracing, video EEG recordings, magnetic resonance imaging (MRI), and seizure course during the follow-up ranging from 2 to 3.5 years.
DNA Analysis
Through the manuscript RefSeq accession numbers NG_016260.1and NM_021140.4 were used for the KDM6A gene, NM_001165963.4 were used for the SCN1A gene, and NG_008143.1 and NM_021007.3 for SCN2A.
An NGS-based custom gene panel of 2088 genes associated with epilepsy using genomic DNA was performed on Illumina NextSeq 500 instrument in 2 × 151 bp paired-end mode to an average depth of minimum 98.7x. The libraries were prepared and enriched using Illumina Nextera Rapid Capture Exome Kit v1.2. All candidate variants were validated using Sanger sequencing.
X-Inactivation Analysis at the HUMARA Locus
The unequal X-chromosome inactivation pattern was analyzed according to Allen et al. (1992). For this purpose, the X-linked HUMARA polymorphic repeat (CAG) n in AR gene exon 1 methylation pattern was detected using methyl sensitive quantitative fluorescent PCR (QF-PCR) with subsequent fragmentary analysis on the ABI3130xl Genetic Analyzer (“Applied Biosystems”, United States). The inactivated X-chromosome-carrying (with or without the mutant allele) cell percentage (XCI ratio) was evaluated according to the formula proposed by Bolduc et al. (2008).
Minigene Splicing Assay
Exon 9, intron 9, and exon 10 with the adjusted intronic regions of the SCN2A gene were amplified from the proband genomic DNA. The PCR product was cloned into a pSpl3-Flu2 plasmid vector as previously described (Sparber et al., 2020). Sanger sequencing was used for the selection of clones carrying wild-type (WT) and NG_008143.1 (NM_021007.2):c.1035–7A>G variant (MUT). WT and MUT plasmids were transfected into HEK293T cells using Metafectene (Biontex) according to the manufacturer’s instructions. Total RNA was isolated from the cells 48 h after transfection using the standard Trizol-based method, treated with DNAse I (Thermo Scientific), and reverse transcribed using ImProm-II™ Reverse Transcription System (Promega). Plasmid-specific primers were used in PCR for the detection of possible splicing alteration. PCR products were analyzed by denaturing PAGE with 8M urea with further Sanger sequencing.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, SUB11135283, and SUB11135320.
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional Review Board of the Research Centre for Medical Genetics. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
AS—conceptualization, methodology, design, and writing—original draft preparation; PS—methodology, design, writing—original draft preparation, and experimental work; AS and DP—methodology and data curation; SK—methodology, data curation, and supervision; and MS—conceptualization, methodology, supervision, and writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Ministry of Science and Higher Education of the Russian Federation (the Federal Scientific-Technical Programme for Genetic Technologies Development for 2019–2027, agreement Nos. 075-15-2021-1061 and RF 193021X0029).
Conflict of Interest
AS, DP, and SK were employed by Genomed Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank the reported family for participation in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.888481/full#supplementary-material
References
Adam, M. P., Banka, S., Bjornsson, H. T., Bodamer, O., Chudley, A. E., Harris, J., et al. (2019). Kabuki Syndrome: International Consensus Diagnostic Criteria. J. Med. Genet. 56, 89–95. doi:10.1136/jmedgenet-2018-105625
Allen, R. C., Zoghbi, H. Y., Moseley, A. B., Rosenblatt, H. M., and Belmont, J. W. (1992). Methylation of HpaII and HhaI Sites Near the Polymorphic CAG Repeat in the Human Androgen-Receptor Gene Correlates with X Chromosome Inactivation. Am. J. Hum. Genet. 51, 1229–1239.
Bertelsen, E. N., Larsen, J. T., Petersen, L., Christensen, J., and Dalsgaard, S. (2016). Childhood Epilepsy, Febrile Seizures, and Subsequent Risk of ADHD. Pediatrics 138, e20154654. doi:10.1542/peds.2015-4654
Boiko, T., Van Wart, A., Caldwell, J. H., Levinson, S. R., Trimmer, J. S., and Matthews, G. (2003). Functional Specialization of the Axon Initial Segment by Isoform-specific Sodium Channel Targeting. J. Neurosci. 23, 2306–2313. doi:10.1523/jneurosci.23-06-02306.2003
Bolduc, V., Chagnon, P., Provost, S., Dubé, M.-P., Belisle, C., Gingras, M., et al. (2008). No Evidence that Skewing of X Chromosome Inactivation Patterns Is Transmitted to Offspring in Humans. J. Clin. Invest. 118, 333–341. doi:10.1172/jci33166
Boniel, S., Szymańska, K., Śmigiel, R., and Szczałuba, K. (2021). Kabuki Syndrome-Clinical Review with Molecular Aspects. Genes (Basel) 12, 468. doi:10.3390/genes12040468
Brunklaus, A., Du, J., Steckler, F., Ghanty, I. I., Johannesen, K. M., Fenger, C. D., et al. (2020). Biological Concepts in Human Sodium Channel Epilepsies and Their Relevance in Clinical Practice. Epilepsia 61, 387–399. doi:10.1111/epi.16438
Carroll, L. S., Woolf, R., Ibrahim, Y., Williams, H. J., Dwyer, S., Walters, J., et al. (2016). Mutation Screening of SCN2A in Schizophrenia and Identification of a Novel Loss-Of-Function Mutation. Psychiatr. Genet. 26, 60–65. doi:10.1097/ypg.0000000000000110
Cetica, V., Chiari, S., Mei, D., Parrini, E., Grisotto, L., Marini, C., et al. (2017). Clinical and Genetic Factors Predicting Dravet Syndrome in Infants with SCN1A Mutations. Neurology 88, 1037–1044. doi:10.1212/wnl.0000000000003716
Dadali, E. L., Sharkov, A. A., Sharkova, I. V., Kanivets, I. V., Konovalov, F. A., and Akimova, I. A. (2016). Hereditary Diseases and Syndromes Accompanied by Febrile Convulsions: Clinical and Genetic Characteristics and Diagnostic Procedures. Rus. Ž. Det. Nevrol. 11, 33–41. doi:10.17650/2073-8803-2016-11-2-33-41
Deng, H., Zheng, W., and Song, Z. (2018). The Genetics and Molecular Biology of Fever-Associated Seizures or Epilepsy. Expert Rev. Mol. Med. 20, e3. doi:10.1017/erm.2018.2
Desmet, F.-O., Hamroun, D., Lalande, M., Collod-Béroud, G., Claustres, M., and Béroud, C. (2009). Human Splicing Finder: an Online Bioinformatics Tool to Predict Splicing Signals. Nucleic Acids Res. 37, e67. doi:10.1093/nar/gkp215
Djémié, T., Weckhuysen, S., Von Spiczak, S., Carvill, G. L., Jaehn, J., Anttonen, A.-K., et al. (2016). Pitfalls in Genetic Testing: the Story of Missed SCN1A Mutations. Mol. Genet. Genomic Med. 4, 457–464. doi:10.1002/mgg3.217
Fernández-Marmiesse, A., Roca, I., Díaz-Flores, F., Cantarín, V., Pérez-Poyato, M. S., Fontalba, A., et al. (2019). Rare Variants in 48 Genes Account for 42% of Cases of Epilepsy with or without Neurodevelopmental Delay in 246 Pediatric Patients. Front. Neurosci. 13, 1135. doi:10.3389/fnins.2019.01135
Gataullina, S., and Dulac, O. (2017). From Genotype to Phenotype in Dravet Disease. Seizure 44, 58–64. doi:10.1016/j.seizure.2016.10.014
Jaganathan, K., Kyriazopoulou Panagiotopoulou, S., Mcrae, J. F., Darbandi, S. F., Knowles, D., Li, Y. I., et al. (2019). Predicting Splicing from Primary Sequence with Deep Learning. Cell 176, 535–548. doi:10.1016/j.cell.2018.12.015
Khodaeian, M., Jafarinia, E., Bitarafan, F., Shafeii, S., Almadani, N., Daneshmand, M. A., et al. (2021). Kabuki Syndrome: Identification of Two Novel Variants in KMT2D and KDM6A. Mol. Syndromol. 12, 118–126. doi:10.1159/000513199
Kim, J., and Lee, C. G. (2017). Coinheritance of Novel Mutations in SCN1A Causing GEFS+ and in KDM6A Causing Kabuki Syndrome in a Family. Ann. Clin. Lab. Sci. 47, 229–235.
Korvigo, I., Afanasyev, A., Romashchenko, N., and Skoblov, M. (2018). Generalising Better: Applying Deep Learning to Integrate Deleteriousness Prediction Scores for Whole-Exome SNV Studies. PLoS One 13, e0192829. doi:10.1371/journal.pone.0192829
Leach, E. L., Van Karnebeek, C. D. M., Townsend, K. N., Tarailo-Graovac, M., Hukin, J., and Gibson, W. T. (2016). Episodic Ataxia Associated with a De Novo SCN2A Mutation. Eur. J. Paediatr. Neurology 20, 772–776. doi:10.1016/j.ejpn.2016.05.020
Lederer, D., Grisart, B., Digilio, M. C., Benoit, V., Crespin, M., Ghariani, S. C., et al. (2012). Deletion of KDM6A, a Histone Demethylase Interacting with MLL2, in Three Patients with Kabuki Syndrome. Am. J. Hum. Genet. 90, 119–124. doi:10.1016/j.ajhg.2011.11.021
Lederer, D., Shears, D., Benoit, V., Verellen-Dumoulin, C., and Maystadt, I. (2014). A Three Generation X-Linked Family with Kabuki Syndrome Phenotype and a Frameshift Mutation inKDM6A. Am. J. Med. Genet. 164, 1289–1292. doi:10.1002/ajmg.a.36442
Lindy, A. S., Stosser, M. B., Butler, E., Downtain‐Pickersgill, C., Shanmugham, A., Retterer, K., et al. (2018). Diagnostic Outcomes for Genetic Testing of 70 Genes in 8565 Patients with Epilepsy and Neurodevelopmental Disorders. Epilepsia 59, 1062–1071. doi:10.1111/epi.14074
Liu, P., Meng, L., Normand, E. A., Xia, F., Song, X., Ghazi, A., et al. (2019). Reanalysis of Clinical Exome Sequencing Data. N. Engl. J. Med. 380, 2478–2480. doi:10.1056/nejmc1812033
Matsumoto, N., and Niikawa, N. (2003). Kabuki Make-Up Syndrome: a Review. Am. J. Med. Genet. 117C, 57–65. doi:10.1002/ajmg.c.10020
Minardi, R., Licchetta, L., Baroni, M. C., Pippucci, T., Stipa, C., Mostacci, B., et al. (2020). Whole‐exome Sequencing in Adult Patients with Developmental and Epileptic Encephalopathy: It Is Never Too Late. Clin. Genet. 98, 477–485. doi:10.1111/cge.13823
Quang, D., Chen, Y., and Xie, X. (2015). DANN: a Deep Learning Approach for Annotating the Pathogenicity of Genetic Variants. Bioinformatics 31, 761–763. doi:10.1093/bioinformatics/btu703
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Sanders, S. J., Campbell, A. J., Cottrell, J. R., Moller, R. S., Wagner, F. F., Auldridge, A. L., et al. (2018). Progress in Understanding and Treating SCN2A-Mediated Disorders. Trends Neurosci. 41, 442–456. doi:10.1016/j.tins.2018.03.011
Schwarz, J. M., Cooper, D. N., Schuelke, M., and Seelow, D. (2014). MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 11, 361–362. doi:10.1038/nmeth.2890
Sharkov, A. A. (2020). Genetic Epilepsy with Febrile Seizures Plus (GEFS+). Èpilepsiâ paroksizmalʹnye sostoâ. 12, 50–56. doi:10.17749/2077-8333.2020.12.1s.s50-s56
Shi, X., Yasumoto, S., Nakagawa, E., Fukasawa, T., Uchiya, S., and Hirose, S. (2009). Missense Mutation of the Sodium Channel Gene SCN2A Causes Dravet Syndrome. Brain Dev. 31, 758–762. doi:10.1016/j.braindev.2009.08.009
Shibata, M., Ishii, A., Goto, A., and Hirose, S. (2021). Comparative Characterization of PCDH19 Missense and Truncating Variants in PCDH19-Related Epilepsy. J. Hum. Genet. 66, 569–578. doi:10.1038/s10038-020-00880-z
Sim, N.-L., Kumar, P., Hu, J., Henikoff, S., Schneider, G., and Ng, P. C. (2012). SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic Acids Res. 40, W452–W457. doi:10.1093/nar/gks539
Sparber, P., Filatova, A., Anisimova, I., Markova, T., Voinova, V., Chuhrova, A., et al. (2020). Various Haploinsufficiency Mechanisms in Pitt-Hopkins Syndrome. Eur. J. Med. Genet. 63, 104088. doi:10.1016/j.ejmg.2020.104088
Sundaram, L., Gao, H., Padigepati, S. R., Mcrae, J. F., Li, Y., Kosmicki, J. A., et al. (2018). Predicting the Clinical Impact of Human Mutation with Deep Neural Networks. Nat. Genet. 50, 1161–1170. doi:10.1038/s41588-018-0167-z
Symonds, J. D., Zuberi, S. M., Stewart, K., Mclellan, A., O‘Regan, M., Macleod, S., et al. (2019). Incidence and Phenotypes of Childhood-Onset Genetic Epilepsies: a Prospective Population-Based National Cohort. Brain 142, 2303–2318. doi:10.1093/brain/awz195
Syrbe, S., Zhorov, B. S., Bertsche, A., Bernhard, M. K., Hornemann, F., Mütze, U., et al. (2016). Phenotypic Variability from Benign Infantile Epilepsy to Ohtahara Syndrome Associated with a Novel Mutation in SCN2A. Mol. Syndromol. 7, 182–188. doi:10.1159/000447526
Verbeek, N. E., Van Der Maas, N. A. T., Jansen, F. E., Van Kempen, M. J. A., Lindhout, D., and Brilstra, E. H. (2013). Prevalence of SCN1A-Related Dravet Syndrome Among Children Reported with Seizures Following Vaccination: a Population-Based Ten-Year Cohort Study. PLoS One 8, e65758. doi:10.1371/journal.pone.0065758
Wai, H. A., Lord, J., Lyon, M., Gunning, A., Kelly, H., Cibin, P., et al. (2020). Blood RNA Analysis Can Increase Clinical Diagnostic Rate and Resolve Variants of Uncertain Significance. Genet. Med. 22, 1005–1014. doi:10.1038/s41436-020-0766-9
Wang, J.-w., Shi, X.-y., Kurahashi, H., Hwang, S.-K., Ishii, A., Higurashi, N., et al. (2012). Prevalence of SCN1A Mutations in Children with Suspected Dravet Syndrome and Intractable Childhood Epilepsy. Epilepsy Res. 102, 195–200. doi:10.1016/j.eplepsyres.2012.06.006
Wang, J., Lin, Z.-J., Liu, L., Xu, H.-Q., Shi, Y.-W., Yi, Y.-H., et al. (2017). Epilepsy-associated Genes. Seizure 44, 11–20. doi:10.1016/j.seizure.2016.11.030
Wirrell, E. C., Laux, L., Donner, E., Jette, N., Knupp, K., Meskis, M. A., et al. (2017). Optimizing the Diagnosis and Management of Dravet Syndrome: Recommendations from a North American Consensus Panel. Pediatr. Neurol. 68, 18–34. doi:10.1016/j.pediatrneurol.2017.01.025
Wolff, M., Brunklaus, A., and Zuberi, S. M. (2019). Phenotypic Spectrum and Genetics of SCN2A-Related Disorders, Treatment Options, and Outcomes in Epilepsy and beyond. Epilepsia 60 (Suppl. 3), S59–S67. doi:10.1111/epi.14935
Wolff, M., Johannesen, K. M., Hedrich, U. B. S., Masnada, S., Rubboli, G., Gardella, E., et al. (2017). Genetic and Phenotypic Heterogeneity Suggest Therapeutic Implications in SCN2A-Related Disorders. Brain 140, 1316–1336. doi:10.1093/brain/awx054
Wu, Y. W., Sullivan, J., Mcdaniel, S. S., Meisler, M. H., Walsh, E. M., Li, S. X., et al. (2015). Incidence of Dravet Syndrome in a US Population. Pediatrics 136, e1310–e1315. doi:10.1542/peds.2015-1807
Xu, J., Deng, X., Watkins, R., and Disteche, C. M. (2008). Sex-specific Differences in Expression of Histone Demethylases Utx and Uty in Mouse Brain and Neurons. J. Neurosci. 28, 4521–4527. doi:10.1523/jneurosci.5382-07.2008
Xu, X., Yang, X., Wu, Q., Liu, A., Yang, X., Ye, A. Y., et al. (2015). Amplicon Resequencing Identified Parental Mosaicism for Approximately 10% of "De Novo" SCN1A Mutations in Children with Dravet Syndrome. Hum. Mutat. 36, 861–872. doi:10.1002/humu.22819
Yokoi, T., Enomoto, Y., Tsurusaki, Y., Naruto, T., and Kurosawa, K. (2018). Nonsyndromic Intellectual Disability with Novel Heterozygous SCN2A Mutation and Epilepsy. Hum. Genome Var. 5, 20. doi:10.1038/s41439-018-0019-5
Keywords: SCN2A, functional analysis, deep phenotyping, Dravet syndrome, Kabuki syndrome
Citation: Sharkov A, Sparber P, Stepanova A, Pyankov D, Korostelev S and Skoblov M (2022) Case Report: Phenotype-Driven Diagnosis of Atypical Dravet-Like Syndrome Caused by a Novel Splicing Variant in the SCN2A Gene. Front. Genet. 13:888481. doi: 10.3389/fgene.2022.888481
Received: 02 March 2022; Accepted: 19 April 2022;
Published: 31 May 2022.
Edited by:
Daniel Grinberg, University of Barcelona, SpainReviewed by:
Jinchen Li, Central South University, ChinaAlison Anderson, Monash University, Australia
Copyright © 2022 Sharkov, Sparber, Stepanova, Pyankov, Korostelev and Skoblov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Artem Sharkov, a.a.sharkov@yandex.ru