 Yue Yang1
Yue Yang1 Shijie Zhang2
Shijie Zhang2 Wenming Yang2,3*
Wenming Yang2,3* Taohua Wei2Wenjie Hao1Ting Cheng4Jiuxiang Wang2Wei Dong1,2Nannan Qian1
Taohua Wei2Wenjie Hao1Ting Cheng4Jiuxiang Wang2Wei Dong1,2Nannan Qian1- 1Graduate School of Anhui University of Chinese Medicine, Hefei, China
- 2The First Affiliated Hospital of Anhui University of Chinese Medicine, Hefei, China
- 3Xin’an Medical Education Ministry Key Laboratory, Hefei, China
- 4Clinical School of Anhui Medical University, Hefei, China
Background: Mitochondrial membrane protein–associated neurodegeneration (MPAN) mostly arises as an autosomal recessive disease and is caused by variants in the chromosome 19 open reading frame 12 (C19orf12) gene. However, a few C19orf12 monoallelic truncating de novo variants have been reported and segregated as autosomal dominant traits in some cases.
Methods: We performed whole-exome sequencing and analyzed genes related to neurodegeneration associated with brain iron accumulation for pathogenic variants. The identified variants were confirmed by Sanger sequencing and tested using in silico tools.
Results: The patient had an onset of depression at the age of 22 years, which rapidly progressed to severe dystonia, dementia, and bladder and bowel incontinence. Neuroimaging showed hypointensity in the substantia nigra and the globus pallidum, with additional frontotemporal atrophy. Genetic analysis revealed a single complex de novo variant [c.336_338delinsCACA (p.Trp112CysfsTer40)] in the C19orf12 gene.
Conclusion: This study enriches the genetic spectrum and clinical features of C19orf12 variants and provides additional evidence of the variable inheritance pattern of MPAN.
Introduction
Mitochondrial membrane protein–associated neurodegeneration (MPAN; OMIM: 614298) is a subtype of neurodegeneration with brain iron accumulation (NBIA) characterized by dystonia, spastic paraparesis with muscle weakness, cognitive decline progressing to dementia, neuropsychiatric symptoms, and optic atrophy (Olgiati et al., 2017). Brain magnetic resonance imaging (MRI) showed an excessive accumulation of iron in the basal ganglia and substantia nigra of an NBIA patient (Svetel et al., 2021). Usually, chromosome 19 open reading frame 12 (C19orf12) displays an entirely autosomal recessive pattern of inheritance in well-characterized cases of MPAN (Hartig et al., 2011); however, a few apparent monoallelic truncating variants in C19orf12 exon 3 have been reported (Hogarth et al., 2013; Deutschländer et al., 2017; Monfrini et al., 2018).
This report describes a female MPAN patient experiencing onset of depression and identified with a single complex de novo variant [c.336_338delinsCACA (p.Trp112CysfsTer40)] in C19orf12. This case broadens the mutation spectrum and clinical phenotypes associated with C19orf12 variants and provides additional evidence of the variable inheritance pattern of MPAN.
Case Presentation
The patient was born to a non-consanguineously married couple, met the normal developmental milestones, and had an unremarkable family history (Figure 1A). At the age of 22 years, she presented with a depressed mood and was referred to a psychiatrist. She was subsequently diagnosed with depression; however, antidepressants were ineffective. At 23, she began complaining of a mild tremor in both upper limbs and had difficulty with fine motor skills. A computed tomography scan of the brain showed hyperdensity in the bilateral globus pallidum. At 24, she manifested with gait imbalance, slowed movements, and frequent falls. The symptoms were progressive, and by 26 years, she presented with cognitive decline, dystonia, rigidity, and spasticity. There were no seizures or optic atrophy, and a neurological examination showed moderate dysarthria, cervical dystonia, severe upper limb tremors, spastic limb hypertonia and muscle weakness, patellar hyperreflexia, and bilateral Babinski sign. An MRI of the brain on T2WI and FLAIR showed hypointensity in the substantia nigra and globus pallidum and hyperintense streaking of the medial medullary lamina between the globus pallidum internus and externus (Figure 1B). After a 2-year follow-up, she was immobile and non-verbal, developed obvious bladder and bowel incontinence, was in a wheelchair, and was unable to perform activities of daily living.
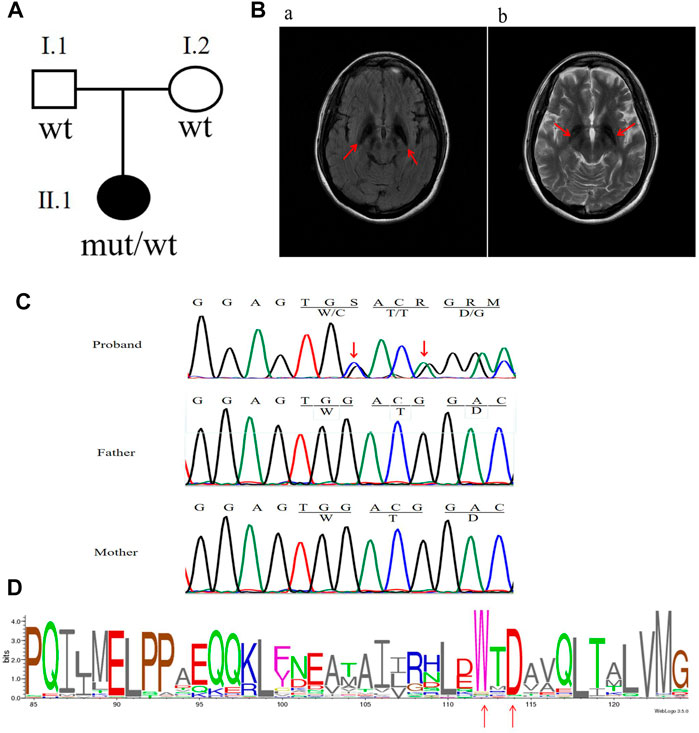
FIGURE 1. Clinical characteristics and mutation analysis of the family in this study. (A) Pedigree of the family in this study. Black symbol denotes the proband. (B) Brain MRI results: red arrows show hypointensity in the substantia nigra and globus pallidum along with additional frontotemporal atrophy. (C) Sanger sequencing results of the C19orf12 gene: red arrows indicate the locations of the mutations. (D) C19orf12 variants in the MPAN proband and frequency diagram of protein mutations. The locations of the W112 and D114 sites are indicated via red arrows.
Materials and Methods
Ethical Compliance
This study was approved by the Medical Ethics Committee of The First Affiliated Hospital of Anhui University of Chinese Medicine (Hefei, China). Informed written consent was obtained from all participants.
Whole-Exome Sequencing and Validation
Genomic DNA of the patient and her parents was extracted from EDTA-anticoagulated blood, whole-exome sequencing was performed, and genes related to NBIA were analyzed for pathogenic variants. The identified variants were confirmed by Sanger sequencing. To determine whether C19orf12 variants were located in two different alleles, a fragment containing the two variants was cloned into the pEGFP-C1 vector, and 10 different clones were sent for sequencing. The primer sequences are shown in Supplementary Table S1.
Variant Interpretation
C19orf12 variants were compared with the list of reported pathogenic variants in the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/gene.php), gnomAD (v3.1.1; http://gnomad-sg.org/), and Ensembl Blast/BLAT (http://asia.ensembl.org/index.html). The pathogenicity of the variants are shown in Supplementary Table S2, which was assessed with SIFT (http://provean.jcvi.org/protein_batch_submit.php), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), and MutationTaster (https://www.mutationtaster.org/). Homology comparisons were conducted by VarSite (https://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/VarSite/GetPage.pl?home=TRUE) to explore whether the mutated region was conserved.
Results
The analysis of whole-exome sequencing data is shown in Supplementary Table S3, which identified two novel variants [c.336G > C (p.W112C) and c.338_339insA (p.D114Gfs*38)] in C19orf12 (NM_001031726) that were not present in gnomAD. Furthermore, neither of the variants were found in the parents of the patient (Figure 1C). VarSite showed that the variants were located in the highly conserved region of the C19orf12 gene across multiple species (Figure 1D). Supplementary Table S2 shows additional evidence for the pathogenicity of the two novel variations. According to the American College of Medical Genetics and Genomics guidelines, the frameshift variant c.338-339insA and missense variant c.336G > C can be classified as pathogenic (PVS1, PS2, PM2, PP3) and likely pathogenic (PS2, PM2, PP3), respectively.
Sanger sequencing confirmed that the two variants were located in the same allele (Supplementary Figure S1). However, given the unlikelihood that two de novo variants occur on the same allele, we treated these variants as a single complex de novo variant [c.336_338delinsCACA (p.Trp112CysfsTer40)].
Discussion
MPAN is a classical form of NBIA and characterized by juvenile-onset and as a slowly progressive phenotype with dystonia, speech disturbances, cognitive decline, optic atrophy, and spastic paraparesis. However, psychiatric disturbances are relatively rare (Hartig et al., 2013; Schulte et al., 2013). Brain MRI results for MPAN are characterized by hypointensity in the substantia nigra and globus palladium (Hogarth et al., 2013), and neuroimaging confirmed hyperintense streaking in the medial medullary lamina, which is typical of MPAN and present in this case. Moreover, we found that additional frontotemporal atrophy was also present, which is an imaging feature associated with the end stages in MPAN individuals. In the present case, MPAN onset included depression at age 22, which progressed rapidly to severe dystonia, dementia, bladder and bowel incontinence, and brain atrophy within 4 years. Additionally, this patient did not have optic atrophy or blurred vision as compared with reports from other studies (de Vries et al., 2021; Langwinska-Wosko et al., 2016).
C19orf12 is a mitochondrial protein with a complex isoform involved in maintaining lipid homeostasis and membrane remodeling, with mutated variants capable of inducing oxidative stress and causing neuroinflammation (Hinarejos et al., 2020). However, it remains unclear how C19orf12 variants induce iron accumulation and orchestrate the MPAN phenotype. Ferroptosis may be a novel theory for the pathophysiologic mechanism of MPAN and might be related to lipid peroxidation and mitochondrial perturbation (Wang et al., 2019). To date, >50 C19orf12 variants have been reported in MPAN cases according to the Human Gene Mutation Database, and abundant evidence supports an autosomal recessive pattern. However, Monfrini et al. (2018) proposed a dominant negative effect associated with a C19orf12 variant in a patient with MPAN who carried a de novo heterozygous mutation. Additionally, Gregory et al. (2019) provided evidence of the molecular mechanism, suggesting the presence of a functional region in C19orf12 that contained a glycine zipper motif that could have a multimerization function (Gagliardi et al., 2015). The C19orf12 variant resulting in a protein truncated after amino acid 79 may retain the ability to multimerize with the protein from the wild-type allele; however, this version of the protein may still cause damage or induce degradation of the resulting protein complex, resulting in loss of function (Gregory et al., 2019). However, this would not explain a protein variant truncated after amino acid 76 (p.Met76Thrfs*3; NM_00103176.3) in patients with autosomal dominant MPAN. Thus, Rickman et al. (2021) considered “haploinsufficiency of isoform 3” as a potential mechanism of monoallelic MPAN, which results in a protein truncated after amino acid 75 and lacking the putative transmembrane region. Other studies subsequently confirmed an autosomal dominant mode of inheritance in MPAN (Fraser et al., 2021; Rickman et al., 2021).
A literature review focusing on age at onset, clinical features, and C19orf12 variant status [homozygous/compound heterozygous (Supplementary Table S1) and heterozygous patients (Table 1)] in MPAN cases indicated that 57 of 70 of cases presented with clinical signs of neurodegeneration before age 18. Moreover, we performed the analysis (Mann–Whitney test) and revealed homozygous/compound heterozygous C19orf12 mutations associated with MPAN cases had an earlier age at onset than heterozygous cases [biallelic vs. monoallelic: 9 ± 4 (range: 3–29 years) vs. 19 ± 16 (range: 1–55 years), p = 0.042]. Independent of the inheritance pattern, most MPAN cases are clinically similar, with the most common presenting symptoms being cognitive decline, gait difficulties, and optic atrophy, which occurred in 38, 34, and 21 of 70 cases, respectively.

TABLE 1. Heterozygous C19orf12 mutations associated with MPAN cases in the medical literature.
As of December 2021, 17 monoallelic truncating variants have been described in patients with MPAN (Figure 2). In this case report, genetic analysis indicated a single complex variant of c.336_338delinsCACA (p.Trp112CysfsTer40) in C19orf12, which represents a de novo occurrence. This C19orf12 variant occurs at position 112 and subsequent codons, resulting in a series of 40 amino acid substitutions and causing early translation interruption during protein translation. Therefore, abnormal protein products are likely to escape nonsense-mediated mRNA decay and may have a significant negative impact on normally translated C19orf12 proteins.
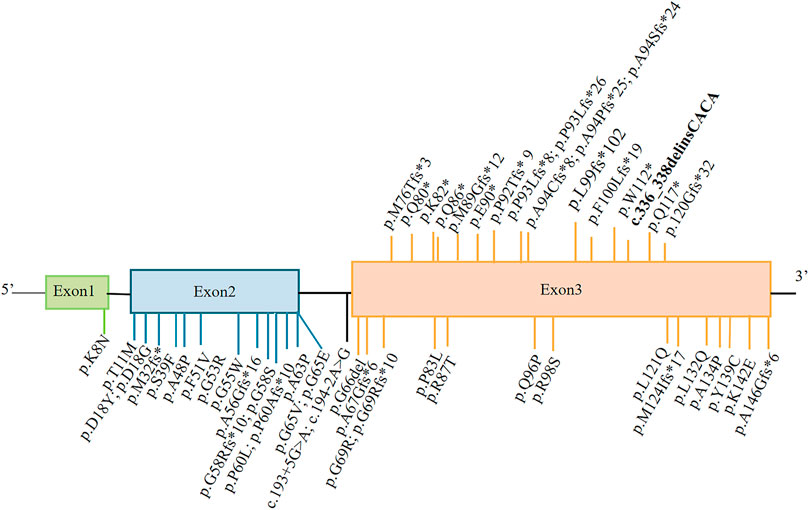
FIGURE 2. Representation of MPAN-related C19orf12 variants. The variants shown above the gene structure are monoallelic, and those shown below are biallelic. The variant reported in this study is shown in bold.
In summary, we identified a single complex de novo variant of C19orf12 [c.336_338delinsCACA (p.Trp112CysfsTer40)] in an MPAN patient with a history of onset of depression that rapidly progressed to severe dystonia, dementia, and bladder and bowel incontinence. Additionally, a review of previously reported MPAN cases supported the novelty of this variant and provided additional evidence of the variable inheritance pattern associated with MPAN.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Medical Ethics Committee of The First Affiliated Hospital of Anhui University of Chinese Medicine. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
WY designed the study supporting the case report; YY and SZ performed data analysis and wrote the article; TW, TC, WH, WD, JW, and NQ were responsible for sample collection and identifying clinical information; WY and SZ revised the article; and all authors reviewed and agreed to publication of the final version of the article.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 81973825 and 81903895), the Collaborative Innovation Project of Anhui Colleges and Universities (No. GXXT-2020-025), the Anhui Provincial Natural Science Foundation of China (No. 2108085QH367), the Open Fund Project of Key Laboratory of Xin’An Medicine of Ministry of Education (No. 2020xayx12), and the Natural Science Research Project of Anhui Universities (No. KJ 2021A0555).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the patient and her family for their participation in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.852374/full#supplementary-material
References
de Vries, R. J., Hellebrekers, D. M. E. I., Reneman, L., Verhamme, C., Smeets, H. J. M., van Maarle, M. C., et al. (2021). Distal Muscle Weakness and Optic Atrophy without central Nervous System Involvement in a Patient with a Homozygous Missense Mutation in the C19ORF12-Gene. Clin. Neurol. Neurosurg. 206, 106637. doi:10.1016/j.clineuro.2021.106637
Deschauer, M., Gaul, C., Behrmann, C., Prokisch, H., Zierz, S., and Haack, T. B. (2012). C19orf12 Mutations in Neurodegeneration with Brain Iron Accumulation Mimicking Juvenile Amyotrophic Lateral Sclerosis. J. Neurol. 259, 2434–2439. doi:10.1007/s00415-012-6521-7
Deutschländer, A., Konno, T., and Ross, O. A. (2017). Mitochondrial Membrane Protein-Associated Neurodegeneration. Parkinsonism Relat. Disord. 39, 1–3. doi:10.1016/j.parkreldis.2017.03.014
Dušek, P., Školoudík, D., Roth, J., and Dušek, P. (2018). Mitochondrial Membrane Protein-Associated Neurodegeneration: a Case Report and Literature Review. Neurocase 24, 161–165. doi:10.1080/13554794.2018.1506038
Fraser, S., Koenig, M., Farach, L., Mancias, P., and Mowrey, K. (2021). A De Novo Case of Autosomal Dominant Mitochondrial Membrane Protein‐associated Neurodegeneration. Mol. Genet. Genomic Med. 9, e1706. doi:10.1002/mgg3.1706
Gagliardi, M., Annesi, G., Lesca, G., Broussolle, E., Iannello, G., Vaiti, V., et al. (2015). C19orf12 Gene Mutations in Patients with Neurodegeneration with Brain Iron Accumulation. Parkinsonism Relat. Disord. 21, 813–816. doi:10.1016/j.parkreldis.2015.04.009
Gowda, V. K., Patil, A., Srinivasan, V. M., and Kathrani, N. (2019). Mitochondrial Membrane Protein Associated Neurodegeneration (MPAN) with a Novel C19orf12 Mutation in the First Decade of Life. Indian J. Pediatr. 86, 746–748. doi:10.1007/s12098-019-02903-w
Gregory, A., Lotia, M., Jeong, S. Y., Fox, R., Zhen, D., Sanford, L., et al. (2019). Autosomal Dominant Mitochondrial Membrane Protein‐associated Neurodegeneration (MPAN). Mol. Genet. Genomic Med. 7. doi:10.1002/mgg3.736
Hartig, M. B., Iuso, A., Haack, T., Kmiec, T., Jurkiewicz, E., Heim, K., et al. (2011). Absence of an Orphan Mitochondrial Protein, C19orf12, Causes a Distinct Clinical Subtype of Neurodegeneration with Brain Iron Accumulation. Am. J. Hum. Genet. 89, 543–550. doi:10.1016/j.ajhg.2011.09.007
Hartig, M., Prokisch, H., Meitinger, T., and Klopstock, T. (2013). Mitochondrial Membrane Protein-Associated Neurodegeneration (MPAN). Int. Rev. Neurobiol. 110, 73–84. doi:10.1016/B978-0-12-410502-7.00004-1
Hinarejos, I., Machuca, C., Sancho, P., and Espinós, C. (2020). Mitochondrial Dysfunction, Oxidative Stress and Neuroinflammation in Neurodegeneration with Brain Iron Accumulation (NBIA). Antioxidants 9, 1020. doi:10.3390/antiox9101020
Hogarth, P., Gregory, A., Kruer, M. C., Sanford, L., Wagoner, W., Natowicz, M. R., et al. (2013). New NBIA Subtype: Genetic, Clinical, Pathologic, and Radiographic Features of Mpan. Neurology 80, 268–275. doi:10.1212/WNL.0b013e31827e07be
Incecik, F., Herguner, O., and Bisgin, A. (2019). Mitochondrial Membrane Protein-Associated Neurodegeneration: a Case Series of Six Children. Ann. Indian Acad. Neurol. 23, 802–804. doi:10.4103/aian.AIAN_268_19
Kasapkara, Ç. S., Tümer, L., Gregory, A., Ezgü, F., İnci, A., Derinkuyu, B. E., et al. (2019). A New NBIA Patient from Turkey with Homozygous C19ORF12 Mutation. Acta Neurol. Belg. 119, 623–625. doi:10.1007/s13760-018-1026-5
Langwinska-Wosko, E., Skowronska, M., Kmiec, T., and Czlonkowska, A. (2016). Retinal and Optic Nerve Abnormalities in Neurodegeneration Associated with Mutations in C19orf12 (MPAN). J. Neurol. Sci. 370, 237–240. doi:10.1016/j.jns.2016.09.046
Li, S. J., Wang, L. L., Qin, L. Z., Wang, X. J., Zhang, J. W., and Li, W. (2019). Pedigree Analysis of C19ORF12 p.Asp18Tyr Mutation in a Family with Mitochondrial Membrane Protein Associated Neurodegeneration. Zhonghua yi xue za zhi 99, 2926–2931. doi:10.3760/cma.j.issn.0376-2491.2019.37.011
Monfrini, E., Melzi, V., Buongarzone, G., Franco, G., Ronchi, D., Dilena, R., et al. (2018). A De Novo C19orf12 Heterozygous Mutation in a Patient with MPAN. Parkinsonism Relat. Disord. 48, 109–111. doi:10.1016/j.parkreldis.2017.12.025
Nagarjunakonda, S., Daggumati, R., Uppala, V., Gajula, R., and Amalakanti, S. (2019). A Novel Mutation in Neurodegeneration with Brain Iron Accumulation - A Case Report. Neurol. India 67, 1341–1343. doi:10.4103/0028-3886.271257
Olgiati, S., Doğu, O., Tufekcioglu, Z., Diler, Y., Saka, E., Gultekin, M., et al. (2017). The p.Thr11Met Mutation in C19orf12 Is Frequent Among Adult Turkish Patients with MPAN. Parkinsonism Relat. Disord. 39, 64–70. doi:10.1016/j.parkreldis.2017.03.012
Panteghini, C., Zorzi, G., Venco, P., Dusi, S., Reale, C., Brunetti, D., et al. (2012). C19orf12 and Fa2h Mutations Are Rare in Italian Patients with Neurodegeneration with Brain Iron Accumulation. Semin. Pediatr. Neurol. 19, 75–81. doi:10.1016/j.spen.2012.03.006
Rickman, O. J., Salter, C. G., Gunning, A. C., Fasham, J., Voutsina, N., Leslie, J. S., et al. (2021). Dominant Mitochondrial Membrane Protein-Associated Neurodegeneration (MPAN) Variants Cluster within a Specific C19orf12 Isoform. Parkinsonism Relat. Disord. 82, 84–86. doi:10.1016/j.parkreldis.2020.10.041
Schulte, E. C., Claussen, M. C., Jochim, A., Haack, T., Hartig, M., Hempel, M., et al. (2013). Mitochondrial Membrane Protein Associated Neurodegenration: a Novel Variant of Neurodegeneration with Brain Iron Accumulation. Mov Disord. 28, 224–227. doi:10.1002/mds.25256
Selikhova, M., Fedotova, E., Wiethoff, S., Schottlaender, L. V., Klyushnikov, S., Illarioshkin, S. N., et al. (2017). A 30-year History of MPAN Case from Russia. Clin. Neurol. Neurosurg. 159, 111–113. doi:10.1016/j.clineuro.2017.05.025
Skowronska, M., Kmiec, T., Kurkowska-Jastrzębska, I., and Czlonkowska, A. (2015). Eye of the Tiger Sign in a 23year Patient with Mitochondrial Membrane Protein Associated Neurodegeneration. J. Neurol. Sci. 352, 110–111. doi:10.1016/j.jns.2015.03.019
Sparber, P., Marakhonov, A., Filatova, A., Sharkova, I., and Skoblov, M. (2018). Novel Case of Neurodegeneration with Brain Iron Accumulation 4 (NBIA4) Caused by a Pathogenic Variant Affecting Splicing. Neurogenetics 19, 257–260. doi:10.1007/s10048-018-0558-4
Svetel, M., Dragašević, N., Petrović, I., Novaković, I., Tomić, A., Kresojević, N., et al. (2021). NBIA Syndromes: a Step Forward from the Previous Knowledge. Neurol. India 69, 1380–1388. doi:10.4103/0028-3886.329603
Tariq, H., Butt, J. U. R., Houlden, H., and Naz, S. (2019). Are Some C19orf12 Variants Monoallelic for Neurological Disorders? Parkinsonism Relat. Disord. 65, 267–269. doi:10.1016/j.parkreldis.2019.05.020
Keywords: mitochondrial membrane protein–associated neurodegeneration, C19orf12, whole-exome sequencing, iron accumulation, de novo variant
Citation: Yang Y, Zhang S, Yang W, Wei T, Hao W, Cheng T, Wang J, Dong W and Qian N (2022) Case Report: Identification of a De novo C19orf12 Variant in a Patient With Mitochondrial Membrane Protein–Associated Neurodegeneration. Front. Genet. 13:852374. doi: 10.3389/fgene.2022.852374
Received: 11 January 2022; Accepted: 18 February 2022;
Published: 30 March 2022.
Edited by:
Enrico Baruffini, University of Parma, ItalyReviewed by:
Alessio Di Fonzo, IRCCS Ca’ Granda Foundation Maggiore Policlinico Hospital, ItalyFélix Javier Jiménez-Jiménez, Hospital Universitario del Sureste, Spain
Grazia Annesi, National Research Council, Italy
Copyright © 2022 Yang, Zhang, Yang, Wei, Hao, Cheng, Wang, Dong and Qian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenming Yang, yangwm8810@126.com