Plastin 3 in X-Linked Osteoporosis: Imbalance of Ca2+-Dependent Regulation Is Equivalent to Protein Loss
 Christopher L. Schwebach
Christopher L. Schwebach Elena Kudryashova
Elena Kudryashova Dmitri S. Kudryashov
Dmitri S. Kudryashov- Department of Chemistry and Biochemistry, The Ohio State University, Columbus, OH, United States
Osteogenesis imperfecta is a genetic disorder disrupting bone development and remodeling. The primary causes of osteogenesis imperfecta are pathogenic variants of collagen and collagen processing genes. However, recently variants of the actin bundling protein plastin 3 have been identified as another source of osteogenesis imperfecta. Plastin 3 is a highly conserved protein involved in several important cellular structures and processes and is controlled by intracellular Ca2+ which potently inhibits its actin-bundling activity. The precise mechanisms by which plastin 3 causes osteogenesis imperfecta remain unclear, but recent advances have contributed to our understanding of bone development and the actin cytoskeleton. Here, we review the link between plastin 3 and osteogenesis imperfecta highlighting in vitro studies and emphasizing the importance of Ca2+ regulation in the localization and functionality of plastin 3.
Introduction
Osteogenesis imperfecta (OI) is a genetic form of skeletal dysplasia affecting 1 in 10,000–20,000 live births and resulting in a broad array of clinical manifestations (Lim et al., 2017; Marini et al., 2017; Palomo et al., 2017). OI, also known as brittle bone disease, is characterized by low bone mass and increased bone fragility with severity ranging from asymptomatic to perinatal lethality (Robinson and Rauch, 2019; Rossi et al., 2019). The primary cause of most OI forms is an impaired extracellular matrix resulting from defects in either quantity/quality or assembly of collagen type I (Nijhuis et al., 2019). Approximately 85–90% of OI cases are a result of pathogenic variants of either the COL1A1 or COL1A2 genes (Lindahl et al., 2015). The remaining OI cases stem from variants of 18 other identified genes, the majority of which are involved in collagen type I processing (Tauer et al., 2019). Interestingly, pathogenic variants of three genes (CRTAP, P3H1, and PPIB) coding for the components of the type I collagen 3-hydroxylation complex are associated with an altered expression of lamin A/C and cofilin 1, suggesting that nucleoskeletal and cytoskeletal abnormalities may be involved in the pathogenesis of the disease (Gagliardi et al., 2017). A more direct link to the cytoskeleton is observed in X-linked OI caused by pathogenic variants in the PLS3 gene located on the X chromosome and coding for the cytoskeletal actin-bundling protein plastin 3 (PLS3; aka T-plastin). To date, PLS3 is the only OI-linked gene not known to affect collagen processing.
PLS3 Structure and Function
Plastins (aka fimbrins) are a highly conserved family of actin-bundling proteins, which are expressed in a tissue-specific manner (Lin et al., 1993; Shinomiya, 2012). Of the three plastin isoforms expressed in vertebrates, PLS3 is the most ubiquitous isoform expressed in all solid tissues (Lin et al., 1988, 1993, 1999), while PLS1 (aka I-plastin) and PLS2 (aka LCP1, LPL, or L-plastin) are primarily found in intestinal epithelium and hematopoietic tissues, respectively. Plastins contain two actin-binding domains (ABD1 and ABD2), each comprising two calponin-homology (CH) domains (Figure 1A). Binding of both ABDs to actin filaments (F-actin) brings the filaments together into a tight bundle (Figure 1B). F-actin bundling is critical to cellular structures (such as stress fibers, filopodia, focal adhesions, and microvilli) and processes (such as cell migration, cytokinesis, endo-, and exocytosis) and appears to be the primary function of plastins (Skau et al., 2011; Schwebach et al., 2020). F-actin bundling by plastins is tightly regulated by Ca2+ binding to plastins' N-terminal regulatory domain (RD), which contains two Ca2+-binding EF-hand motifs (Figure 1A). Upon binding of Ca2+, the RD potently inhibits the bundling activity of plastins by disabling ABD2 (Figure 1B) (Schwebach et al., 2017, 2020).
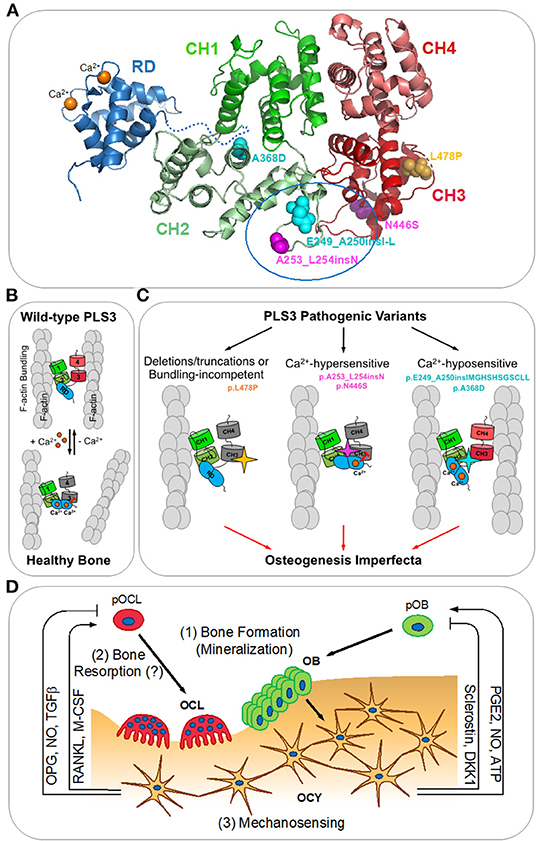
Figure 1. PLS3 in osteogenesis imperfecta. (A) A homology model of human PLS3 built using Phyre2.0 (Kelley et al., 2015) is colored according to the domain organization. Calponin-homology domains 1-2 (CH1 and CH2) and 3-4 (CH3 and CH4) form actin-binding domains 1 and 2 (ABD1 and ABD2), respectively. The N-terminal regulatory domain (RD; PDB:5JOJ; Ishida et al., 2017) is comprised of two Ca2+-binding EF-hand domains and positioned arbitrarily as its precise orientation relative to the actin-binding core is not known. OI-linked mutations resulting in the full-length protein are indicated as spheres: Ca2+-hypersensitive mutations are in magenta, Ca2+-hyposensitive mutations are in cyan, the bundling-disabling mutation is in yellow. A tentative RD docking site outlined by a blue oval at the ABD1-ABD2 interface has been determined by the location of the OI mutations disrupting Ca2+-sensitivity and reduced accessibility for labeling in the presence of the RD (Schwebach et al., 2020). (B) Actin bundle formation by the wild-type PLS3 is regulated by calcium. At low Ca2+ concentrations (nanomolar range), PLS3 forms actin bundles by binding simultaneously to two actin filaments (F-actin) via ABD1 and ABD2; saturation of the EF-hands in the RD with Ca2+ inhibits the ability of ABD2 to bind to F-actin and disassembles the bundle (Schwebach et al., 2017). (C) Finely tuned Ca2+ regulation of F-actin bundling by PLS3 is essential for proper bone formation (Schwebach et al., 2020). OI-causative pathogenic variants of PLS3 disrupting its normal functioning can be classified in three groups: (1) F-actin bundling-incompetent (deletions/truncations of PLS3 or p.L478P directly disrupting the actin-binding site of ABD2); (2) inhibiting ABD2 by sub-physiological Ca2+ concentrations (p.N446S and p.A253_L254insN); (3) disabling the ABD2 inhibition by physiological Ca2+ concentrations (p.A368D and p.E249_A250insIMGHSHSGSCLL). (D) Bone remodeling (reviewed in Niedzwiedzki and Filipowska, 2015): osteocytes (OCY) orchestrate bone lysis by osteoclasts (OCL) and bone formation by osteoblasts (OBL) via coordinated secretion of RANKL (receptor-activator of NFκβ ligand), M-CSF (macrophage colony stimulating factor), OPG (osteoprotegerin), NO (nitric oxide), TGFβ (transforming growth factor beta), PGE2 (prostaglandin E2), ATP, sclerostin, and DKK1 (Dickkopf-1). pOCL and pOBL – osteoclast and osteoblast precursors, respectively. PLS3 has been proposed to be involved in: (1) insufficient mineralization by OBL; (2) increased bone resorption by OCL; (3) dysregulation of mechanosensing by OCY leading to misbalance between bone resorption and formation.
PLS3 contributes to a variety of cellular activities and appears to be particularly important for shaping the actin cytoskeleton near membranes. Thus, PLS3 is found associated with the lamellipodium (Garbett et al., 2020; Schwebach et al., 2020) and focal adhesions in fibroblasts and osteoblasts (Schwebach et al., 2020). PLS3 has recently been shown to contribute to cell migration by strengthening and stabilizing membrane protrusions (Garbett et al., 2020). S-nitrosylation of PLS3 in endothelial cells in response to angiotensin II has been implicated in weakening adherence junctions but promoting migration and tube formation (Pan et al., 2020). Similarly, PLS2 is well-recognized to contribute to the migratory capabilities of immune and cancerous cells (Schaffner-Reckinger and Machado, 2020).
Both PLS3 and PLS2 interact with activated Rab5 and their expression is accompanied by increased fluid-phase endocytosis (Hagiwara et al., 2011). Plastin's ortholog in yeast (fimbrin) is localized almost exclusively to endocytic actin patches and is essential for endocytosis (Skau et al., 2011). Improved endocytosis has also been proposed to account for the PLS3's role as a protective modifier of spinal muscular atrophy (Ackermann et al., 2013; Lyon et al., 2014; Kaifer et al., 2017; Alrafiah et al., 2018). PLS3 also mediates membrane trafficking in hypoxia (Wottawa et al., 2017) and in the ectoplasmic specialization- testis-specific hybrid cell-cell junctions between Sertoli cells and spermatids. Localization of PLS3 with these contacts correlates with a highly dynamic reorganization of the membrane-associated actin cytoskeleton in spermatogenesis (Li et al., 2016).
A role of plastins in cytokinesis has also been revealed but appears to be less universal. In Caenorhabditis elegans zygotes, plastin increases cortical contractility and helps to stabilize myosin at the equatorial cortex (Ding et al., 2017; Leite et al., 2020), whereas the role of the fission yeast ortholog fimbrin in cytokinesis is controversial (Laporte et al., 2012; Christensen et al., 2019). Interestingly, plastin and myosin cooperate in cytokinesis (Leite et al., 2020) and epidermal morphogenesis/basement membrane assembly (Dor-On et al., 2017), while demonstrating mutually exclusive subcellular localization (Garbett et al., 2020), which can result from direct competition between myosin and plastin for binding sites on actin (Behrmann et al., 2012; Schwebach et al., 2020). Additional explanation for this antagonism is competition with tropomyosin, which has been demonstrated for yeast fimbrin (Christensen et al., 2019), but not yet for mammalian plastin and tropomyosin isoforms.
Surprisingly, PLS3 expression has also been shown to correlate with robust DNA repair (Hisano et al., 1996; Higuchi et al., 1998; Sasaki et al., 2002; Ikeda et al., 2005). Accordingly, PLS3 has been found to be an important marker in predicting the effectiveness of chemotherapeutics in the treatment of cancers (Hisano et al., 1996; Higuchi et al., 1998; Xin et al., 2020).
Despite significant advancements over the years, a comprehensive understanding of the PLS3 structure and biomechanics remains elusive. Of particular interest is obtaining a complete high-resolution structure of plastins including both the RD and actin-binding core, as this discovery would aid in deciphering a detailed molecular mechanism of PLS3 regulation by Ca2+. Furthering our understanding of the molecular and structural aspects of PLS3 bundling and regulation will be critical to uncover the role(s) of PLS3 in cells and tissues and shed light on how pathogenic variants of PLS3 lead to OI. It remains intriguing and obscure how mutations in the ubiquitously expressed cytoskeletal protein PLS3 result in pathogenic phenotype exclusively in bone tissue. A possible explanation is partial functional redundancy with other plastin isoforms (or other actin-bundling proteins). In this regard, it will be important to analyze the expression patterns of the less ubiquitous PLS1 and PLS2 isoforms in different tissues in response to the impact from the pathogenic PLS3 variants.
PLS3 Pathogenic Variants Identified in X-Linked Osteogenesis Imperfecta and Their Clinical Manifestations
To date, 27 OI-associated pathogenic variants (including coding mutations and gene deletions/truncations) have been identified in the PLS3 gene located on the X chromosome (Table 1). The majority of these are expected to result in the absence of functional protein: four full or partial PLS3 gene deletions (Kämpe et al., 2017a; Kannu et al., 2017; Lv et al., 2017), seven frame-shift mutations resulting in truncated mRNA constructs (Fahiminiya et al., 2014; Nishi et al., 2016; Kämpe et al., 2017a,b; Lv et al., 2017), six nonsense mutations (Fahiminiya et al., 2014; Kämpe et al., 2017b; Balasubramanian et al., 2018; Chen et al., 2018; Wang et al., 2020), and one aberrant splicing variant (Cao et al., 2019). All but two of the resulting truncated mRNA molecules appear to be the canonical substrates for degradation by nonsense-mediated mRNA decay (NMD) (Miller and Pearce, 2014; Kishor et al., 2019). Indeed, in several tested cases, the pathogenic variants of PLS3 have not been detected at the protein level in tissues from OI patients (Van Dijk et al., 2013; Wang et al., 2020). The two variants, which do not meet the criteria for degradation by the NMD pathway may generate truncated proteins, which, however, are highly unstable upon expression in Escherichia coli and are unlikely to be functional when expressed in vivo (Schwebach et al., 2020).
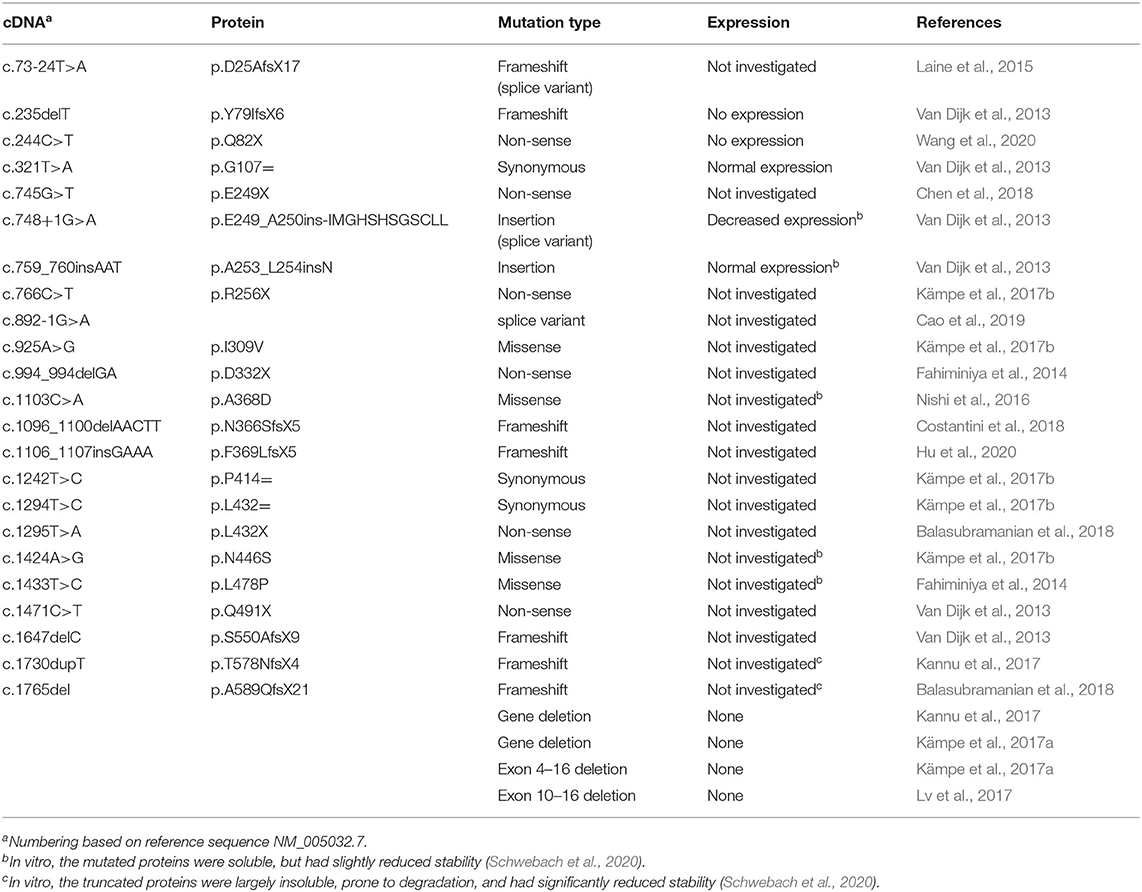
Table 1. List of identified OI-linked PLS3 variants.
Of particular interest in further understanding of PLS3's role in OI are variants that generate full-length mutated proteins. In two cases, the nucleotide insertions do not result in a frame shift and give rise to full-length PLS3 protein variants containing one (p.A253_L254insN) or twelve (p.E249_A250insIMGHSHSGSCLL) extra amino acids (Van Dijk et al., 2013). In three other cases, full-length OI-linked proteins result from missense mutations p.A368D, p.N446S, and p.L478P (Fahiminiya et al., 2014; Nishi et al., 2016; Kämpe et al., 2017b). Structural and functional consequences of these variants are discussed below.
The severity of OI clinical manifestations does not appear to directly correlate with a specific pathogenic PLS3 variant and varies greatly from the absence of physical symptoms to severe osteoporosis and dysmorphia (Makitie et al., 2019). While all patients have classical signs of osteoporosis with thin trabeculae and scarce osteoid seams, in general, hemizygous males are more affected than heterozygous females (Makitie et al., 2019). Phenotypes vary in females from mild to severe with variations existing within families (Kämpe et al., 2017b), which may be explained by variability in inactivation of the mutated X chromosome. Low bone mineral density (BMD) is common for all PLS3 OI variants, however its extent varies. Bone fractures are a functional consequence of decreased BMD and constitute another universal trait in PLS3-related OI. Vertebrae compression fractures, as well as long bone fractures resulting from minor trauma are common. In more severe cases, phenotypes can include facial dismorphism, clumsy gait, joint hyperlaxity, kyphosis, deafness, and blue sclerae (Fahiminiya et al., 2014; Nishi et al., 2016; Kämpe et al., 2017a,b; Lv et al., 2017; Chen et al., 2018; Costantini et al., 2018; Cao et al., 2019; Hu et al., 2020).
Importance of PLS3 for Proper Bone Formation
Animal model studies have emphasized an importance of PLS3 in bone tissue development and support of healthy bone architecture. A Pls3 knock down in zebrafish manifests in significant skeletal and muscular abnormalities (Van Dijk et al., 2013), while Pls3 knock out mice show significant osteoporosis and decreased bone strength mirroring human OI symptoms (Neugebauer et al., 2018; Yorgan et al., 2019). Concordantly, PLS3 over expression in mice has resulted in increased bone strength and a thickened cortical bone (Neugebauer et al., 2018). However, the molecular mechanisms underlying the PLS3 function in bone tissue and whether PLS3 actin-bundling ability is important for bone homeostasis remain elusive.
To date, few studies have addressed the mechanisms by which PLS3 variants cause OI. Strong hypomineralization of the bone matrix is a common finding but is not thought to be due to increased bone turnover, albeit evidence of increased bone resorption has been reported (Laine et al., 2015). Bone shows reduced mineralizing surface and adjusted mineral apposition rate, while mineralization lag time is significantly increased (Laine et al., 2015; Kämpe et al., 2017a). A link between PLS3 and mineralization has been supported by increased expression of PLS3 in osteoblastic MC3T3-E1 cells upon mineralization (Fahiminiya et al., 2014) as well as the presence of PLS3 in matrix vesicles (Thouverey et al., 2011; Kim et al., 2013) generated from the apical microvilli of osteoblasts during mineralization (Thouverey et al., 2011). The presence of PLS3 in matrix vesicles correlates with its function as a regulator of apical microvilli (Fath and Burgess, 1995; Volkmann et al., 2001; Delanote et al., 2005) and membrane trafficking (Hagiwara et al., 2011; Wottawa et al., 2017). However, it remains unknown whether PLS3 contributes to the role of matrix vesicles in bone mineralization.
Calcium Regulation Is Key to PLS3 Activity and Drives Its Localization in Cells
A recent study has revealed the molecular consequences of OI-linked full-length PLS3 variants (p.E249_A250insIMGHSHSGSCLL, p.A253_L254insN, p.A368D, p.N446S, and p.L478P) at the protein and cellular levels (Schwebach et al., 2020). These variants segregate into three groups (Figure 1C) with distinct biochemical properties and intracellular localization. The p.L478P variant has been found to bind actin through intact ABD1 while being unable to bundle actin filaments in vitro. High-resolution cryo-EM reconstruction of ABD2 bound to F-actin has provided structural insights for this deficiency: the L478P mutation disrupts the structure of a conserved loop involved in F-actin binding. In cells, wild-type PLS3 has been shown to localize to actin-rich structures in lamellipodia and focal adhesions. This localization is severely affected for the bundling-deficient p.L478P, which, despite actin binding via ABD1, failed to associate with the actin cytoskeleton. This finding implies that F-actin bundling, rather than binding, is the primary function of PLS3 required for its proper intracellular localization including its association with the branched actin network at the leading edge. Therefore, similar to other loss-of-function variants (frame-shift, non-sense, and PLS3 gene deletions), the L478P missense mutation results in non-functional (i.e., non-bundling) PLS3 as a likely cause of OI.
More intriguingly, not only is PLS3 actin-bundling activity important, but its finely tuned Ca2+ regulation is essential for proper bone formation (Schwebach et al., 2020). Indeed, it has been demonstrated that the remaining full-length OI-causative PLS3 variants are capable to bundle actin, but their response to Ca2+ is perturbed toward either hypersensitivity (p.A253_L254insN and p.N446S) or hyposensitivity (p.E249_A250insIMGHSHSGSCLL and p.A368D). The altered sensitivity to Ca2+ affects the localization of PLS3 within cells: variants hypersensitive to Ca2+ associate more strongly with the branched actin network in the lamellipodia and are largely excluded from focal adhesions, while Ca2+-insensitive variants are depleted from the lamellipodia and localize primarily at significantly enlarged focal adhesions and in stress fibers. Moreover, such differential localization of PLS3 is Ca2+-dependent, as Ca2+ depletion from the cell culture medium has resulted in redistribution of PLS3 from the lamellipodia to focal adhesion sites, implying that Ca2+ aids in cycling of PLS3 between aligned actin bundles in focal adhesions and the branched actin network at the leading edge. This suggests that fine regulation of PLS3 by Ca2+ is critical to bone formation as its imbalance in either direction results in OI, comparable to that resulting from the loss of PLS3 (Figure 1C).
It should be restated that to date, no complete structure of any plastin/fimbrin has been described and the location of the RD relative to the core remained unknown until recently. While the structures of the actin-binding core (Klein et al., 2004) and the regulatory domain (Ishida et al., 2017) have been solved, a molecular model of how they are associated with each other has not been produced. Analysis of four distinct OI-linked PLS3 variants resulting in opposing Ca2+ sensitivities has suggested that the RD is positioned at the interface between ABD1 and ABD2. Homology modeling suggests that flexible loops between CH1-CH2 (containing residues E249-A250) and CH2-CH3 reside in proximity and together likely represent the RD docking site (Figure 1A). This hypothesis has been confirmed by differential labeling of two designed cysteine residues in the absence and presence of the RD (Schwebach et al., 2020).
Molecular dynamics simulations of one PLS3 variant, p.E249_A250insIMGHSHSGSCLL, has predicted the conformational changes in a loop between CH1 and CH2 in ABD1 (Chen et al., 2018). Chen and coauthors suggest that, despite the addition of 12 amino acids, the mutant showed less conformational change than the wild-type protein and a more compact structure via a tighter association of the loop with CH3 of ABD2 (Chen et al., 2018). The simulation, however, used a homologous model of the protein and, therefore, must be revised after the high-resolution structure becomes available.
Potential Mechanisms by Which PLS3 Pathogenic Variants Perturb Osteogenesis
The mechanisms by which PLS3 affects osteogenesis/bone remodeling remain largely unknown. Three main hypotheses of PLS3-related osteogenesis imperfecta mechanisms have emerged (Figure 1D): (1) insufficient mineralization by osteoblasts (Fahiminiya et al., 2014), (2) increased bone resorption by osteoclasts (Neugebauer et al., 2018), and (3) dysregulation of osteocyte mechanosensing leading to misbalance between bone resorption and formation (Van Dijk et al., 2013).
(1) The effect of PLS3 on bone mineralization by osteoblasts has been inferred in multiple studies focusing on the level of bone mineralization in patients. Fahiminiya and colleagues found that in vitro differentiation of cultured mouse cranial MC3T3-E1 osteoblasts correlated with the increased expression of PLS3 (Fahiminiya et al., 2014), indirectly implying that PLS3 may be involved in bone mineralization. PLS3 deletions have been shown to cause significant hypomineralization of the bone matrix (Kämpe et al., 2017a). This is in contrast to OI caused by COL1A1 or COL1A2 variants, which result in hypermineralized bone matrix (Roschger et al., 2008). Regulation of intracellular vesicles in late osteoblasts transitioning to early osteocytes is thought to drive bone mineralization (Hearn and Russell, 1981; Barragan-Adjemian et al., 2006). PLS3 is recognized as a regulator of vesicle trafficking (Hagiwara et al., 2011; Wottawa et al., 2017) and is upregulated in matrix vesicles and microvilli of osteoblasts upon mineralization (Thouverey et al., 2011; Kim et al., 2013) further suggesting a link between PLS3 variants and altered bone matrix mineralization.
(2) Contribution of PLS3 to bone resorption would be surprising given that osteoclasts as all cells of hematopoietic origin express PLS2 as the primary plastin isoform (Ma et al., 2010; Si et al., 2018; Li et al., 2020). Thus far, endogenous expression of PLS3 has not been clearly demonstrated in osteoclasts in vivo. Accordingly, PLS3 has been shown to have no effect on osteoclast activity including bone resorption (Fahiminiya et al., 2014; Kämpe et al., 2017a). Yet, more recent work has proposed a role for PLS3 in osteoclast activity through the regulation of podosomes by NFκB signaling (Neugebauer et al., 2018). Since these findings are based on either overexpression or knockout of PLS3 in mouse models, evaluation of the effects of endogenously expressed pathogenic variants of PLS3 would be required to verify the validity of this hypothesis.
(3) Involvement of PLS3 in mechanosensing by osteocytes has also been hypothesized (Van Dijk et al., 2013; Laine et al., 2015). Recent studies demonstrating the importance of Ca2+ regulation in the PLS3 actin-bundling activity (Schwebach et al., 2017, 2020) have added further credence to this hypothesis. Embedded in bone matrix, osteocytes orchestrate bone remodeling by coordinated secretion of factors regulating both bone lysis by osteoclasts and bone construction by osteoblasts (reviewed in Niedzwiedzki and Filipowska, 2015) (Figure 1D). Therefore, being the major plastin isoform in osteocytes, PLS3 could contribute to both osteogenesis and osteolysis. Intriguingly, PLS3 is enriched in dendrites, and specifically at their branching points (Kamioka et al., 2004), which are recognized as the structures by which osteocytes sense their environment (Kamioka et al., 2004; Galli et al., 2010). Furthermore, the activity of integrin/adhesion-dependent voltage-gated calcium channels is a strong candidate for the driving force of bone formation and remodeling (O'Neill and Galasko, 2000; Li et al., 2002; Cao et al., 2019; Sun et al., 2019). The findings that Ca2+ is involved in the redistribution of PLS3 from focal adhesions to the leading edge (Schwebach et al., 2020) represents a strong link between the activities of PLS3 and the machinery thought to drive bone mechanosensing and reorganization. Future work in osteocytes is required to determine the specific role of PLS3 and its regulation by Ca2+ in these activities.
Conclusion
PLS3-mediated osteogenesis imperfecta is an exciting new field of study with implications in the clinic as well as the fields of bone development and actin biochemistry. Uncovering the mechanisms by which PLS3 causes OI has already contributed to our understanding of PLS3's roles and regulation in cells as well as guiding ongoing structural work. Future discoveries testing the hypotheses described above should reveal not only the mechanisms of X-linked osteoporosis but more broadly answer lingering questions about bone mineralization and mechanosensing. Integration of future PLS3 biochemistry and bone development studies could lead to targeted therapeutics for X-linked OI.
Author Contributions
CLS designed and drafted the manuscript and prepared the table. EK and DSK edited the manuscript and prepared the figures. All authors revised the manuscript and approved the submitted version.
Funding
This work was supported by the NIH GM114666 grant (to DSK) and the Ohio State University Cancer Comprehensive Center Pelotonia Graduate Fellowship (to CLS). Any opinions, findings, and conclusions expressed in this material are those of the authors and do not necessarily reflect those of NIH and the Pelotonia Fellowship Program.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ackermann, B., Krober, S., Torres-Benito, L., Borgmann, A., Peters, M., Hosseini Barkooie, S. M., et al. (2013). Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum. Mol. Genet. 22, 1328–1347. doi: 10.1093/hmg/dds540
Alrafiah, A., Karyka, E., Coldicott, I., Iremonger, K., Lewis, K. E., Ning, K., et al. (2018). Plastin 3 promotes motor neuron axonal growth and extends survival in a mouse model of spinal muscular atrophy. Mol. Ther. Methods Clin. Dev. 9, 81–89. doi: 10.1016/j.omtm.2018.01.007
Balasubramanian, M., Fratzl-Zelman, N., O'Sullivan, R., Bull, M., Peel, N. F. A., Pollitt, R. C., et al. (2018). Novel PLS3 variants in X-linked osteoporosis: exploring bone material properties. Am. J. Med. Genet. 176, 1578–1586. doi: 10.1002/ajmg.a.38830
Barragan-Adjemian, C., Nicolella, D., Dusevich, V., Dallas, M. R., Eick, J. D., and Bonewald, L. F. (2006). Mechanism by which MLO-A5 late osteoblasts/early osteocytes mineralize in culture: similarities with mineralization of lamellar bone. Calcif. Tissue Int. 79, 340–353. doi: 10.1007/s00223-006-0107-2
Behrmann, E., Muller, M., Penczek, P. A., Mannherz, H. G., Manstein, D. J., and Raunser, S. (2012). Structure of the rigor actin-tropomyosin-myosin complex. Cell 150, 327–338. doi: 10.1016/j.cell.2012.05.037
Cao, Y. J., Zhang, H., and Zhang, Z. L. (2019). Novel Mutations in the Wnt1, Tmem38b, P4hb, and Pls3 genes in four unrelated chinese families with osteogenesis imperfecta. Endocr. Pract. 25, 230–241. doi: 10.4158/EP-2018-0443
Chen, T., Wu, H., Zhang, C., Feng, J., Chen, L., Xie, R., et al. (2018). Clinical, genetics, and bioinformatic characterization of mutations affecting an essential region of PLS3 in patients with BMND18. Int. J. Endocrinol. 2018, 1–9. doi: 10.1155/2018/8953217
Christensen, J. R., Homa, K. E., Morganthaler, A. N., Brown, R. R., Suarez, C., Harker, A. J., et al. (2019). Cooperation between tropomyosin and alpha-actinin inhibits fimbrin association with actin filament networks in fission yeast. Elife 8:e47279. doi: 10.7554/eLife.47279
Costantini, A., Krallis, P. N., Kämpe, A., Karavitakis, E. M., Taylan, F., Mäkitie, O., et al. (2018). A novel frameshift deletion in PLS3 causing severe primary osteoporosis. J. Hum. Genet. 63, 923–926. doi: 10.1038/s10038-018-0472-5
Delanote, V., Vandekerckhove, J., and Gettemans, J. (2005). Plastins: versatile modulators of actin organization in (patho)physiological cellular processes. Acta Pharmacol. Sin. 26, 769–779. doi: 10.1111/j.1745-7254.2005.00145.x
Ding, W. Y., Ong, H. T., Hara, Y., Wongsantichon, J., Toyama, Y., Robinson, R. C., et al. (2017). Plastin increases cortical connectivity to facilitate robust polarization and timely cytokinesis. J. Cell Biol. 216, 1371–1386. doi: 10.1083/jcb.201603070
Dor-On, E., Raviv, S., Cohen, Y., Adir, O., Padmanabhan, K., and Luxenburg, C. (2017). T-plastin is essential for basement membrane assembly and epidermal morphogenesis. Sci. Signal. 10:eaal3154. doi: 10.1126/scisignal.aal3154
Fahiminiya, S., Majewski, J., Al-Jallad, H., Moffatt, P., Mort, J., Glorieux, F. H., et al. (2014). Osteoporosis caused by mutations in PLS3: clinical and bone tissue characteristics. J. Bone Miner. Res. 29, 1805–1814. doi: 10.1002/jbmr.2208
Fath, K. R., and Burgess, D. R. (1995). Microvillus assembly. Not actin alone. Curr. Biol. 5, 591–593. doi: 10.1016/S0960-9822(95)00117-5
Gagliardi, A., Besio, R., Carnemolla, C., Landi, C., Armini, A., Aglan, M., et al. (2017). Cytoskeleton and nuclear lamina affection in recessive osteogenesis imperfecta: a functional proteomics perspective. J. Proteomics 167, 46–59. doi: 10.1016/j.jprot.2017.08.007
Galli, C., Passeri, G., and Macaluso, G. M. (2010). Osteocytes and WNT: the mechanical control of bone formation. J. Dent. Res. 89, 331–343. doi: 10.1177/0022034510363963
Garbett, D., Bisaria, A., Yang, C., McCarthy, D. G., Hayer, A., Moerner, W. E., et al. (2020). T-Plastin reinforces membrane protrusions to bridge matrix gaps during cell migration. Nat. Commun. 11:4818. doi: 10.1038/s41467-020-18586-3
Hagiwara, M., Shinomiya, H., Kashihara, M., Kobayashi, K., Tadokoro, T., and Yamamoto, Y. (2011). Interaction of activated Rab5 with actin-bundling proteins, L- and T-plastin and its relevance to endocytic functions in mammalian cells. Biochem. Biophys. Res. Commun. 407, 615–619. doi: 10.1016/j.bbrc.2011.03.082
Hearn, P. R., and Russell, R. G. G. (1981). The formation and orientation of brush border vesicles from rat duodenal mucosa. J. Cell Sci. 47, 227–236.
Higuchi, Y., Kita, K., Hakanishi, H., Wang, X. L., Sugaya, S., Tanzawa, H., et al. (1998). Search for genes involved in UV-resistance in human cells by mRNA differential display: increased transcriptional expression of nucleophosmin and T-plastin genes in association with the resistance. Biochem. Biophys. Res. Commun. 248, 597–602. doi: 10.1006/bbrc.1998.8978
Hisano, T., Ono, M., Nakayama, M., Naito, S., Kuwano, M., and Wada, M. (1996). Increased expression of T-plastin gene in cisplatin-resistant human cancer cells: identification by mRNA differential display. FEBS Lett. 397, 101–107. doi: 10.1016/S0014-5793(96)01150-7
Hu, J., Li, L. J., Zheng, W. B., Zhao, D. C., Wang, O., Jiang, Y., et al. (2020). A novel mutation in PLS3 causes extremely rare X-linked osteogenesis imperfecta. Mol. Genet. Genomic Med. 8:e1525. doi: 10.1002/mgg3.1525
Ikeda, H., Sasaki, Y., Kobayashi, T., Suzuki, H., Mita, H., Toyota, M., et al. (2005). The role of T-fimbrin in the response to DNA damage: silencing of T-fimbrin by small interfering RNA sensitizes human liver cancer cells to DNA-damaging agents. Int. J. Oncol. 27, 933–940. doi: 10.3892/ijo.27.4.933
Ishida, H., Jensen, K. V., Woodman, A. G., Hyndman, M. E., and Vogel, H. J. (2017). The calcium-dependent switch helix of L-plastin regulates actin bundling. Sci. Rep. 7, 1–12. doi: 10.1038/srep40662
Kaifer, K. A., Villalon, E., Osman, E. Y., Glascock, J. J., Arnold, L. L., Cornelison, D. D. W., et al. (2017). Plastin-3 extends survival and reduces severity in mouse models of spinal muscular atrophy. JCI Insight 2:e89970. doi: 10.1172/jci.insight.89970
Kamioka, H., Sugawara, Y., Honjo, T., Yamashiro, T., and Takano-Yamamoto, T. (2004). Terminal differentiation of osteoblasts to osteocytes is accompanied by dramatic changes in the distribution of actin-binding proteins. J. Bone Miner. Res. 19, 471–478. doi: 10.1359/JBMR.040128
Kämpe, A. J., Costantini, A., Levy-Shraga, Y., Zeitlin, L., Roschger, P., Taylan, F., et al. (2017a). PLS3 deletions lead to severe spinal osteoporosis and disturbed bone matrix mineralization. J. Bone Miner. Res. 32, 2394–2404. doi: 10.1002/jbmr.3233
Kämpe, A. J., Costantini, A., Mäkitie, R. E., Jäntti, N., Valta, H., Mäyränpää, M., et al. (2017b). PLS3 sequencing in childhood-onset primary osteoporosis identifies two novel disease-causing variants. Osteoporosis Int. 28, 3023–3032. doi: 10.1007/s00198-017-4150-9
Kannu, P., Mahjoub, A., Babul-Hirji, R., Carter, M. T., and Harrington, J. (2017). PLS3 mutations in X-linked osteoporosis: clinical and bone characteristics of two novel mutations. Horm. Res. Paediatr. 88, 298–304. doi: 10.1159/000477242
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015.053
Kim, J. M., Kim, J., Kim, Y. H., Kim, K. T., Ryu, S. H., Lee, T. G., et al. (2013). Comparative secretome analysis of human bone marrow-derived mesenchymal stem cells during osteogenesis. J. Cell. Physiol. 228, 216–224. doi: 10.1002/jcp.24123
Kishor, A., Fritz, S. E., and Hogg, J. R. (2019). Nonsense-mediated mRNA decay: the challenge of telling right from wrong in a complex transcriptome. Wiley Interdiscip. Rev. RNA 10:e1548. doi: 10.1002/wrna.1548
Klein, M. G., Shi, W., Ramagopal, U., Tseng, Y., Wirtz, D., Kovar, D. R., et al. (2004). Structure of the actin crosslinking core of fimbrin. Structure 12, 999–1013. doi: 10.1016/j.str.2004.04.010
Laine, C. M., Wessman, M., Toiviainen-Salo, S., Kaunisto, M. A., Mäyränpää, M. K., Laine, T., et al. (2015). A novel splice mutation in PLS3 causes X-linked early onset low-turnover osteoporosis. J. Bone Miner. Res. 30, 437–445. doi: 10.1002/jbmr.2355
Laporte, D., Ojkic, N., Vavylonis, D., and Wu, J. Q. (2012). α-Actinin and fimbrin cooperate with myosin II to organize actomyosin bundles during contractile-ring assembly. Mol. Biol. Cell 23, 3094–3110. doi: 10.1091/mbc.e12-02-0123
Leite, J., Chan, F. Y., Osorio, D. S., Saramago, J., Sobral, A. F., Silva, A. M., et al. (2020). Equatorial non-muscle myosin II and plastin cooperate to align and compact F-actin bundles in the cytokinetic ring. Front. Cell Dev. Biol. 8:573393. doi: 10.3389/fcell.2020.573393
Li, J., Duncan, R. L., Burr, D. B., and Turner, C. H. (2002). L-type calcium channels mediate mechanically induced bone formation in vivo. J. Bone Miner. Res. 17, 1795–1800. doi: 10.1359/jbmr.2002.17.10.1795
Li, N., Wong, C. K., and Cheng, C. Y. (2016). Plastins regulate ectoplasmic specialization via its actin bundling activity on microfilaments in the rat testis. Asian J. Androl. 18, 716–722. doi: 10.4103/1008-682X.166583
Li, X., Wang, L., Huang, B., Gu, Y., Luo, Y., Zhi, X., et al. (2020). Targeting actin-bundling protein L-plastin as an anabolic therapy for bone loss. Sci. Adv. 6:eabb7135. doi: 10.1126/sciadv.abb7135
Lim, J., Grafe, I., Alexander, S., and Lee, B. (2017). Genetic causes and mechanisms of Osteogenesis Imperfecta. Bone 102, 40–49. doi: 10.1016/j.bone.2017.02.004
Lin, C. S., Aebersold, R. H., Kent, S. B., Varma, M., and Leavitt, J. (1988). Molecular cloning and characterization of plastin, a human leukocyte protein expressed in transformed human fibroblasts. Mol. Cell. Biol. 8, 4659–4668. doi: 10.1128/MCB.8.11.4659
Lin, C. S., Lau, A., Huynh, T., and Lue, T. F. (1999). Differential regulation of human T-plastin gene in leukocytes and non-leukocytes: identification of the promoter, enhancer, and CpG Island. DNA Cell Biol. 18, 27–37. doi: 10.1089/104454999315592
Lin, C. S., Park, T., Chen, Z. P., and Leavitt, J. (1993). Human Plastin Genes: comparative gene structure, chromosome location, and differential expression in normal and neoplastic cells. J. Biol. Chem. 268, 2781–2792.
Lindahl, K., Åström, E., Rubin, C. J., Grigelioniene, G., Malmgren, B., Ljunggren, Ö., et al. (2015). Genetic epidemiology, prevalence, and genotype-phenotype correlations in the Swedish population with osteogenesis imperfecta. Eur. J. Hum. Genet. 23, 1042–1050. doi: 10.1038/ejhg.2015.81
Lv, F., Ma, M., Liu, W., Xu, X., Song, Y., Li, L., et al. (2017). A novel large fragment deletion in PLS3 causes rare X-linked early-onset osteoporosis and response to zoledronic acid. Osteoporosis Int. 28, 2691–2700. doi: 10.1007/s00198-017-4094-0
Lyon, A. N., Pineda, R. H., Hao Le, T., Kudryashova, E., Kudryashov, D. S., and Beattie, C. E. (2014). Calcium binding is essential for plastin 3 function in Smn-deficient motoneurons. Hum. Mol. Genet. 23, 1990–2004. doi: 10.1093/hmg/ddt595
Ma, T., Sadashivaiah, K., Madayiputhiya, N., and Chellaiah, M. A. (2010). Regulation of sealing ring formation by L-plastin and cortactin in osteoclasts. J. Biol. Chem. 285, 29911–29924. doi: 10.1074/jbc.M109.099697
Makitie, R. E., Costantini, A., Kampe, A., Alm, J. J., and Makitie, O. (2019). New insights into monogenic causes of osteoporosis. Front. Endocrinol. 10:70. doi: 10.3389/fendo.2019.00070
Marini, J. C., Forlino, A., Bachinger, H. P., Bishop, N. J., Byers, P. H., Paepe, A., et al. (2017). Osteogenesis imperfecta. Nat. Rev. Dis. Primers 3:17052. doi: 10.1038/nrdp.2017.52
Miller, J. N., and Pearce, D. A. (2014). Nonsense-mediated decay in genetic disease: friend or foe? Mutat. Res. Rev. Mutat. Res. 762, 52–64. doi: 10.1016/j.mrrev.2014.05.001
Neugebauer, J., Heilig, J., Hosseinibarkooie, S., Ross, B. C., Mendoza-Ferreira, N., Nolte, F., et al. (2018). Plastin 3 influences bone homeostasis through regulation of osteoclast activity. Hum. Mol. Genet. 27, 4249–4262. doi: 10.1093/hmg/ddy318
Niedzwiedzki, T., and Filipowska, J. (2015). Bone remodeling in the context of cellular and systemic regulation: the role of osteocytes and the nervous system. J. Mol. Endocrinol. 55, R23–R36. doi: 10.1530/JME-15-0067
Nijhuis, W. H., Eastwood, D. M., Allgrove, J., Hvid, I., Weinans, H. H., Bank, R. A., et al. (2019). Current concepts in osteogenesis imperfecta: bone structure, biomechanics and medical management. J. Child. Orthop. 13, 1–11. doi: 10.1302/1863-2548.13.180190
Nishi, E., Masuda, K., Arakawa, M., Kawame, H., Kosho, T., Kitahara, M., et al. (2016). Exome sequencing-based identification of mutations in non-syndromic genes among individuals with apparently syndromic features. Am. J. Med. Genet. 170, 2889–2894. doi: 10.1002/ajmg.a.37826
O'Neill, C. A., and Galasko, C. S. (2000). Calcium mobilization is required for spreading in human osteoblasts. Calcif. Tissue Int. 67, 53–59. doi: 10.1007/s00223001097
Palomo, T., Vilaca, T., and Lazaretti-Castro, M. (2017). Osteogenesis imperfecta: diagnosis and treatment. Curr. Opin. Endocrinol. Diabetes Obes. 24, 381–388. doi: 10.1097/MED.0000000000000367
Pan, L., Lin, Z., Tang, X., Tian, J., Zheng, Q., Jing, J., et al. (2020). S-nitrosylation of plastin-3 exacerbates thoracic aortic dissection formation via endothelial barrier dysfunction. Arterioscler. Thromb. Vasc. Biol. 40, 175–188. doi: 10.1161/ATVBAHA.119.313440
Robinson, M. E., and Rauch, F. (2019). Mendelian bone fragility disorders. Bone 126, 11–17. doi: 10.1016/j.bone.2019.04.021
Roschger, P., Fratzl-Zelman, N., Misof, B. M., Glorieux, F. H., Klaushofer, K., and Rauch, F. (2008). Evidence that abnormal high bone mineralization in growing children with osteogenesis imperfecta is not associated with specific collagen mutations. Calcif. Tissue Int. 82, 263–270. doi: 10.1007/s00223-008-9113-x
Rossi, V., Lee, B., and Marom, R. (2019). Osteogenesis imperfecta: advancements in genetics and treatment. Curr. Opin. Pediatr. 31, 708–715. doi: 10.1097/MOP.0000000000000813
Sasaki, Y., Itoh, F., Kobayashi, T., Kikuchi, T., Suzuki, H., Toyota, M., et al. (2002). Increased expression of T-fimbrin gene after DNA damage in CHO cells and inactivation of T-fimbrin by CpG methylation in human colorectal cancer cells. Int. J. Cancer 97, 211–216. doi: 10.1002/ijc.1587
Schaffner-Reckinger, E., and Machado, R. A. C. (2020). The actin-bundling protein L-plastin-A double-edged sword: Beneficial for the immune response, maleficent in cancer. Int. Rev. Cell. Mol. Biol. 355, 109–154. doi: 10.1016/bs.ircmb.2020.05.004
Schwebach, C. L., Agrawal, R., Lindert, S., Kudryashova, E., and Kudryashov, D. S. (2017). The roles of actin-binding domains 1 and 2 in the calcium-dependent regulation of actin filament bundling by human plastins. J. Mol. Biol. 429, 2490–2508. doi: 10.1016/j.jmb.2017.06.021
Schwebach, C. L., Kudryashova, E., Zheng, Q., Orchard, M., Smith, H., Runyan, L. A., et al. (2020). Osteogenesis imperfecta mutations in plastin 3 lead to impaired calcium regulation of actin bundling. Bone Res. 8, 1–13. doi: 10.1038/s41413-020-0095-2
Shinomiya, H. (2012). Plastin family of actin-bundling proteins: its functions in leukocytes, neurons, intestines, and cancer. Int. J. Cell Biol. 2012, 1–8. doi: 10.1155/2012/213492
Si, M., Goodluck, H., Zeng, C., Pan, S., Todd, E. M., Morley, S. C., et al. (2018). LRRK1 regulation of actin assembly in osteoclasts involves serine 5 phosphorylation of L-plastin. J. Cell. Biochem. 119, 10351–10357. doi: 10.1002/jcb.27377
Skau, C. T., Courson, D. S., Bestul, A. J., Winkelman, J. D., Rock, R. S., Sirotkin, V., et al. (2011). Actin filament bundling by fimbrin is important for endocytosis, cytokinesis, and polarization in fission yeast. J. Biol. Chem. 286, 26964–26977. doi: 10.1074/jbc.M111.239004
Sun, Z., Costell, M., and Fassler, R. (2019). Integrin activation by talin, kindlin and mechanical forces. Nat. Cell Biol. 21, 25–31. doi: 10.1038/s41556-018-0234-9
Tauer, J. T., Robinson, M. E., and Rauch, F. (2019). Osteogenesis imperfecta: new perspectives from clinical and translational research. JBMR Plus 3:e10174. doi: 10.1002/jbm4.10174
Thouverey, C., Malinowska, A., Balcerzak, M., Strzelecka-Kiliszek, A., Buchet, R., Dadlez, M., et al. (2011). Proteomic characterization of biogenesis and functions of matrix vesicles released from mineralizing human osteoblast-like cells. J. Proteomics 74, 1123–1134. doi: 10.1016/j.jprot.2011.04.005
Van Dijk, F. S., Zillikens, M. C., Micha, D., Riessland, M., Marcelis, C. L., De Die-Smulders, C. E., et al. (2013). PLS3 mutations in X-linked osteoporosis with fractures. N. Engl. J. Med. 369, 1529–1536. doi: 10.1056/NEJMoa1308223
Volkmann, N., Derosier, D., Matsudaira, P., and Hanein, D. (2001). An atomic model of actin filaments cross-linked by fimbrin and its implications for bundle assembly and function. J. Cell Biol. 153, 947–956. doi: 10.1083/jcb.153.5.947
Wang, L., Bian, X., Cheng, G., Zhao, P., Xiang, X., Tian, W., et al. (2020). A novel nonsense variant in PLS3 causes X-linked osteoporosis in a Chinese family. Ann. Hum. Genet. 84, 92–96. doi: 10.1111/ahg.12344
Wottawa, M., Naas, S., Böttger, J., Van Belle, G. J., Möbius, W., Revelo, N. H., et al. (2017). Hypoxia-stimulated membrane trafficking requires T-plastin. Acta Physiol. 221, 59–73. doi: 10.1111/apha.12859
Xin, Z., Li, D., Mao, F., Du, Y., Wang, X., Xu, P., et al. (2020). PLS3 predicts poor prognosis in pancreatic cancer and promotes cancer cell proliferation via PI3K/AKT signaling. J. Cell Physiol. 235, 8416–8423. doi: 10.1002/jcp.29685
Keywords: plastin 3, X-linked osteoporosis, osteogenesis imperfecta, bone development, actin bundling, Ca2+-dependent regulation, PLS3, T-Plastin
Citation: Schwebach CL, Kudryashova E and Kudryashov DS (2021) Plastin 3 in X-Linked Osteoporosis: Imbalance of Ca2+-Dependent Regulation Is Equivalent to Protein Loss. Front. Cell Dev. Biol. 8:635783. doi: 10.3389/fcell.2020.635783
Received: 30 November 2020; Accepted: 28 December 2020;
Published: 21 January 2021.
Edited by:
Lei-Miao Yin, Shanghai University of Traditional Chinese Medicine, ChinaReviewed by:
Gerard Pals, Amsterdam University Medical Center, NetherlandsBrunhilde Wirth, University of Cologne, Germany
Roberta Besio, University of Pavia, Italy
Copyright © 2021 Schwebach, Kudryashova and Kudryashov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dmitri S. Kudryashov, kudryashov.1@osu.edu