Co-Translational Folding of Multi-Domain Proteins
 Nandakumar Rajasekaran1
Nandakumar Rajasekaran1  Christian M. Kaiser2,3*
Christian M. Kaiser2,3*- 1CMDB Graduate Program, Johns Hopkins University, Baltimore, MD, United States
- 2Department of Biology, Johns Hopkins University, Baltimore, MD, United States
- 3Department of Biophysics, Johns Hopkins University, Baltimore, MD, United States
The majority of proteins in nature are composed of multiple domains connected in a single polypeptide. How these long sequences fold into functional structures without forming toxic misfolds or aggregates is poorly understood. Their folding is inextricably linked to protein synthesis and interactions with cellular machinery, making mechanistic studies challenging. Recent progress has revealed critical features of multi-domain protein folding in isolation and in the context of translation by the ribosome. In this review, we discuss challenges and progress in understanding multi-domain protein folding, and highlight how molecular interactions shape folding and misfolding pathways. With the development of new approaches and model systems, the stage is now set for mechanistically exploring the folding of large multi-domain proteins.
Multi-domain Proteins
Proteins exhibit an astonishing functional diversity, playing essential roles in all cellular processes. Strikingly, this remarkable diversity is achieved with a surprisingly small number of building blocks. So far, approximately 1,500 unique folds have been identified in all known structures (Andreeva et al., 2020). The vast diversity observed in extant proteomes is achieved by combining domains (Doolittle, 1995) into a single polypeptide. The majority of proteins in all organisms are composed of multiple constituent domains, with some eukaryotic proteomes having their proportion of multi-domain proteins reach as much as four fifths (Han et al., 2007). Stringing together multiple domains into a single polypeptide chain therefore appears to be a successful evolutionary strategy to create proteins with novel functions and might also be attractive for designing new functions for synthetic applications (Poust et al., 2014; Kim et al., 2015a).
Interactions between domains in a large protein (Figure 1) are conceptually similar to the protein-protein interactions that, with their finely tuned specificities and affinities, are a main driver of biological complexity at the molecular level (Huttlin et al., 2021). This complexity makes detailed biophysical studies of multi-domain protein folding and dynamics challenging. Among the best-characterized models for interactions between domains that are connected by a linker are the eye lens crystallins, for which domain coupling has been elucidated in detail [reviewed in (Jaenicke, 1999)]. Recently, it was also shown that the folding of gamma-B crystallin is directed by early co-translational events (Buhr et al., 2016).
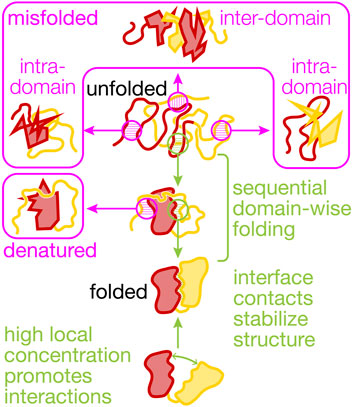
FIGURE 1. Productive and non-productive interactions between domains in a protein. The cartoons visualize non-productive interactions (magenta) and productive interactions (green) within and between two domains (N-terminal, red, and C-terminal, yellow) that shape multi-domain (mis)folding and dynamics. Co-translational folding abets sequential domain-wise folding, reducing inter-domain misfolding. Interactions with the ribosome and molecular chaperones further reduce misfolding and the destabilizing interactions between the unfolded yellow and the folded red domain that results in denaturation. Non-native states are prone to irreversible aggregation (not shown here). Interface contacts between the folded domains can be mutually stabilizing and are promoted by high local concentrations that result from covalently linking the domains in a single polypeptide chain.
Connecting domains covalently into a single polypeptide results in very high local concentrations. For example, two domains connected by a flexible glycine-serine linker of 20 amino acids have an effective concentration, defined as the concentration required to achieve the same encounter rate between two untethered domains, of 4 mM (Sørensen and Kjaergaard, 2019). The identity of the intervening linker sequence affects the local concentration effect (Sørensen and Kjaergaard, 2019; Kjaergaard, 2022), providing a way of potentially modulating these interactions, as has also been shown for the crystallins (Jaenicke, 1999). Combining two domains together thus increases their likelihood of interacting between two domains (Sivaramakrishnan and Spudich, 2011), which may tune the rate of catalyzed reactions (Dyla and Kjaergaard, 2020). The enhanced encounter rate may be crucial for stabilizing protein domains (Robinson and Sauer, 1998; Jäger and Plückthun, 1999) and to promote proximity between functional units.
Experimental Approaches
One potential evolutionary limitation of stringing together domains into proteins with novel functions is the requirement that the resulting multi-domain protein must be able to fold into a functional structure (Han et al., 2007). Indeed, misfolding between unfolded domains has been demonstrated to significantly challenge the refolding of multi-domain proteins in vitro (Fernandez et al., 1999; Borgia et al., 2011; Jahn et al., 2016; Liu et al., 2017), making experimental studies challenging. Misfolding, aggregation and the formation of partially folded intermediates result in complex measured signals that are difficult to resolve with the traditional methods that have been highly successful for studying small proteins or individual domains (Bartlett and Radford, 2009). In addition, averaging over ensembles of stochastically folding molecules makes it challenging to resolve individual steps in the multi-step folding pathways of large proteins. Single-molecule approaches are a particularly promising avenue toward resolving the folding of larger multi-domain proteins.
Following the folding of individual molecules circumvents the problems of aggregation and ensemble averaging. Single-molecule fluorescence methods have allowed direct visualization of different conformations of multi-domain proteins at equilibrium (Huang et al., 2009) and measurement of global refolding kinetics (Borgia et al., 2011). Force spectroscopy approaches, with their ability to selectively perturb the stability of specific domains while leaving the rest of the protein folded, are ideally suited to dissect multi-domain protein folding. Single-molecule manipulation with optical tweezers permits the detailed characterization of energetics and kinetics (Bustamante et al., 2020). Importantly, it also provides a means of observing the folding of a protein of interest against the complex backdrop of the molecular machinery that is crucial for efficient folding of multi-domain proteins.
Co-Translational Folding
The mode of cellular protein synthesis is a key factor for the folding of multi-domain proteins and, therefore, their evolution. Phillips noted five decades ago that “it seems useful to note that protein molecules are synthesized from their terminal amino ends [...] and to suggest that the folding process may begin during synthesis.” (Phillips, 1967). At this time, co-translational folding (and assembly into a functional tetramer) had already been suggested by detecting enzymatic activity of ribosome-bound β-galactosidase (Zipser and Perrin, 1963). Subsequently, co-translational folding was demonstrated for a number of proteins, including immunoglobulin domains (Bergman and Kuehl, 1979) and firefly luciferase (Frydman et al., 1994) (see Fedorov and Baldwin, 1997 for a review). The slow addition of amino acids by the ribosome during translation of the genetic information gives the nascent protein ample time to populate stably folded intermediates. Timely structuring of emerging nascent chain segments reduces the accumulation of unfolded domains, thus enabling temporal sequestration of the folding of individual domains and counteracting misfolding. Recently, interactions with the ribosome and with molecular chaperones were shown to be crucial for efficiently producing functional multi-domain proteins (Deuerling et al., 2019; Kramer et al., 2019; Liutkute et al., 2020a; Cassaignau et al., 2020; Maciuba et al., 2021). Experimentally studying the process requires overcoming significant challenges (see below), and the number of folding studies analyzing multi-domain proteins is still small compared to those investigating single-domain proteins (Braselmann et al., 2013). Here, we discuss some of the recent progress that has been made toward defining principles of multi-domain protein folding both in the context of translation by the ribosome and in isolation.
The ribosome is itself a large complex of several megadaltons made of RNA and protein. Most spectroscopic techniques cannot discern the signal from the folding nascent chain against the backdrop of the ribosome. New NMR methodologies employing selective isotope labeling of nascent proteins and novel pulse sequences (Cassaignau et al., 2016) have provided high-resolution structural information of nascent chain folding and dynamics (Waudby et al., 2013). However, they require high sample concentrations and are thus far limited to relatively short nascent proteins. Fluorescence methods such as FRET have allowed direct visualization of different conformations of the nascent chain populated during translation and resolve folding kinetics. Combined FRET and PET measurements were used to show that nascent chains can form secondary structural elements even inside the exit tunnel (Holtkamp et al., 2015; Liutkute et al., 2020b). Force spectroscopy techniques also allow detection of nascent chain folding on the ribosome. In addition, application of defined, variable mechanical force can be used to selectively destabilize nascent protein structure, keeping interacting partners such as the ribosome intact, in contrast to globally acting chemical or thermal denaturation. Force spectroscopy can thus be used to drive non-equilibrium transitions and to study the folding dynamics of nascent chains in the presence of biologically relevant interaction partners.
Because of the substantial challenges associated with implementing, conducting, and analyzing experiments of co-translational folding, initial studies were carried out using single-domain proteins with artificial C-terminal extensions that allowed their full extrusion from the ribosome (Kaiser et al., 2011; Knight et al., 2013; Samelson et al., 2016; Guinn et al., 2018; Waudby et al., 2018; Liu et al., 2019a). While these experiments did not capture the complexity of naturally occurring large proteins, they yielded several key insights about the interactions of emerging domains with the ribosome that are directly relevant to multi-domain protein folding. In particular, single-molecule optical tweezers experiments revealed that interactions with the ribosome generally decelerate and destabilize nascent chain folding, presumably due to electrostatic interactions with the surface of the ribosome that decay as the nascent chain is elongated (Kaiser et al., 2011). The distance-dependent deceleration of domain folding is likely the result of competition between mutually exclusive interactions of the nascent chain with itself (resulting in folding) and with the ribosome surface (preventing folding). These interactions may help to stabilize a folding-competent conformation of the nascent protein during translation before a foldable unit has been synthesized and extruded from the ribosome.
The ability to precisely monitor changes in molecular length with optical tweezers enables real time observation of nascent chain elongation and folding. The addition of individual amino acids is difficult to resolve because the change in molecular extension per amino acid added is very small (less than ∼2 Å at forces below 10 pN) relative to the noise of an optical tweezers measurement. However, overall chain elongation is apparent as an increase in length of the tethered molecule. Gradual hydrophobic collapse and partial folding was observed during synthesis of a small single-domain protein in the optical tweezers (Wruck et al., 2017). Studies of the calcium-binding protein calerythrin showed that the protein populates a misfolded state on stalled ribosomes in a few seconds. When the elongation and folding of the nascent chain is monitored in real time, the protein does not populate the misfolded state until tens of seconds have elapsed (Alexander et al., 2019). This study illustrates that the nascent chain does not necessarily equilibrate rapidly, and that non-equilibrium effects may suppress formation of misfolded states.
Misfolding Interactions
Inter-Domain Misfolding
Interactions between residues in separate unfolded domains can lead to misfolding (Figure 1). A tandem-repeat protein constructed from the titin I27 domain exhibits inter-domain misfolding after mechanical unfolding (Fernandez et al., 1999). In an artificial construct with repeats of maltose-binding protein, stable misfolds involving multiple domains are observed to form that require high forces to unfold (Mashaghi et al., 2016). Repeats sharing a high sequence similarity are particularly prone to forming very stable misfolded structures, having a lifetime of up to several days (Borgia et al., 2011). At least for the small Ig-like beta-sandwich domains like I27 that have been studied in detail, these structures can form when strands from different domains are swapped between domains. Contiguous domains having lower similarity, however, are less likely to form misfolded structures.
However, inter-domain misfolding is not limited to the special case of repeat proteins. Even relatively small proteins, such as the calcium-binding protein calmodulin and NCS-1, populate non-native structures that compete with productive folding (Stigler et al., 2011; Heidarsson et al., 2014). The three domains of 709-residue long Hsp90 form several inter-domain misfolded structures (Jahn et al., 2016). The structurally complex luciferase protein also populates misfolded states (Scholl et al., 2014). Elongation factor G, a 704-residue long protein consisting of five non-homologous domains forms many stable non-native structures upon refolding (Liu et al., 2017). At least some of these structures involve residues from multiple domains that misfold into mechanically labile, molten globule-like structures. Even in truncated constructs containing only the first two domains, inter-domain misfolding is observed (Liu et al., 2019a). Only a small number of mechanistic studies are available to date. However, inter-domain misfolding into loosely structured misfolded states (for natural multi-domain proteins) appears to be a general phenomenon (Zheng et al., 2013; Tian and Best, 2016).
The results outlined in the preceding paragraphs were obtained with isolated proteins and suggest a general propensity of large proteins to populate non-native structures. Nascent chains appear to be at least partially protected from this type of misfolding. For EF-G, misfolding interactions are greatly reduced by the ribosome, promoting efficient sequential folding of the two N-terminal domains G and II (Liu et al., 2019a) (Figure 2). The importance of productive co-translational folding is illustrated by the fact that cells have evolved not only to contain an armory of molecular chaperones that facilitate folding (Balchin et al., 2016; Koubek et al., 2021), but also mechanisms for detecting misfolded states that result in nascent chain degradation (Brandman et al., 2012; Wang et al., 2013) or termination of translation (Zhao et al., 2021). The temporal sequestration of folding during synthesis allows domains to fold into stable structures and prevents non-native interactions. Translation elongation has been suggested to be slow at domain boundaries (Komar, 2009), which would amplify this effect. More generally, translation elongation is likely tuned to allow productive folding (Komar, 2016), as indicated by pronounced effects that synonymous codon changes can have on protein structure, stability and, potentially, function (Kimchi-Sarfaty et al., 2007; Komar, 2009; Zhou et al., 2013; Sander et al., 2014; Kim et al., 2015b; Buhr et al., 2016; Walsh et al., 2020) (see (Liu et al., 2021) for a recent review).
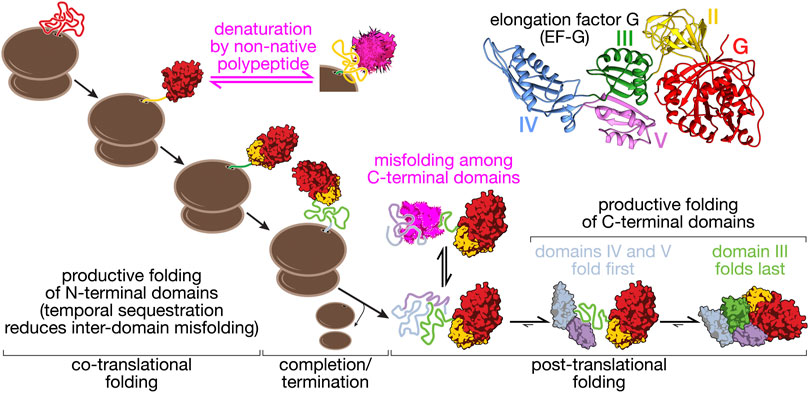
FIGURE 2. Folding pathway of elongation factor G (EF-G). EF-G is a five-domain protein (structure, top right: pdb 4v9p) that illustrates some of the core aspects of multi-domain protein folding. The N-terminal domains (G, II) fold co-translationally, with G-domain folding being a prerequisite for domain II folding. Productive folding competes with denaturation of the G-domain through interactions with unfolded domain II. This complication of co-translational folding is mitigated by a nascent chain-binding chaperone (Liu et al., 2019a). Because domain III requires stabilizing contacts with domains IV and V, co-translational folding is disrupted. Post-translational folding of the C-terminal domains (III–V) results in the accumulation of unfolded polypeptide during EF-G synthesis that has a high propensity to form off-pathway misfolded states (Liu et al., 2019b). EF-G thus provides an example of how overall folding pathways are shaped by domain interactions in large proteins.
Intra-Domain Misfolding
In addition to inter-domain misfolding interactions, constituent domains in complex proteins can suffer from internal misfolding (Figure 1). Modeling suggests how tuning of elongation rates by natural selection can help co-translational folding by circumventing kinetic traps (Bitran et al., 2020). Experimentally, individual domains of Hsp90 form misfolded states, in addition to forming inter-domain misfolds (Jahn et al., 2016). The glucocorticoid receptor ligand binding domain populates an ensemble of misfolded states which compete with productive folding (Suren et al., 2018). Application of mechanical force to the molecule reduces misfolding, presumably because mechanically stretching the protein disfavors intra-chain interactions that result in misfolding. Interestingly, folding of nascent proteins close to the ribosomal exit tunnel generates mechanical force (Goldman et al., 2015; Maciuba et al., 2021). The tension generated in the process could similarly destabilize misfolded states and promote the formation of native structures, although this possibility remains to be experimentally tested.
The G-domain of EF- G populates unstable molten-globule like states and intermediate structures upon refolding. When the protein is synthesized, however, the nascent chain remains unfolded until the entire domain is extruded from the ribosome, avoiding the formation of non-native structures (Chen et al., 2020). The resulting directional folding in the C- to N-terminal direction may be prevalent for domains that are prone to misfolding, as illustrated by several examples. The N-terminal domain of calerythrin folds into stable structures in isolation but fails to fold on the ribosome until the entire protein is synthesized (Alexander et al., 2019). The nucleotide binding domain of Hsp70 folds through a folding nucleus involving the C-terminal lobes (Bauer et al., 2018) and may thus remain unstructured until the entire domain is fully extruded from the ribosome during translation. Other domains fold in the opposite direction, such as the nucleotide binding domain of CFTR (Khushoo et al., 2011; Kim et al., 2015b).
Intermediates are commonly thought to be populated on the way to native state, even for relatively small domains (Brockwell and Radford, 2007). It is intuitive to assume that the sequential addition of amino acids to the growing polypeptide will result in gradual compaction and formation of hierarchical structures but, as the examples discussed above demonstrate, the directionality of folding can run counter to that of synthesis. It is interesting to note that folding of the nucleotide binding domains appears to nucleate around the ligand binding site for both CFTR and Hsp70. However, the nucleation sites are located at the N-terminus and C-terminus of the domains, respectively, which might explain why they exhibit differences in their co-translational folding properties. Regardless of the underlying causes, the examples discussed above (EF-G, Hsp70, and calerythrin) strongly suggest that caution is in order when relating folding pathways determined with full-length domains in vitro to the co-translational pathways that are likely relevant in vivo.
Stabilizing Interactions
Protein segments rich in low complexity sequences remain intrinsically disordered in solution (van der Lee et al., 2014), unlike globular proteins that are thought to mostly adopt more or less well-defined structures. IDPs have defined sequence features distinct from non-IDPs. However, whether a member of either class is structured or unstructured can depend on context and interactions. Domains in larger globular proteins may not be stable by themselves, and IDPs may adopt defined structures. For instance, binding of intrinsically disordered proteins to their ligands or interacting partners promotes structure formation (Wright and Dyson, 2009). The phosphorylated kinase inducible activation domain of the transcription factor CREB is intrinsically disordered in solution and forms alpha helical structure upon binding to the KIX domain (Sugase et al., 2007). The nuclear coactivator-binding domain of CBP adopts stable structures by interactions with ACTR domain (Zosel et al., 2018). The close association through a stabilizing interface favors the folded conformations of the disordered domains.
The domain interfaces in a multi-domain protein provide opportunities for favorable interactions that stabilize unstable structures (Figure 1). Such stabilizing effects can even promote structure formation in otherwise unstructured domains. For instance, the isolated C-terminal domain of phosphoglycerate kinase remains disordered in solution but is stably folded in the presence of the N-terminal domain (Young et al., 2007). In EF-G, domain II appears to rely on contacts with the N-terminal G-domain for stability (Liu et al., 2019a). In another example, also from EF-G, the central domain III does not appear to be stably folded in isolation, lacking stable tertiary structure (Liu et al., 2019b). Domain III forms extensive interface with its neighboring domains, IV and V. These interfacial contacts stabilize the folded state relative to the unfolded state and the domain is fully folded in the presence of folded domains IV and V (Liu et al., 2019b) (Figure 2).
This stabilization by interaction may be analogous to the ‘binding and folding’ model observed for several disordered proteins. For example, ACTR is largely unstructured in solution and binding to its partner, NCBD, enables it to adopt tertiary structure (Demarest et al., 2002; Marino et al., 2018; Zosel et al., 2018). Another example is the PUMA protein that adopts a helical structure only when binding to MCL-1 (Rogers et al., 2014). A folded domain may also provide an interaction surface that accelerates the folding of a yet unstructured neighbor. The spectrin repeat R17 folds faster when it is stabilized by the upstream domain R16 whereas it folds more slowly in isolation (Batey et al., 2006). Conversely, the unfolding rate of R17 is slower by two orders of magnitude in the presence of stabilizing interactions with R16. The scaffolding effect provided by domain interfaces and modulation of folding rates may aid in productive structure formation. Interestingly, some protein complexes appear to require assembly in a sequential order (Marsh et al., 2013). It seems possible that the folding order prescribed by the directionality of protein synthesis may enforce a similarly ordered formation of domain interfaces in multi-domain proteins that ultimately serves efficient folding.
Not all domain-domain interfaces have stabilizing effects. An extensive interface of domain III in EF-G with its N-terminal neighbors (domains G and II) does not contribute measurably to stability (Liu et al., 2019b). The implications of this complex energetic dependency to the biogenesis of the protein are two-fold. First, domain III will be synthesized before the emergence of domains IV-V, shifting the folding from co- to post- translational regime (Figure 2). Second, the presence of a disordered sequence during synthesis increases the probability of misfolding of emerging nascent chain segments, thus requiring the aid of chaperones. The fusion of multiple domains in a multi-domain protein may stabilize structures that are unstable in isolation. In some cases, like that of domain III of EFG, the energetic dependencies may have evolved to meet functional needs for flexibility. The complex energetic interactions between different domains may pose problems to co-translational folding.
Denaturation and Destabilizing Interactions
Nascent proteins can begin to acquire secondary and (limited) tertiary structure even before their emergence from the ribosome exit tunnel (Nilsson et al., 2015; Marino et al., 2016; Tian et al., 2018; Agirrezabala et al., 2022). These early structures might be important stepping stones toward the native state. Large structures cannot be accommodated inside the ribosome exit tunnel. Because protein folding is typically cooperative, these structures are fully formed only when an entire domain has emerged from the ribosome. Non-native nascent chain segments therefore are inevitably present during protein synthesis. It has been shown that they can destabilize already folded domains (Figure 1). Unfolded domain II of EF-G destabilizes the folded G-domain, ultimately resulting in its denaturation (Liu et al., 2019a) (Figure 2). Sequestering the unfolded segments near the ribosomal surface may help to reduce denaturing interactions. However, in the case of nascent EF-G, the ribosome does not prevent destabilization of the G-domain by domain II. However, trigger factor, a bacterial nascent chain binding chaperone, binds to and sequesters domain II, protecting the folded state of the G-domain (Liu et al., 2019a). Controlled release of domain II once it is fully synthesized and capable of productive folding may then ensure correct structure acquisition.
It is unclear how general the denaturing effect of an unfolded domain on a folded neighbor is. As for other aspects of co-translational folding, computer simulations and theoretical models (reviewed elsewhere, see ref. (Trovato and O'Brien, 2016)) are poised to complement experimental observations in dissecting mechanistic details. Domains G and II from EF-G so far represent the only experimental example of destabilization by unfolded polypeptide in a natural multi-domain protein. However, destabilizing effects that are frequently observed in fusion proteins may be manifestations of this effect. Fluorescent proteins fused to other proteins like engrailed homeodomain or phosphoglycerate kinase have been observed to reduce the stability of the protein (Sokolovski et al., 2015; Dave et al., 2016). C-terminal disordered regions show destabilizing effects in several proteins. For instance, a tail present in the UGDH enzyme dimer reduces thermal stability by 3.6 °C (Keul et al., 2018). The presence of extended C-terminal tail also increases conformational flexibility and destabilizes the UVR8 plant photoreceptor protein (Camacho et al., 2019). Destabilizing effects are also well-known for signal peptides of secreted proteins, which effectively prevent the formation of stable tertiary structures (Randall and Hardy, 1986; Park et al., 1988; Singh et al., 2013). It thus seems possible that destabilization of folded domains by unfolded neighbors is a general phenomenon.
Conclusion
Although multi-domain proteins make up a large proportion of proteomes, a mechanistic understanding of their unique folding pathways is still lacking, in part due to experimental challenges. Methodological advances in the past few years have fueled progress in our understanding of how large proteins fold, illustrated by the examples highlighted here. Some of the complexity that results from interactions of folded and unfolded domains during multi-domain protein biogenesis is now beginning to be unraveled. The recently developed approach of following protein folding in vivo using arrest peptides (Goldman et al., 2015; Marino et al., 2016; Chen et al., 2020) offers exciting opportunities for complementing biophysical in vitro experiments. However, despite recent advances, many questions regarding multi-domain protein folding remain: How are protein synthesis and folding tuned to each other to avoid the formation of non-functional or toxic products? How do the functional requirements for conformational flexibility compete with folding and interdomain interactions? And how do molecular chaperones rescue misfolded states of multi-domain proteins? Studies on diverse sets of authentic multi-domain proteins in vivo and in vitro will be required to define general principles of their folding.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by a Grant from the National Institutes of Health (5R01GM121567) to CK.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation with the authors NR/CK at time of review.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Agirrezabala, X., Samatova, E., Macher, M., Liutkute, M., Maiti, M., Gil‐Carton, D., et al. (2022). A Switch from α‐helical to β‐strand Conformation during Co‐translational Protein Folding. EMBO J. 41 (4), e109175. PubMed PMID: 34994471; PMCID: PMC8844987. doi:10.15252/embj.2021109175
Alexander, L. M., Goldman, D. H., Wee, L. M., and Bustamante, C. (2019). Non-equilibrium Dynamics of a Nascent Polypeptide during Translation Suppress its Misfolding. Nat. Commun. 10 (1), 2709. PubMed PMID: 31221966; PMCID: PMC6586675. doi:10.1038/s41467-019-10647-6
Andreeva, A., Kulesha, E., Gough, J., and Murzin, A. G. (2020). The SCOP Database in 2020: Expanded Classification of Representative Family and Superfamily Domains of Known Protein Structures. Nucleic Acids Res. 48 (D1), D376–D382. PubMed PMID: 31724711; PMCID: PMC7139981. doi:10.1093/nar/gkz1064
Balchin, D., Hayer-Hartl, M., and Hartl, F. U. (2016). In Vivo aspects of Protein Folding and Quality Control. Science 353 (6294), aac4354. PubMed PMID: 27365453. doi:10.1126/science.aac4354
Bartlett, A. I., and Radford, S. E. (2009). An Expanding Arsenal of Experimental Methods Yields an Explosion of Insights into Protein Folding Mechanisms. Nat. Struct. Mol. Biol. 16 (6), 582–588. doi:10.1038/nsmb.1592
Batey, S., Scott, K. A., and Clarke, J. (2006). Complex Folding Kinetics of a Multi-Domain Protein. Biophysical J. 90 (6), 2120–2130. PubMed PMID: 16387757; PMCID: PMC1386790. doi:10.1529/biophysj.105.072710
Bauer, D., Meinhold, S., Jakob, R. P., Stigler, J., Merkel, U., Maier, T., et al. (2018). A Folding Nucleus and Minimal ATP Binding Domain of Hsp70 Identified by Single-Molecule Force Spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 115 (18), 4666–4671. PubMed PMID: 29669923; PMCID: PMC5939067. doi:10.1073/pnas.1716899115
Bergman, L. W., and Kuehl, W. M. (1979). Formation of an Intrachain Disulfide Bond on Nascent Immunoglobulin Light Chains. J. Biol. Chem. 254 (18), 8869–8876. Epub 1979/09/25. PubMed PMID: 113402. doi:10.1016/s0021-9258(19)86780-7
Bitran, A., Jacobs, W. M., Zhai, X., and Shakhnovich, E. (2020). Cotranslational Folding Allows Misfolding-Prone Proteins to Circumvent Deep Kinetic Traps. Proc. Natl. Acad. Sci. U.S.A. 117 (3), 1485–1495. PubMed PMID: 31911473; PMCID: PMC6983386. doi:10.1073/pnas.1913207117
Borgia, M. B., Borgia, A., Best, R. B., Steward, A., Nettels, D., Wunderlich, B., et al. (2011). Single-molecule Fluorescence Reveals Sequence-specific Misfolding in Multi-Domain Proteins. Nature 474 (7353), 662–665. PubMed PMID: 21623368; PMCID: 3160465. doi:10.1038/nature10099
Brandman, O., Stewart-Ornstein, J., Wong, D., Larson, A., Williams, C. C., Li, G.-W., et al. (2012). A Ribosome-Bound Quality Control Complex Triggers Degradation of Nascent Peptides and Signals Translation Stress. Cell 151 (5), 1042–1054. PubMed PMID: 23178123; PMCID: PMC3534965. doi:10.1016/j.cell.2012.10.044
Braselmann, E., Chaney, J. L., and Clark, P. L. (2013). Folding the Proteome. Trends Biochemical Sciences 38 (7), 337–344. PubMed PMID: 23764454; PMCID: 3691291. doi:10.1016/j.tibs.2013.05.001
Brockwell, D. J., and Radford, S. E. (2007). Intermediates: Ubiquitous Species on Folding Energy Landscapes? Curr. Opin. Struct. Biol. 17 (1), 30–37. Epub 2007/01/24. S0959-440X(07)00004-8 [pii]. doi:10.1016/j.sbi.2007.01.003
Buhr, F., Jha, S., Thommen, M., Mittelstaet, J., Kutz, F., Schwalbe, H., et al. (2016). Synonymous Codons Direct Cotranslational Folding toward Different Protein Conformations. Mol. Cel 61 (3), 341–351. PubMed PMID: 26849192; PMCID: PMC4745992. doi:10.1016/j.molcel.2016.01.008
Bustamante, C., Alexander, L., Maciuba, K., and Kaiser, C. M. (2020). Single-Molecule Studies of Protein Folding with Optical Tweezers. Annu. Rev. Biochem. 89, 443–470. PubMed PMID: 32569525. doi:10.1146/annurev-biochem-013118-111442
Camacho, I. S., Theisen, A., Johannissen, L. O., Díaz-Ramos, L. A., Christie, J. M., Jenkins, G. I., et al. (2019). Native Mass Spectrometry Reveals the Conformational Diversity of the UVR8 Photoreceptor. Proc. Natl. Acad. Sci. U.S.A. 116 (4), 1116–1125. PubMed PMID: 30610174; PMCID: PMC6347689. doi:10.1073/pnas.1813254116
Cassaignau, A. M. E., Cabrita, L. D., and Christodoulou, J. (2020). How Does the Ribosome Fold the Proteome? Annu. Rev. Biochem. 89, 389–415. PubMed PMID: 32569518. doi:10.1146/annurev-biochem-062917-012226
Cassaignau, A. M. E., Launay, H. M. M., Karyadi, M.-E., Wang, X., Waudby, C. A., Deckert, A., et al. (2016). A Strategy for Co-translational Folding Studies of Ribosome-Bound Nascent Chain Complexes Using NMR Spectroscopy. Nat. Protoc. 11 (8), 1492–1507. PubMed PMID: 27466710. doi:10.1038/nprot.2016.101
Chen, X., Rajasekaran, N., Liu, K., and Kaiser, C. M. (2020). Synthesis Runs Counter to Directional Folding of a Nascent Protein Domain. Nat. Commun. 11 (1), 5096. doi:10.1038/s41467-020-18921-8
Dave, K., Gelman, H., Thu, C. T. H., Guin, D., and Gruebele, M. (2016). The Effect of Fluorescent Protein Tags on Phosphoglycerate Kinase Stability Is Nonadditive. J. Phys. Chem. B 120 (11), 2878–2885. PubMed PMID: 26923443. doi:10.1021/acs.jpcb.5b11915
Demarest, S. J., Martinez-Yamout, M., Chung, J., Chen, H., Xu, W., Dyson, H. J., et al. (2002). Mutual Synergistic Folding in Recruitment of CBP/p300 by P160 Nuclear Receptor Coactivators. Nature 415 (6871), 549–553. PubMed PMID: 11823864. doi:10.1038/415549a
Deuerling, E., Gamerdinger, M., and Kreft, S. G. (2019). Chaperone Interactions at the Ribosome. Cold Spring Harb Perspect. Biol. 11, a033977. PubMed PMID: 30833456. doi:10.1101/cshperspect.a033977
Doolittle, R. F. (1995). The Multiplicity of Domains in Proteins. Annu. Rev. Biochem. 64, 287–314. PubMed PMID: 7574483. doi:10.1146/annurev.bi.64.070195.001443
Dyla, M., and Kjaergaard, M. (2020). Intrinsically Disordered Linkers Control Tethered Kinases via Effective Concentration. Proc. Natl. Acad. Sci. U.S.A. 117 (35), 21413–21419. PubMed PMID: 32817491; PMCID: PMC7474599. doi:10.1073/pnas.2006382117
Fedorov, A. N., and Baldwin, T. O. (1997). Cotranslational Protein Folding. J. Biol. Chem. 272 (52), 32715–32718. PubMed PMID: 9407040. doi:10.1074/jbc.272.52.32715
Fernandez, J. M., Oberhauser, A. F., Marszalek, P. E., and Carrion-Vazquez, M. (1999). Single Protein Misfolding Events Captured by Atomic Force Microscopy. Nat. Struct. Biol. 6 (11), 1025–1028. PubMed PMID: 10542093. doi:10.1038/14907
Frydman, J., Nimmesgern, E., Ohtsuka, K., and Hartl, F. U. (1994). Folding of Nascent Polypeptide Chains in a High Molecular Mass Assembly with Molecular Chaperones. Nature 370 (6485), 111–117. PubMed PMID: 8022479. doi:10.1038/370111a0
Goldman, D. H., Kaiser, C. M., Milin, A., Righini, M., Tinoco, I., and Bustamante, C. (2015). Mechanical Force Releases Nascent Chain-Mediated Ribosome Arrest In Vitro and In Vivo. Science 348 (6233), 457–460. PubMed PMID: 25908824. doi:10.1126/science.1261909
Guinn, E. J., Tian, P., Shin, M., Best, R. B., and Marqusee, S. (2018). A Small Single-Domain Protein Folds through the Same Pathway on and off the Ribosome. Proc. Natl. Acad. Sci. U.S.A. 115 (48), 12206–12211. PubMed PMID: 30409803; PMCID: PMC6275501. doi:10.1073/pnas.1810517115
Han, J.-H., Batey, S., Nickson, A. A., Teichmann, S. A., and Clarke, J. (2007). The Folding and Evolution of Multi-Domain Proteins. Nat. Rev. Mol. Cel Biol 8 (4), 319–330. PubMed PMID: 17356578. doi:10.1038/nrm2144
Heidarsson, P. O., Naqvi, M. M., Otazo, M. R., Mossa, A., Kragelund, B. B., and Cecconi, C. (2014). Direct Single-Molecule Observation of Calcium-dependent Misfolding in Human Neuronal Calcium Sensor-1. Proc. Natl. Acad. Sci. U.S.A. 111 (36), 13069–13074. PubMed PMID: 25157171; PMCID: PMC4246975. doi:10.1073/pnas.1401065111
Holtkamp, W., Kokic, G., Jager, M., Mittelstaet, J., Komar, A. A., and Rodnina, M. V. (2015). Cotranslational Protein Folding on the Ribosome Monitored in Real Time. Science 350 (6264), 1104–1107. PubMed PMID: 26612953. doi:10.1126/science.aad0344
Huang, F., Rajagopalan, S., Settanni, G., Marsh, R. J., Armoogum, D. A., Nicolaou, N., et al. (2009). Multiple Conformations of Full-Length P53 Detected with Single-Molecule Fluorescence Resonance Energy Transfer. Proc. Natl. Acad. Sci. U.S.A. 106 (49), 20758–20763. PubMed PMID: 19933326; PMCID: PMC2791586. doi:10.1073/pnas.0909644106
Huttlin, E. L., Bruckner, R. J., Navarrete-Perea, J., Cannon, J. R., Baltier, K., Gebreab, F., et al. (2021). Dual Proteome-Scale Networks Reveal Cell-specific Remodeling of the Human Interactome. Cell 184 (11), 3022–3040 e28. PubMed PMID: 33961781; PMCID: PMC8165030. doi:10.1016/j.cell.2021.04.011
Jaenicke, R. (1999). Stability and Folding of Domain Proteins. Prog. Biophys. Mol. Biol. 71 (2), 155–241. PubMed PMID: 10097615. doi:10.1016/s0079-6107(98)00032-7
Jäger, M., and Plückthun, A. (1999). Domain Interactions in Antibody Fv and scFv Fragments: Effects on Unfolding Kinetics and Equilibria. FEBS Lett. 462 (3), 307–312. PubMed PMID: 10622716. doi:10.1016/s0014-5793(99)01532-x
Jahn, M., Buchner, J., Hugel, T., and Rief, M. (2016). Folding and Assembly of the Large Molecular Machine Hsp90 Studied in Single-Molecule Experiments. Proc. Natl. Acad. Sci. U.S.A. 113 (5), 1232–1237. PubMed PMID: 26787848; PMCID: PMC4747692. doi:10.1073/pnas.1518827113
Kaiser, C. M., Goldman, D. H., Chodera, J. D., Tinoco, I., and Bustamante, C. (2011). The Ribosome Modulates Nascent Protein Folding. Science 334 (6063), 1723–1727. Epub 2011/12/24. doi:10.1126/science.1209740
Keul, N. D., Oruganty, K., Beattie, E. T. N. R., KadirvelrajMcDonald, R., Gross, M. L., Phillips, R. S., et al. (2018). The Entropic Force Generated by Intrinsically Disordered Segments Tunes Protein Function. Nature 563 (7732), 584–588. PubMed PMID: 30420606; PMCID: PMC6415545. doi:10.1038/s41586-018-0699-5
Khushoo, A., Yang, Z., Johnson, A. E., and Skach, W. R. (2011). Ligand-Driven Vectorial Folding of Ribosome-Bound Human CFTR NBD1. Mol. Cel 41 (6), 682–692. Epub 2011/03/23S1097-2765(11)00171-7 [pii]. doi:10.1016/j.molcel.2011.02.027
Kim, E., Moore, B. S., and Yoon, Y. J. (2015). Reinvigorating Natural Product Combinatorial Biosynthesis with Synthetic Biology. Nat. Chem. Biol. 11 (9), 649–659. PubMed PMID: 26284672; PMCID: PMC4757526. doi:10.1038/nchembio.1893
Kim, S. J., Yoon, J. S., Shishido, H., Yang, Z., Rooney, L. A., Barral, J. M., et al. (2015). Translational Tuning Optimizes Nascent Protein Folding in Cells. Science 348 (6233), 444–448. PubMed PMID: 25908822. doi:10.1126/science.aaa3974
Kimchi-Sarfaty, C., Oh, J. M., Kim, I.-W., Sauna, Z. E., Calcagno, A. M., Ambudkar, S. V., et al. (2007). A "Silent" Polymorphism in the MDR 1 Gene Changes Substrate Specificity. Science 315 (5811), 525–528. Epub 2006/12/23. 1135308 [pii]. doi:10.1126/science.1135308
Kjaergaard, M. (2022). Estimation of Effective Concentrations Enforced by Complex Linker Architectures from Conformational Ensembles. Biochemistry 61, 171–182. PubMed PMID: 35061369. doi:10.1021/acs.biochem.1c00737
Knight, A. M., Culviner, P. H., Kurt-Yilmaz, N., Zou, T., Ozkan, S. B., and Cavagnero, S. (2013). Electrostatic Effect of the Ribosomal Surface on Nascent Polypeptide Dynamics. ACS Chem. Biol. 8 (6), 1195–1204. PubMed PMID: 23517476. doi:10.1021/cb400030n
Komar, A. A. (2009). A Pause for Thought along the Co-translational Folding Pathway. Trends Biochem. Sci. 34 (1), 16–24. PubMed PMID: 18996013. doi:10.1016/j.tibs.2008.10.002
Komar, A. A. (2016). The Yin and Yang of Codon Usage. Hum. Mol. Genet. 25 (R2), R77–R85. doi:10.1093/hmg/ddw207
Koubek, J., Schmitt, J., Galmozzi, C. V., and Kramer, G. (2021). Mechanisms of Cotranslational Protein Maturation in Bacteria. Front. Mol. Biosci. 8, 689755. PubMed PMID: 34113653; PMCID: PMC8185961. doi:10.3389/fmolb.2021.689755
Kramer, G., Shiber, A., and Bukau, B. (2019). Mechanisms of Cotranslational Maturation of Newly Synthesized Proteins. Annu. Rev. Biochem. 88, 337–364. PubMed PMID: 30508494. doi:10.1146/annurev-biochem-013118-111717
Liu, K., Chen, X., and Kaiser, C. M. (2019). Energetic Dependencies Dictate Folding Mechanism in a Complex Protein. Proc. Natl. Acad. Sci. U.S.A. 116 (51), 25641–25648. PubMed PMID: 31776255; PMCID: PMC6925980. doi:10.1073/pnas.1914366116
Liu, K., Maciuba, K., and Kaiser, C. M. (2019). The Ribosome Cooperates with a Chaperone to Guide Multi-Domain Protein Folding. Mol. Cel 74 (2), 310–319 e7. PubMed PMID: 30852061. doi:10.1016/j.molcel.2019.01.043
Liu, K., Rehfus, J. E., Mattson, E., and Kaiser, C. M. (2017). The Ribosome Destabilizes Native and Non-native Structures in a Nascent Multidomain Protein. Protein Sci. 26 (7), 1439–1451. PubMed PMID: 28474852; PMCID: PMC5477528. doi:10.1002/pro.3189
Liu, Y., Yang, Q., and Zhao, F. (2021). Synonymous but Not Silent: The Codon Usage Code for Gene Expression and Protein Folding. Annu. Rev. Biochem. 90, 375–401. PubMed PMID: 33441035. doi:10.1146/annurev-biochem-071320-112701
Liutkute, M., Maiti, M., Samatova, E., Enderlein, J., and Rodnina, M. V. (2020). Gradual Compaction of the Nascent Peptide during Cotranslational Folding on the Ribosome. Elife 9. PubMed PMID: 33112737; PMCID: PMC7593090. doi:10.7554/eLife.60895
Liutkute, M., Samatova, E., and Rodnina, M. V. (2020). Cotranslational Folding of Proteins on the Ribosome. Biomolecules 10 (1). PubMed PMID: 31936054; PMCID: 97. doi:10.3390/biom10010097
Maciuba, K., Rajasekaran, N., Chen, X., and Kaiser, C. M. (2021). Co‐translational Folding of Nascent Polypeptides: Multi‐layered Mechanisms for the Efficient Biogenesis of Functional Proteins. Bioessays 43, 2100042. PubMed PMID: 33987870. doi:10.1002/bies.202100042
Marino, J., Buholzer, K. J., Zosel, F., Nettels, D., and Schuler, B. (2018). Charge Interactions Can Dominate Coupled Folding and Binding on the Ribosome. Biophysical J. 115 (6), 996–1006. PubMed PMID: 30173887; PMCID: PMC6139605. doi:10.1016/j.bpj.2018.07.037
Marino, J., von Heijne, G., and Beckmann, R. (2016). Small Protein Domains Fold inside the Ribosome Exit Tunnel. FEBS Lett. 590 (5), 655–660. PubMed PMID: 26879042. doi:10.1002/1873-3468.12098
Marsh, J. A., Hernández, H., Hall, Z., Ahnert, S. E., Perica, T., Robinson, C. V., et al. (2013). Protein Complexes Are under Evolutionary Selection to Assemble via Ordered Pathways. Cell 153 (2), 461–470. PubMed PMID: 23582331; PMCID: PMC4009401. doi:10.1016/j.cell.2013.02.044
Mashaghi, A., Bezrukavnikov, S., Minde, D. P., Wentink, A. S., Kityk, R., Zachmann-Brand, B., et al. (2016). Alternative Modes of Client Binding Enable Functional Plasticity of Hsp70. Nature 539 (7629), 448–451. PubMed PMID: 27783598. doi:10.1038/nature20137
Nilsson, O. B., Hedman, R., Marino, J., Wickles, S., Bischoff, L., Johansson, M., et al. (2015). Cotranslational Protein Folding inside the Ribosome Exit Tunnel. Cel Rep. 12 (10), 1533–1540. PubMed PMID: 26321634; PMCID: 4571824. doi:10.1016/j.celrep.2015.07.065
Park, S., Liu, G., Topping, T. B., Cover, W. H., and Randall, L. L. (1988). Modulation of Folding Pathways of Exported Proteins by the Leader Sequence. Science 239 (4843), 1033–1035. PubMed PMID: 3278378. doi:10.1126/science.3278378
Phillips, D. C. (1967). Hen Egg-White Lysozyme Molecule. P Natl. Acad. Sci. USA 57 (3), 484. PubMed PMID: WOS:A19679098900001. doi:10.1073/pnas.57.3.483
Poust, S., Hagen, A., Katz, L., and Keasling, J. D. (2014). Narrowing the gap between the Promise and Reality of Polyketide Synthases as a Synthetic Biology Platform. Curr. Opin. Biotechnol. 30, 32–39. PubMed PMID: 24816568. doi:10.1016/j.copbio.2014.04.011
Randall, L. L., and Hardy, S. J. S. (1986). Correlation of Competence for export with Lack of Tertiary Structure of the Mature Species: a Study In Vivo of Maltose-Binding Protein in E. coli. Cell 46 (6), 921–928. PubMed PMID: 3530497. doi:10.1016/0092-8674(86)90074-7
Robinson, C. R., and Sauer, R. T. (1998). Optimizing the Stability of Single-Chain Proteins by Linker Length and Composition Mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 95 (11), 5929–5934. PubMed PMID: 9600894; PMCID: PMC34497. doi:10.1073/pnas.95.11.5929
Rogers, J. M., Oleinikovas, V., Shammas, S. L., Wong, C. T., De Sancho, D., Baker, C. M., et al. (2014). Interplay between Partner and Ligand Facilitates the Folding and Binding of an Intrinsically Disordered Protein. Proc. Natl. Acad. Sci. U.S.A. 111 (43), 15420–15425. PubMed PMID: 25313042; PMCID: PMC4217413. doi:10.1073/pnas.1409122111
Samelson, A. J., Jensen, M. K., Soto, R. A., Cate, J. H. D., and Marqusee, S. (2016). Quantitative Determination of Ribosome Nascent Chain Stability. Proc. Natl. Acad. Sci. U.S.A. 113 (47), 13402–13407. PubMed PMID: 27821780; PMCID: PMC5127326. doi:10.1073/pnas.1610272113
Sander, I. M., Chaney, J. L., and Clark, P. L. (2014). Expanding Anfinsen's Principle: Contributions of Synonymous Codon Selection to Rational Protein Design. J. Am. Chem. Soc. 136 (3), 858–861. PubMed PMID: 24392935; PMCID: 3959793. doi:10.1021/ja411302m
Scholl, Z. N., Yang, W., and Marszalek, P. E. (2014). Chaperones rescue Luciferase Folding by Separating its Domains. J. Biol. Chem. 289 (41), 28607–28618. PubMed PMID: 25160632; PMCID: PMC4192510. doi:10.1074/jbc.M114.582049
Singh, P., Sharma, L., Kulothungan, S. R., Adkar, B. V., Prajapati, R. S., Ali, P. S. S., et al. (2013). Effect of Signal Peptide on Stability and Folding of Escherichia coli Thioredoxin. PLoS One 8 (5), e63442. PubMed PMID: 23667620; PMCID: PMC3646739. doi:10.1371/journal.pone.0063442
Sivaramakrishnan, S., and Spudich, J. A. (2011). Systematic Control of Protein Interaction Using a Modular ER/K α-helix Linker. Proc. Natl. Acad. Sci. U.S.A. 108 (51), 20467–20472. PubMed PMID: 22123984; PMCID: PMC3251109. doi:10.1073/pnas.1116066108
Sokolovski, M., Bhattacherjee, A., Kessler, N., Levy, Y., and Horovitz, A. (2015). Thermodynamic Protein Destabilization by GFP Tagging: A Case of Interdomain Allostery. Biophysical J. 109 (6), 1157–1162. PubMed PMID: 25998254; PMCID: PMC4576145. doi:10.1016/j.bpj.2015.04.032
Sørensen, C. S., and Kjaergaard, M. (2019). Effective Concentrations Enforced by Intrinsically Disordered Linkers Are Governed by Polymer Physics. Proc. Natl. Acad. Sci. U.S.A. 116 (46), 23124–23131. PubMed PMID: 31659043; PMCID: PMC6859346. doi:10.1073/pnas.1904813116
Stigler, J., Ziegler, F., Gieseke, A., Gebhardt, J. C. M., and Rief, M. (2011). The Complex Folding Network of Single Calmodulin Molecules. Science 334 (6055), 512–516. PubMed PMID: 22034433. doi:10.1126/science.1207598
Sugase, K., Dyson, H. J., and Wright, P. E. (2007). Mechanism of Coupled Folding and Binding of an Intrinsically Disordered Protein. Nature 447 (7147), 1021–1025. PubMed PMID: 17522630. doi:10.1038/nature05858
Suren, T., Rutz, D., Mößmer, P., Merkel, U., Buchner, J., and Rief, M. (2018). Single-molecule Force Spectroscopy Reveals Folding Steps Associated with Hormone Binding and Activation of the Glucocorticoid Receptor. Proc. Natl. Acad. Sci. U.S.A. 115 (46), 11688–11693. PubMed PMID: 30366952; PMCID: PMC6243279. doi:10.1073/pnas.1807618115
Tian, P., and Best, R. B. (2016). Structural Determinants of Misfolding in Multidomain Proteins. Plos Comput. Biol. 12 (5), e1004933. PubMed PMID: 27163669; PMCID: PMC4862688. doi:10.1371/journal.pcbi.1004933
Tian, P., Steward, A., Kudva, R., Su, T., Shilling, P. J., Nickson, A. A., et al. (2018). Folding Pathway of an Ig Domain Is Conserved on and off the Ribosome. Proc. Natl. Acad. Sci. U.S.A. 115 (48), E11284–E93. PubMed PMID: 30413621; PMCID: PMC6275497. doi:10.1073/pnas.1810523115
Trovato, F., and O'Brien, E. P. (2016). Insights into Cotranslational Nascent Protein Behavior from Computer Simulations. Annu. Rev. Biophys. 45, 345–369. PubMed PMID: 27297399. doi:10.1146/annurev-biophys-070915-094153
van der Lee, R., Buljan, M., Lang, B., Weatheritt, R. J., Daughdrill, G. W., Dunker, A. K., et al. (2014). Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 114 (13), 6589–6631. PubMed PMID: 24773235; PMCID: PMC4095912. doi:10.1021/cr400525m
Walsh, I. M., Bowman, M. A., Soto Santarriaga, I. F., Rodriguez, A., and Clark, P. L. (2020). Synonymous Codon Substitutions Perturb Cotranslational Protein Folding In Vivo and Impair Cell Fitness. Proc. Natl. Acad. Sci. U.S.A. 117 (7), 3528–3534. PubMed PMID: 32015130; PMCID: PMC7035613. doi:10.1073/pnas.1907126117
Wang, F., Durfee, L. A., and Huibregtse, J. M. (2013). A Cotranslational Ubiquitination Pathway for Quality Control of Misfolded Proteins. Mol. Cel 50 (3), 368–378. PubMed PMID: 23583076; PMCID: PMC3654026. doi:10.1016/j.molcel.2013.03.009
Waudby, C. A., Launay, H., Cabrita, L. D., and Christodoulou, J. (2013). Protein Folding on the Ribosome Studied Using NMR Spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 74, 57–75. PubMed PMID: 24083462; PMCID: PMC3991860. doi:10.1016/j.pnmrs.2013.07.003
Waudby, C. A., Wlodarski, T., Karyadi, M.-E., Cassaignau, A. M. E., Chan, S. H. S., Wentink, A. S., et al. (2018). Systematic Mapping of Free Energy Landscapes of a Growing Filamin Domain during Biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 115 (39), 9744–9749. PubMed PMID: 30201720; PMCID: PMC6166796. doi:10.1073/pnas.1716252115
Wright, P. E., and Dyson, H. J. (2009). Linking Folding and Binding. Curr. Opin. Struct. Biol. 19 (1), 31–38. PubMed PMID: 19157855; PMCID: PMC2675572. doi:10.1016/j.sbi.2008.12.003
Wruck, F., Katranidis, A., Nierhaus, K. H., Büldt, G., and Hegner, M. (2017). Translation and Folding of Single Proteins in Real Time. Proc. Natl. Acad. Sci. U.S.A. 114, E4399–E407. PubMed PMID: 28507157. doi:10.1073/pnas.1617873114
Young, T. A., Skordalakes, E., and Marqusee, S. (2007). Comparison of Proteolytic Susceptibility in Phosphoglycerate Kinases from Yeast and E. coli: Modulation of Conformational Ensembles without Altering Structure or Stability. J. Mol. Biol. 368 (5), 1438–1447. PubMed PMID: 17397866. doi:10.1016/j.jmb.2007.02.077
Zhao, L., Castanié-Cornet, M.-P., Kumar, S., Genevaux, P., Hayer-Hartl, M., and Hartl, F. U. (2021). Bacterial RF3 Senses Chaperone Function in Co-translational Folding. Mol. Cel 81, 2914–2928. PubMed PMID: 34107307. doi:10.1016/j.molcel.2021.05.016
Zheng, W., Schafer, N. P., and Wolynes, P. G. (2013). Frustration in the Energy Landscapes of Multidomain Protein Misfolding. Proc. Natl. Acad. Sci. U.S.A. 110 (5), 1680–1685. PubMed PMID: 23319605; PMCID: PMC3562767. doi:10.1073/pnas.1222130110
Zhou, M., Guo, J., Cha, J., Chae, M., Chen, S., Barral, J. M., et al. (2013). Non-optimal Codon Usage Affects Expression, Structure and Function of Clock Protein FRQ. Nature 495 (7439), 111–115. PubMed PMID: 23417067; PMCID: PMC3629845. doi:10.1038/nature11833
Zipser, D., and Perrin, D. (1963). Complementation on Ribosomes. Cold Spring Harbor Symposia Quantitative Biol. 28, 533–537. doi:10.1101/sqb.1963.028.01.071
Keywords: co-translational folding, multi-domain proteins, single-molecule methods, optical tweezers, inter-domain interactions, protein misfolding, ribosome
Citation: Rajasekaran N and Kaiser CM (2022) Co-Translational Folding of Multi-Domain Proteins. Front. Mol. Biosci. 9:869027. doi: 10.3389/fmolb.2022.869027
Received: 03 February 2022; Accepted: 21 March 2022;
Published: 20 April 2022.
Edited by:
Stephen D. Fried, Johns Hopkins University, United StatesReviewed by:
Marina V. Rodnina, Max Planck Institute for Biophysical Chemistry, GermanyAnton A. Komar, Cleveland State University, United States
Edward Patrick O’Brien, The Pennsylvania State University (PSU), United States
Copyright © 2022 Rajasekaran and Kaiser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian M. Kaiser, kaiser@jhu.edu