Exploring the Potential Role of Terrestrially Derived Humic Substances in the Marine Biogeochemistry of Iron
 François L. L. Muller
François L. L. Muller- Department of Oceanography, National Sun Yat-sen University, Kaohsiung, Taiwan
There is a widely recognized link between iron bioavailability and phytoplankton growth in large tracts of the World Ocean. Iron bioavailability is thus pivotal to the removal of atmospheric carbon via the ocean’s ‘biological pump.’ To evaluate future scenarios of global climate change, we must therefore understand better how the bioavailability of iron in seawater is linked to processes controlling its supply, chemical speciation and removal from the water column. Much of the research on iron inputs to the open ocean has focused on atmospheric, benthic (shelf sediments) and hydrothermal (mid-ocean) sources of iron. The conventional wisdom has been that riverine iron sources are negligible because many of the major world rivers exhibit extensive removal of dissolved iron by flocculation processes during estuarine mixing. However, recent studies have revealed that a fraction of iron associated with peatland-derived humic and fulvic acids may survive aggregation processes in the freshwater-seawater mixing zone, and thus be exported offshore. This review is a synthesis of available data and information for and against the hypothesis that land-derived humic substances exert a significant control on the marine biogeochemical cycle of iron. From the outset, it is shown that this hypothesis can neither be verified nor disproved at present, in part due to analytical difficulties in characterizing the all-important marine colloidal phase. Evidence is then presented on the likely chemical nature and structure of iron-binding humic ligands along with implications for the lateral transport of iron in surface waters and its participation in carbon stabilization in marine sediments. This is followed by a discussion of photochemically and microbially mediated processes acting on terrestrial humic substances and a discussion of how terrestrial humic substances may influence the biological availability of iron. The review finishes with a presentation of measurement technologies and approaches that could be used to assess (i) how iron-binding ligands in seawater relate to land-derived humic substances and (ii) how terrestrial humics may influence the global carbon cycle indirectly by influencing the processes that control the supply and maintain the pool of ‘dissolved’ iron in the ocean.
How Significant Are River Inputs of Iron and Iron Complexing Ligands to Marine Waters?
Interactions of the Iron and Carbon Cycles
At present, the impact of human disturbances, in particular the perturbations in the flow of CO2 through the ocean-atmosphere system, cannot be predicted because we do not yet understand in adequate detail what processes control the production, transformation and destruction of biomass in the different oceanic compartments (Falkowski, 1997, 2012; Hutchins and Boyd, 2016; Walker et al., 2016). Obtaining such knowledge about the marine carbon cycle presents unique difficulties, not least because a number of biologically essential trace elements are intricately involved in that cycle. For example, there has been a broad recognition over the past two decades of the power of iron, an element which occurs at sub-nanomolar concentrations throughout the world’s oceans, to influence the oceanic biogeochemical cycling of carbon, nitrogen and phosphorus (Pasquier and Holzer, 2017; Tagliabue et al., 2017). Iron availability is a key determinant of the abundance and species of phytoplankton that can flourish in any given location (Sunda and Huntsman, 1995; Hutchins et al., 1998, 1999; Jickells et al., 2005). In return, the balance struck between pico- and nanoplanktonic algal production strongly affects how much carbon (but also nitrogen, phosphorus, and indeed iron) is exported from the ocean surface layer to the deep ocean. An increase in nitrate export results in an alkalinity increase and a pCO2 decrease in surface waters, hence mediating the ocean’s role in climate change (Sigman and Boyle, 2000). Therefore, there are intricate and dynamic relationships between (i) iron supply and availability, (ii) the biological processes that produce and export marine organic matter, (iii) the drawdown of nutrients and carbon from the ocean surface layer, and (iv) global climate. As such, recent modeling studies have highlighted the importance of including up to two classes of iron chelating organic compounds, or ‘ligands,’ acting to stabilize the iron inventory in the deep ocean, in global biogeochemical models of iron (Völker and Tagliabue, 2015; Tagliabue et al., 2017). In these models, the ligands controlling dissolved iron levels according to equilibrium complexation assumptions are assumed to be produced in situ, i.e., through biological processes. While this may be a reasonable assumption to make, we actually know extremely little about the sources, nature and residence times of these ligands.
Another example of how intertwined the iron and carbon cycles are, comes from the finding that iron minerals and organic matter mutually stabilize each other by forming nanosized aggregates in sediments. Indeed, recent work suggests that 21% of all organic carbon buried and preserved in marine sediments is associated with reactive iron minerals (Lalonde et al., 2012). The exact mechanism responsible for this association is unknown but the authors have drawn an interesting parallel with the aggregation of soluble and colloidal organic matter occurring in seawater and resulting in structures where organic molecules are glued together by either ionic bridges provided by Ca2+, Mg2+, or Fe3+ ions (Chin et al., 1998; Muller, 1999; Wells, 2002; Batchelli et al., 2010) or nanophases of iron oxides (Schröder et al., 2016). This means of preserving potentially reactive organic carbon through association with reactive iron phases in sediments may have important consequences for the long-term control (i.e., tens of thousands of years) of atmospheric CO2 (Burdige, 2007; Lalonde et al., 2012; Johnson et al., 2015).
Difficulties in Estimating Global Riverine Inputs and Quantifying Aquatic Humic Substances
The importance of organic ligands for stabilizing iron in solution (or in suspension) is now widely accepted. However, the direct source of these ligands and the role they play in the iron cycle is still not well understood. Recently, new sources and processes have been identified which challenge the conventional understanding of both iron (Conway and John, 2014) and ligand oceanic cycles (Tagliabue et al., 2016). In particular, field and laboratory studies have revealed that high-latitude humic-rich rivers have a high iron-carrying capacity (Dai and Martin, 1995; Batchelli et al., 2010; Krachler et al., 2015, 2016) although they are currently underrepresented in global riverine flux calculations. This implies that global river inputs of iron (Raiswell and Canfield, 2012) may currently be underestimated. Further work based on the separation of soluble (< 1 to 5 kDa) and colloidal fractions of iron (< 0.2 to 1 μm) by ultrafiltration or field-flow fractionation techniques has recently revealed that the soluble fraction invariably shows a linear conservative trend with increasing salinity while the colloidal fraction generally shows a concave trend with increasing salinity (Figures 1, 2). Molecular weight (MW) cut-off values of 1, 3 or 5 kDa have been used to distinguish between soluble and colloidal fractions. It is important to realize that these boundaries are operationally defined and that the size or MW fractionation process may suffer artifacts affecting its selectivity. In addition, chemical (especially humic) species possessing identical properties may occur within a size continuum on both the high molecular weight (HMW) and low molecular weight (LMW) sides of the chosen cut-off. Despite its shortcomings, size fractionation has allowed consistent patterns to emerge in estuarine and coastal datasets, as mentioned earlier. In certain environmental settings, e.g., where the pH of river water is close to that of seawater and there is no proper estuary but instead an open, unconstrained river plume, the entire pool of dissolved iron can be maintained in solution (LMW fraction) and suspension (HMW fraction) across the salinity gradient (Figure 2D).
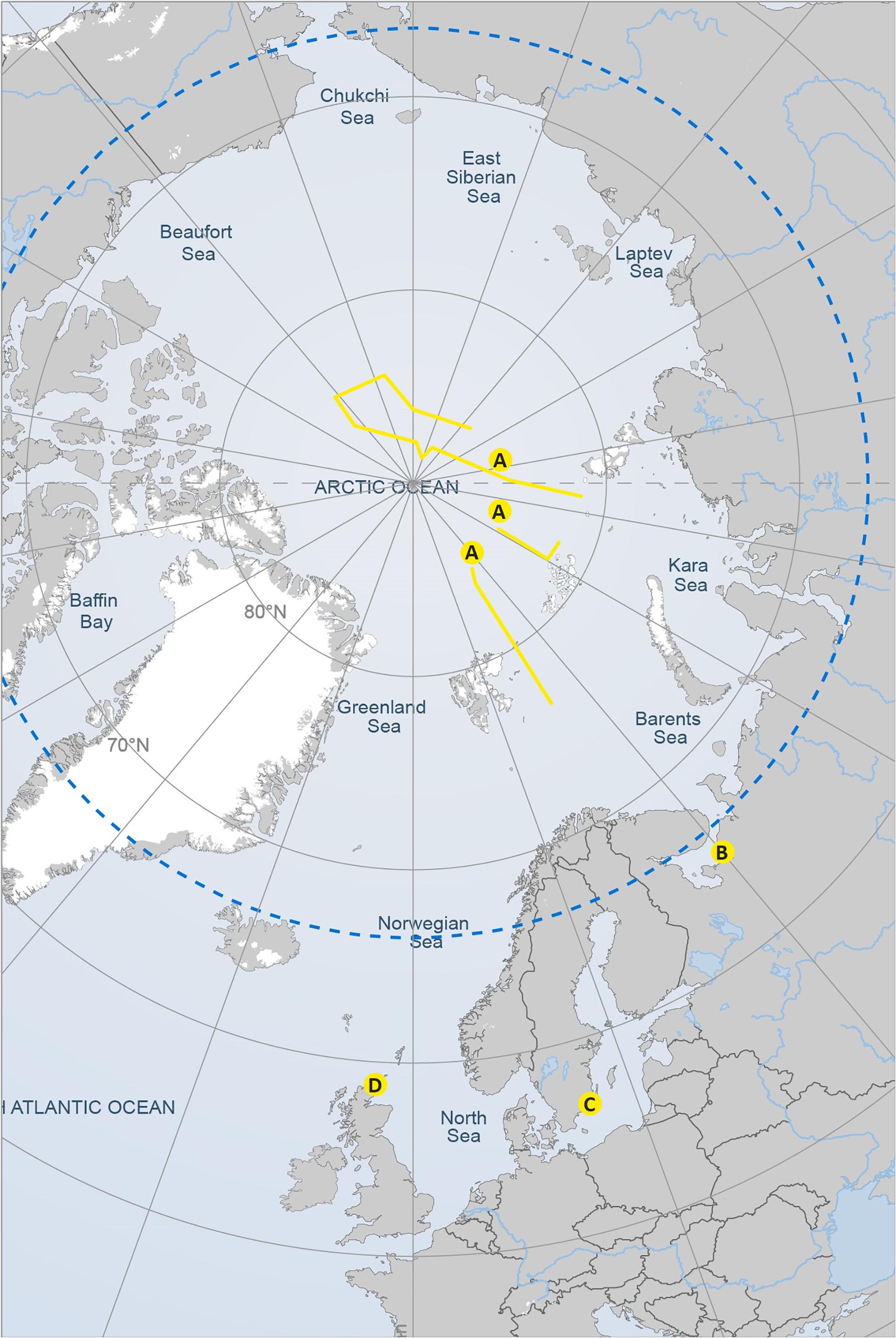
FIGURE 1. Map of the sampling transects and sampling areas corresponding to the four study datasets presented in Figure 2. Reproduced with the permission of the Arctic Portal.
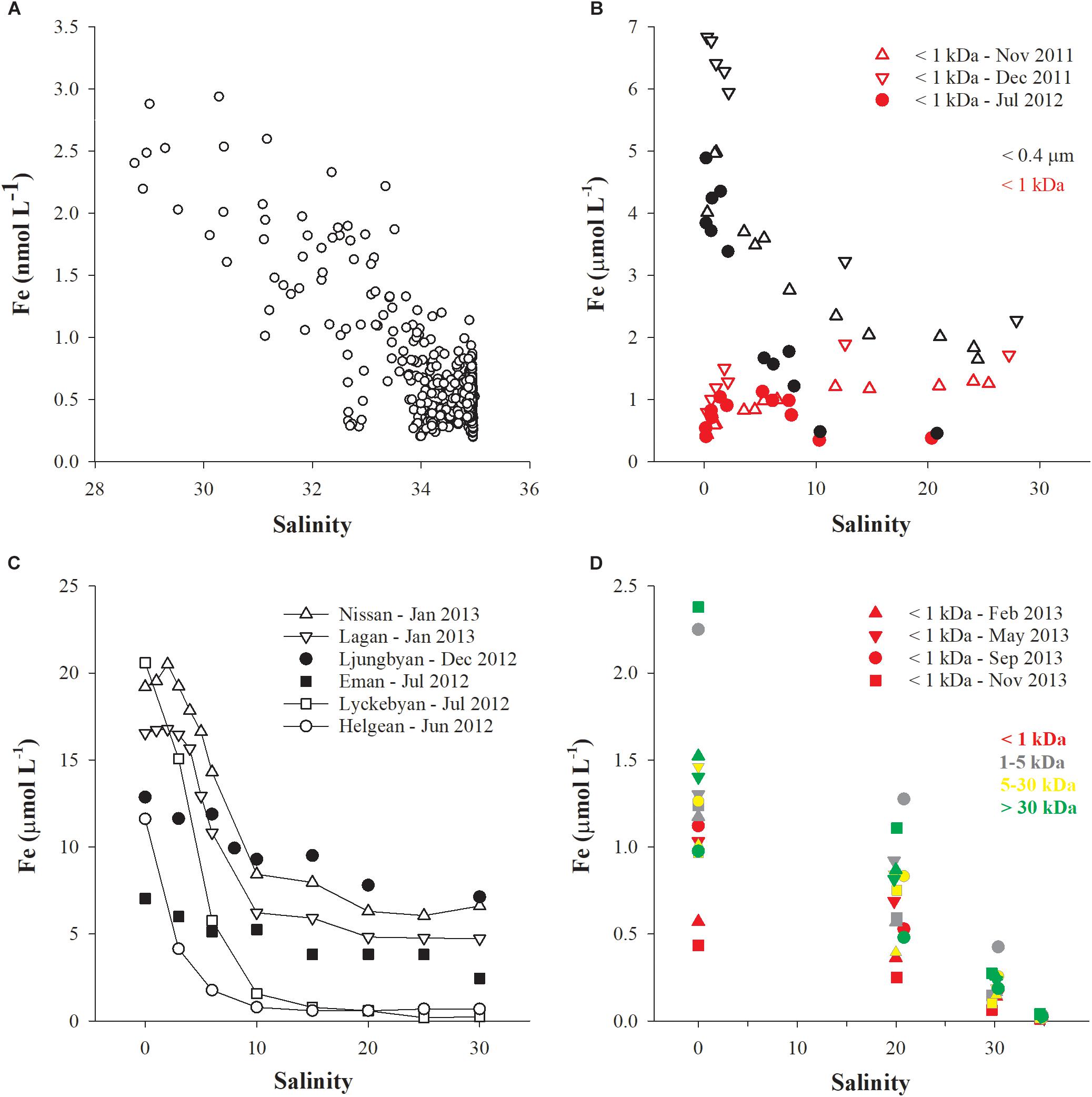
FIGURE 2. The concentrations of dissolved iron, [Fe], present in various size classes of DOM are plotted as a function of salinity in four mid- and high-latitude systems. Data source: (A) Klunder et al. (2012), (B) Pokrovsky et al. (2014), (C) Kritzberg et al. (2014), (D) Muller and Cuscov (2017). In (D), four samples of salinity 0, 20, 30, and 35, respectively, were collected during four seasonal transects of Thurso Bay (denoted by symbol shapes) and all samples were CFF fractionated into four size fractions (denoted by symbol colors).
Recent efforts to quantify the riverine flux of iron to the coastal ocean have thus identified humic substances (the term is used here to designate humic and fulvic acids according to their soil science definition) as the main vector at northern latitudes (Laglera and van den Berg, 2009; Krachler et al., 2010; Crocket et al., 2015). The complex and non-constant composition of humic substances poses a big challenge to marine chemists: they are heterogeneous, multifunctional polyelectrolytes (Buffle and Altmann, 1987) which defy characterization because each analytical technique senses a different property. This leads to a complex physicochemical speciation for iron, with equilibrium complexation reactions alone being unable to represent this complexity (Gledhill and Buck, 2012). As part of this review, I will be highlighting the capabilities and limitations of currently used techniques when it comes to (i) describing the reactivity (complexation, redox, kinetics) of the different physicochemical components of the dissolved iron pool or (ii) tracking terrestrial humic substances along the land-ocean continuum.
Organization of the Review
The Section “How Important is the Distinction Between Soluble and Colloidal Fractions When It Comes to Iron’s Ocean Cycling?” reviews the different lines of evidence pointing to the importance of the colloidal phase in the transport properties and marine geochemical behavior of iron. Section “What Do We Know About the Nature and Structure of Iron-Binding Ligands in Natural Waters?” summarizes our current knowledge on the functional group chemistry and structural constraints that natural iron-binding ligands conform to, then briefly examines how humic compounds meet these constraints. Section “What Happens to Land Derived Humic Substances Moving From River to Ocean?” examines the whole gamut of processes (physicochemical, photochemical, and microbial) that are thought to act on humic substances during their land-ocean transfer and the consequences for iron’s ocean cycling. Section “How Bioavailable Is Humic-Bound Iron in the Marine Environment?” provides examples of processes that facilitate the transfer of iron from the humic (or mixed humic/mineral) phase to the biota. Section “What Is the Current Status of Measurement Technologies for Iron-Humic Studies in Marine Waters?” is devoted to the analytical methods currently in use for the detection of humic substances in marine waters as well as the methods used to probe iron-humic interactions at solid interfaces. Section “What Are the Consequences of Marine and Terrestrial Organic Matter Interacting With Iron Oxides and With Each Other?” considers how iron speciation may be affected by the competitive interactions between marine and terrestrial dissolved organic matter (DOM) in the water column, and how iron and carbon biogeochemical cycles are coupled via their mutual stabilization in marine sediments. Section “Conclusion” discusses future research directions and suggests some lines of investigation that would provide the data necessary to improve our understanding of the role played by terrestrial humic substances in the marine iron cycle.
How Important Is the Distinction Between Soluble and Colloidal Fractions When It Comes to Iron’s Ocean Cycling?
The Nature, Function, and Fate of Marine Colloids
Our recognition of the role played by aquatic colloids as transport vectors and chemical reactants has increased over the last two decades. In particular, there is a broad recognition that organic and mixed mineral/organic colloids are extremely abundant in coastal and oceanic waters (Muller, 1998; Santschi et al., 1998; Guo et al., 2000; Wells, 2002; Stolpe and Hassellöv, 2010). In many coastal waters, the colloidal phase turns out to contain the majority of iron and iron-binding ligands present in these waters (Boye et al., 2010; Muller and Cuscov, 2017). However, the fate of colloidal iron is likely to vary according to the nature and fate of its colloidal carrier phase. The latter may consist of (i) an entirely organic matrix made up of individual humic molecules held together by hydrogen bonds and metal coordination bonds (Muller, 1996, 1999; Batchelli et al., 2010), (ii) products of direct release or degradation of algal material (Boehme and Wells, 2006), (iii) organically coated mineral nanoparticles (Karlsson and Persson, 2010), or (iv) a product of the coprecipitation of iron (oxyhydr)oxides with organic matter (Fritzsche et al., 2015). Some of the most recent research has also suggested that viruses could hold as much as 70% of the colloidal fraction of organically complexed iron in oceanic surface waters (Bonnain et al., 2016), thus representing a very substantial portion of the colloidal iron pool. Specifically, virus-associated iron is concentrated in bacteriophage tails which take advantage of the cellular iron uptake mechanism to infect bacteria. This leads to bacterial cell lysis, followed by release of cellular iron into the water column, of which a significant fraction is already incorporated into new phage tails (Bonnain et al., 2016). This rapid recycling of iron thus produces and maintains a pool of marine colloidal organic matter made up of viruses. As for the non-living forms of colloidal iron, their fate in ocean waters is still a matter of debate. If present as (oxyhydr)oxide nanoparticles, one would expect their most important physical property, in the context of the iron cycle, to be their tendency to aggregate. Their intrinsic stability against aggregation would be expected to depend on the repulsive interactions between similarly charged electrical double layers and on particle-water affinity. The adsorption of humic macromolecules and other polymeric material would promote their stability on both counts and therefore would tend to slow down their aggregation and subsequent sinking. If, on the other hand, iron formed complexes with ligands within an entirely organic colloidal matrix, and if that matrix was held together by a combination of hydrogen bonds and coordination bonds with metals (Mackey and Zirino, 1994), then one would expect aggregation to be governed by chemical equilibrium reactions involving single molecules (i.e., the building blocks), polymers (gel precursors) and particulate gels (Chin et al., 1998; Wells, 2002; Elliott et al., 2014), rather than by any intrinsic instability of the colloidal phase. The fate of iron incorporated during gel formation would then depend on the vertical stability of the gel but also on what goes on within it. For example, it is well known that marine gel draws oceanic microbes together in tight, interactive neighborhoods. In addition, recent evidence suggests that bacteria and viruses represent locally high concentrations of iron. This iron pool might be released as a pulse to the ambient environment when the bacteria are grazed upon by protozoans or attacked by viruses.
It should be added as a note of caution that all the above descriptions are somewhat speculative. Until now, there has not been any joint evaluations of both the physicochemical nature of the colloidal phase and its iron binding properties in the natural environment. On the one hand, the methods used to determine the iron binding strength of organic ligands do not give any information about the nature of the iron-bearing colloidal phase. On the other hand, bulk measurements of the colloidal phase (Belzile and Guo, 2006; Boehme and Wells, 2006; Stolpe et al., 2013; Cuss and Guéguen, 2015), although attractive from a methodological point of view, cannot generally be related to iron-binding properties which are governed by minute concentrations of highly specific ligands. For example, terrigenous humic acids have a density of carboxyl and hydroxyl sites of around 1-4 μmol mg-1 HA (Masini et al., 1998), i.e., two orders of magnitude higher than the density of iron binding sites (Laglera and van den Berg, 2009). More sophisticated approaches are therefore needed to obtain iron binding information in tandem with information on the nature and reactivity of iron’s colloidal carrier phase. Only then will it be possible to assess the impact of global perturbations, e.g., increased humic-bound iron fluxes from northern rivers (Neal et al., 2008) and surface ocean warming and acidification, on the iron biogeochemical cycle (Völker and Tagliabue, 2015).
What Do We Know About the Nature and Structure of Iron-Binding Ligands in Natural Waters?
As we seek to understand the potential role of terrestrially derived humic substances in the ocean, it is worthwhile briefly reminding ourselves of the definition of humic substances as well as reviewing the current state of knowledge about iron-humic binding. Traditionally, the aquatic humic substances we are concerned with here have been considered to comprise two operational fractions distinguished by their solubility and adsorption properties. Humic acids (HA) are soluble in base solutions but insoluble in strong acid solutions. Fulvic acids (FA) are soluble in both base and acid. The method adopted by the International Humic Substance Society (IHSS) for the isolation of humic and fulvic acids is one in which XAD-8 resin is used to concentrate humic substances from acidified (pH 2) samples (Thurman and Malcolm, 1981; Aiken, 1985). The concentrated humic substances are then eluted with 0.1 M NaOH and fractionated into HA (precipitated at pH 1, rinsed and dissolved in NaOH) and FA (soluble at pH 1, adjusted to pH 2 and re-applied to XAD-8, rinsed and eluted with NaOH). A more extensive fractionation scheme based on XAD-4 and XAD-8 resins has also been used successfully to divide a seawater sample into hydrophilic and hydrophobic fractions (Ćosović et al., 2010; Zhang et al., 2017). Finally, size exclusion chromatography has also been applied successfully to the fractionation of the humic material by molecular weight. In combination with organic carbon detection (SEC-LC-OCD) it allows seamless extraction and quantification of humic substances, even in seawater (Hubert et al., 2011).
Until recently, the prevailing view about humic substances was that they were comprised of inherently large molecules with a weight-average MW between 1 and 200 + kDa (Stevenson, 1994). Molecular weights of aquatic FA were reported to be 1–2 kDa (Gilliam and Riley, 1981; Aiken and Malcolm, 1987) and MW of HA between 4 and 200 + kDa (Reid et al., 1990). Recently, new evidence has emerged in support of the argument put forward by Sutton and Sposito (2005) that depicts humic substances as “collections of diverse, relatively low molecular mass compounds forming dynamic associations stabilized by hydrophobic interactions and hydrogen bonds.” For example, there is now experimental proof that the apparently high molecular weights of humic molecules actually reflect supramolecular associations (Batchelli et al., 2009) of small monomer units (< 1 kDa). The resulting associations can be disrupted by the action of weak organic acids (Piccolo et al., 1999) and by ion exchange (Batchelli et al., 2010). Because of this continuum, it seems reasonable to use the term humic, to refer collectively to humic and fulvic acids. Additional support for this terminology comes from recent findings that all humic and fulvic substances appear to contain the same functional groups, i.e., carboxylic, phenolic, and carbonyl, only their abundances differ (Blazevic et al., 2016; Schellekens et al., 2017). On the basis of such findings, Orlowska et al. (2017b) synthesized a range of model compounds in an attempt to represent the main components of humic substances, namely lignin decomposition products. They found that β-O-4 type dilignol compounds, which included catechol moieties, were excellent model ligands for aquatic humic acids. Indeed, the synthesized dilignols were found to have very similar properties, as measured by 1H-NMR and 13C-NMR spectroscopy, EPR, IR and UV-Vis spectroscopies, and ESI-MS. Their iron binding properties and the bioavailability of their iron complexes were also in good agreement, prompting Orlowska et al. (2017b) to suggest that catechol moieties could play a critical role in the stabilization of Fe(III) by humic substances. This is in accordance with earlier findings that Fe binding sites tend to reside in the proximity of aromatic structures (Fujii et al., 2014). Since the latter are more abundant in HA than in FA, it is also consistent with Laglera and van den Berg’s (2009) measurements of the iron binding capacity of the Suwannee River humic acid (32 nmol Fe per mg HA) being higher than fulvic acid (17 nmol Fe per mg FA). Finally, it supports the notion (Aiken et al., 2011; Nuzzo et al., 2013; Fujii et al., 2014; Schellekens et al., 2017) that the iron binding properties of humic substances are driven by recognition chemistry (highly selective binding sites) and structural constraints (molecular architecture) rather than by the abundance of carboxylic and phenolic groups or some other bulk property.
Theoretical approaches to the classification of metals and ligands in solution tell us that Fe3+ is a hard Lewis acid which will tend to form strong complexes with correspondingly hard ligands such as OH- (Morel and Hering, 1993; Stumm and Morgan, 1996; Luther, 2016). Recent XAS analyses of terrigenous humic substances have indeed shown that iron complexation was only dependent on oxygen containing groups such as carbonyl and phenol (Blazevic et al., 2016; Orlowska et al., 2017a). This, in turn, is consistent with the view that lignin products are important in maintaining iron in solution (Murphy et al., 2008; Krachler et al., 2012) and also that functional groups residing in the proximity of aromatic structures within the humics are especially significant (Fujii et al., 2014). Since phenolic groups are redox mediators, there may also be implications for the photochemical reactivity of humic substances in surface waters and the Fe(III)/Fe(II) photochemical cycle. Photochemical processing of humic substances along the land-ocean continuum will be briefly reviewed in the next section. At this point, it is worth mentioning the study by Blazevic et al. (2016) which suggests that the coordination chemistry of iron remains unchanged during the land-ocean transfer of iron-humic complexes while its redox state changes from Fe(III) to Fe(II) in ocean waters. It is also worth comparing the above iron-humic complexation studies with studies of iron binding at the iron oxide-DOM interface, such as occurs in sediments where iron and organic carbon mutually stabilize each other. Such a comparison reveals that the same functional groups (carboxylic, phenolic, catechol) are involved in the stabilization of iron in solution (Blazevic et al., 2016; Orlowska et al., 2017a) as in the stabilization of nanoscale iron and manganese oxides in sediments (Lalonde et al., 2012; Johnson et al., 2015), or indeed in the stabilization of engineered nanomaterials (Chen et al., 2015). Therefore, determining the control that humic ligands exert on the iron cycle would benefit from extensive transfer of information and collaboration opportunities between the seawater chemistry and sedimentary organic geochemistry communities. In order to make an interdisciplinary effort between seawater chemists and organic geochemists work, both must be willing to design some carefully controlled complexation/adsorption experiments on which they can begin to focus. As a first step, model compounds can be used such as compounds with a dilignol backbone (Orlowska et al., 2017b) for terrestrial humics and a melanoidin backbone (Ćosović et al., 2010) for marine humics.
What Happens to Land Derived Humic Substances Moving From River to Ocean?
Environmental and Geochemical Functions of Humic Substances
Humic substances are complex, acidic organic molecules formed by biotic and abiotic processes following the decomposition of plant and microbial material (Aiken, 1985; Stevenson, 1994). They form in soils and in the water columns of lakes and oceans. They tend to resist microbiological attack because of their complex molecular structure (Madsen, 2008). They have many environmental functions, including carbon cycling, light penetration into waters, mediation of light-driven reactions and buffering of nutrient and metal concentrations. In addition to these processes, which are driven by their molecular properties, we have seen that humic substances also have the potential to participate in the geochemical cycling of iron through their colloidal properties. In effect, this means providing iron with a microenvironment different from the ambient aqueous environment in terms of its structural constraints, polarity, light regime and redox chemistry. This colloidal dimension (Buffle et al., 1998; Wells, 2002) can add a great deal of complexity to the speciation and biogeochemical behavior of iron. For example, recent measurements of the partition of iron and iron binding ligands into or onto the colloidal phase have raised the question whether an equilibrium complexation model is even appropriate for colloidal iron binding sites (Batchelli et al., 2010; Fitzsimmons et al., 2015). Regardless of whether LMW or HMW fractions are considered, terrestrial humic substances have a larger number of aromatic moieties and/or redox-active moieties than marine humics (Koch et al., 2005; Colatriano et al., 2018). As such, terrestrial humics would be expected to be particularly effective at bonding with Fe(III), stabilizing it in solution and mediating the photochemically driven Fe(III)/Fe(II) redox cycle. The question is, are they exported to the open ocean in sufficient quantities to play these roles?
The above question eludes consensus at this time, although work that has looked at this issue has largely drawn the conclusion that terrestrial DOM is rapidly degraded during transport to the open ocean. This is because the bulk chemical composition of marine DOM, as indicated by, e.g., lignin phenols (Benner et al., 2005; Louchouarn et al., 2010), δ13C (Hossler and Bauer, 2013), spectral absorbance (Fichot and Benner, 2012) and fluorescence spectra (Green and Blough, 1994) shows little terrestrial character. Indeed, estimates of terrestrial DOC turnover times based on lignin phenol content (20-130 years; Opsahl and Benner, 1998) and water mass age (7±3 years; Hansell, 2002) are consistent with rapid degradation of terrestrial DOM. While this evidence clearly suggests that the bulk of terrestrial DOM, and hence terrestrial humic substances, suffer extensive alterations, the effects of these alterations on iron speciation are largely unknown. For instance, Rathgeb et al. (2017) found that iron-binding lignin and tannin structures were common in humic-rich rivers but were not found in the estuarine mixing zone; nevertheless, they argued that iron could still be maintained in solution as a result of ligand exchange in estuarine waters. Further recent work has demonstrated that while some lignin residues appear to be lost through the sequence river-estuary-ocean (Cao et al., 2018), new compounds are formed during the hydroxy radical alteration of lignin which no longer bear the lignin biomarker signature (Waggoner and Hatcher, 2017). Therefore, there is a strong possibility that structural units or ligands of terrestrial humic origin might persist on timescales long enough to be transported into the ocean. Studies of the nature and extent of terrestrial humic alterations in the coastal zone are therefore urgently required.
Processing of Land Derived Humic Substances in the Estuarine and Coastal Zone
As mentioned in the Introduction, a number of estuarine or river plume systems are particularly effective at transporting terrigenous DOM to the coastal ocean, although it is not immediately clear what they have in common (Ward, 2017). In some cases, the high pH and alkalinity of the river water itself appear to contribute to maintaining humic substances in solution by preconditioning them against flocculation, which would normally be caused by the changing pH and ionic strength (Sholkovitz and Copland, 1981), specific interactions with Ca2+ and Mg2+ cations (Nowostawska et al., 2008), and light-induced aggregation of DOM (Helms et al., 2013; Chen et al., 2014; Sun et al., 2017). In addition, rivers and streams draining intact peatlands have been reported to contain humic constituents that are not prone to flocculation but instead are transported, along with complexed iron, directly to the sea (Batchelli et al., 2009; Pokrovsky et al., 2014; Krachler et al., 2015; Cao et al., 2018). We have yet to understand what controls the stability of these humic constituents as well as their long-term fate in the marine environment. In most river-ocean systems, however, the removal of both DOC and dissolved iron by coagulation and coprecipitation is so extensive that the escape of small amounts of iron-carrying humic constituents (Krachler et al., 2015) has usually escaped detection. In such systems, a well-documented sequence of processes act on humic substances during their land-ocean transfer: (i) coagulation in the low salinity estuarine mixing zone, (ii) photochemical degradation, which is mediated by the colored, light absorbing components of the DOM (CDOM) and tends to lower the mean MW of DOM (including humics), and (iii) carbon uptake by marine microbes.
Key variables responsible for the physical removal of DOC and dissolved iron during estuarine mixing are pH, ionic strength, suspended particulate matter (SPM) and the presence of sunlight. The degree to which, and the conditions under which, each of these factors alters the chemical composition and MW distribution of freshwater DOM is still poorly understood (Helms et al., 2013). Although a general trend toward coagulation and removal of organic compounds and iron can be anticipated, the degree of removal can vary between 0 and 100% depending on the physicochemical forms of organic carbon and iron. For example, 46-99% of riverine dissolved iron is removed across the full salinity gradient in rivers draining into the southern Baltic Sea (Kritzberg et al., 2014) while negligible or no loss of DOC and Fe occurs within the River Thurso mixing plume, off the north coast of Scotland (Muller and Cuscov, 2017). Differing river-borne constituents between the sampled estuaries may explain why DOC and Fe exhibit a conservative or nonconservative mixing behavior. Thus, iron in the Baltic rivers is already present as aggregates of organic matter and Fe oxyhydroxides nanoparticles which are prone to salinity induced flocculation (Asmala et al., 2014b; Du et al., 2018) whereas the entire pool of dissolved iron in the River Thurso is present as organic complexes which survive the electrostatic forcing of increased salinity (Krachler et al., 2010; Crocket et al., 2015). Where iron is distributed in different forms, there is now clear evidence from field-flow fractionation studies with UV or ICPMS detection (Krachler et al., 2015) as well as measurements based on X-ray absorption fine structure (EXAFS) spectroscopy (Herzog et al., 2017) that the unstable forms are removed by aggregation or coprecipitation while the stable humic complexes are much less affected and can be transported to sea. To conclude, although the key variables affecting the stability of specific iron and humic forms in the mixing zone are well known, their effects on the dissolved iron and humic estuarine mixing behavior cannot be predicted in a generalize manner. Instead, the conventional approach has been to use a metric, often a single parameter, which can be used to anticipate mixing behavior in a given estuarine system. Depending on the geochemical setting, the concentration and molecular weight distribution of humic substances (Krachler et al., 2015; Muller and Cuscov, 2017), the strength of Fe-binding organic ligands (Bundy et al., 2015), the Fe/Corg elemental ratio (Kritzberg et al., 2014) or the inorganic-organic partitioning of Fe (Krachler et al., 2015; Herzog et al., 2017) have all been used as predictors of how much dissolved iron is likely to be exported to the coastal ocean.
Photochemical degradation of DOM not only forms inorganic carbon (Benner et al., 2005; Louchouarn et al., 2010; Hossler and Bauer, 2013) but selectively degrades aromatic and phenolic groups and lowers the mean molecular weight of DOM. It is important to note that such transformations are favored by energetic UVB radiation. Due to these compositional changes, photodegradation significantly enhances subsequent degradation by marine microbes (Asmala et al., 2014a; Jones et al., 2016). Photodegradation can also cause an irreversible loss of electron donating capacity of phenol groups in humic substances (Sharpless et al., 2014). As a result, it has been argued that the photochemically altered humic substances are in fact less labile and less involved in colloid-colloid and colloid-particulate interactions (Boehme and Wells, 2006; Cao et al., 2018). Based on this argument, they might be expected to persist long enough to allow their transport into the ocean (Rossel et al., 2013; Cao et al., 2018).
Photoirradiation in the less energetic part of the solar spectrum can be responsible for the formation of new POM and the production of new soluble compounds (Helms et al., 2013; Chen et al., 2014; Sun et al., 2017). Recent experiments conducted with solar simulators (Chen et al., 2014) and UVA lamps (Helms et al., 2013) have produced very complex effects on natural, humic-rich freshwaters. The role of iron and pre-existing colloids appears to be especially complex. However, 3 consistent observations emerge from these experiments: (1) newly formed POM is enriched in nitrogen (Chen et al., 2014), especially amide functionality (Helms et al., 2013; Sun et al., 2017); (2) newly formed POM comprises a mixture of newly formed condensed aromatic compounds resembling black carbon (Chen et al., 2014) and aliphatic molecules of high H/C and low O/C molar ratios (Helms et al., 2013; Chen et al., 2014); (3) aliphatic molecules that are more oxygenated than their POM counterparts are produced in increasing concentrations in the DOM pool with increasing photoirradiation time (Helms et al., 2013; Chen et al., 2014). The latter observation raises the question of whether new iron binding ligands may be produced in the process and whether they may offset the loss of soluble ligands due to the increasing mineralization of DOM. In any case, it is worth noting that the newly produced ligands may no longer be recognizable as ‘terrestrial’ with conventional methods.
The bacterial degradation of terrestrial humic substances is poorly understood. It has been suggested that it does not occur to any appreciable extent in the estuarine environment (Kisand et al., 2008) although more recent work has shown some heterotrophic activity by estuarine bacteria on material which had been predegraded and amended with inorganic nutrients (Asmala et al., 2014a). Humic DOC decomposition by marine microbial communities can be significant (Rossel et al., 2013; Jones et al., 2016). Even less understood are the effects that the combined photochemical and microbial processing of humics may have on iron speciation. One useful approach to examining this question would be to elucidate which properties of the humic substances drive the speciation of iron. This could be achieved by (i) determining properties of humics AND iron speciation in samples drawn from time series experiments and field transects, (ii) measuring the key properties before and after solar irradiation, and before and after controlled exposure to bacterial communities, (iii) evaluating the role of heterotrophic bacteria in transferring iron from the humic to the biogenic phase.
How Bioavailable Is Humic-Bound Iron in the Marine Environment?
How Does the Biological Uptake of Iron Relate to Its Chemical Speciation?
Originally, iron uptake by phytoplankton was thought to be proportional to the concentration of dissolved, unchelated inorganic iron species such as Fe3+, Fe(OH)2+, Fe(OH)3, Fe(OH)4-, Fe2+, FeCl+, Fe(SO4) or Fe(CO3). The inorganic species of Fe(III), which are controlled by hydrolysis products Fe(OH)2+, Fe(OH)3 and Fe(OH)4-, are collectively denoted Fe’. Although Fe’ is the most bioavailable fraction of Fe(III), it is also the least abundant due to the very low solubility of Fe(III) in oxygenated seawater at pH 8. The presence of organic complexes greatly increases the solubility of iron (Kuma et al., 1996) and the possibility for its biological uptake. Iron uptake mechanisms include the direct uptake of the organically complexed iron (Chen and Wang, 2008; Kuhn et al., 2014), e.g., via a siderophore-based transport system (Sunda, 2001; Gledhill and Buck, 2012), or the complex being first reduced to Fe(II) which is then bioavailable (Sunda, 2001; Lis et al., 2015). In addition, studies using ultrafiltration methods have revealed that colloids account for a significant fraction of dissolved iron (Moran et al., 1996; Breitbarth et al., 2010). Further studies have shown that siderophores produced by microorganisms can readily remove Fe(III) from humic substances and so provide an effective mechanism of Fe(III) uptake (Kuhn et al., 2014; Orlowska et al., 2017b). Owing to their surface-active, light-absorption, redox and complexation properties, humic substances have the potential to deliver bioavailable iron via any of these three strategies, functioning alone or in combination. In the next paragraph, several lines of evidence are presented which give credence to this proposition.
Humic Substances as a Versatile and Effective Source of Bioavailable Iron
Table 1 presents measured values of total dissolved iron, [FeT], and calculated values of labile inorganic iron concentrations, [Fe’], in marine waters where humic ligands are likely to control the speciation of iron. The reported [Fe’] values span 3 orders of magnitude. Sub-picomolar values prevail in environments where humic ligand concentrations, [LT], clearly exceeds [FeT] and thus buffer [Fe’] at very low values. Sub-nanomolar concentrations are found in environments where humic ligands are saturated with Fe, and the excess Fe augments Fe’ by a corresponding amount. We note that, in either case, uncomplexed iron (Fe’) is very small compared to organically complexed iron (FeL). Recent phytoplankton culture experiments have revealed that iron bioavailability to different species of eukaryotic phytoplankton (Lis et al., 2014) as well as cyanobacteria (Lis et al., 2015) reflects a mixed pool including both complexed (FeL) and uncomplexed iron (Fe’). Among all the complexing organic substrate tested, Lis et al. (2014) found that humic acids had the highest bioavailability. Furthermore, they found that the iron uptake rate for the Fe-humic substrate was greater or equal than the uptake rate for the uncomplexed iron Fe’. No other substrate, including iron siderophores, produced such high bioavailability! These results can be explained by a combination of the surface-active and photochemical properties of humics: these properties effectively enable iron photoreduction to take place at the phytoplankton cell surface, prior to its transport across the cell membrane. As a result, a steady-state pool of unchelated iron is present at the cell-solution interface so that its transport is limited by transmembrane reductase transport rather than diffusion from the bulk solution. These results are consistent with those of Maldonado et al. (2006) showing that iron reduction also takes place at the cell surface, involving transmembrane reductases. This was also the case in the iron uptake experiments of Chen and Wang (2008) in which the highest uptake rates by diatoms and cyanobacteria were found for inorganic and humic-bound iron, as opposed to iron bound to desferrioxamine or ferrichrome. These results are in agreement with a range of findings concerning the role of both terrestrial (Blazevic et al., 2016) and freshly produced marine humic substances (Barbeau et al., 2001) as electron donors to Fe(III) when exposed to sunlight. Moreover, they fit well with the presence of a steady state pool of Fe(II) postulated by Blazevic et al. (2016) and Krachler et al. (2016). The prevalence of the reductive iron uptake mechanism among marine cyanobacteria and eukaryotic phytoplankton is not a new finding (Sunda, 2001). Nor is the maintenance of Fe(II) in oxygenated waters by organic ligands (Croot et al., 2001), including rainwater humic-like ligands (Willey et al., 2008). However, only recently have XAS studies shown that the seawater resistant component of terrigenous humic substances also undergo photochemical reactions that result in reduction of Fe(III) to Fe(II). In addition, it would appear that the coordination geometry (octahedral FeIIIO6) of the Fe-humic complex remains unchanged during transport from freshwaters to marine waters (Blazevic et al., 2016).
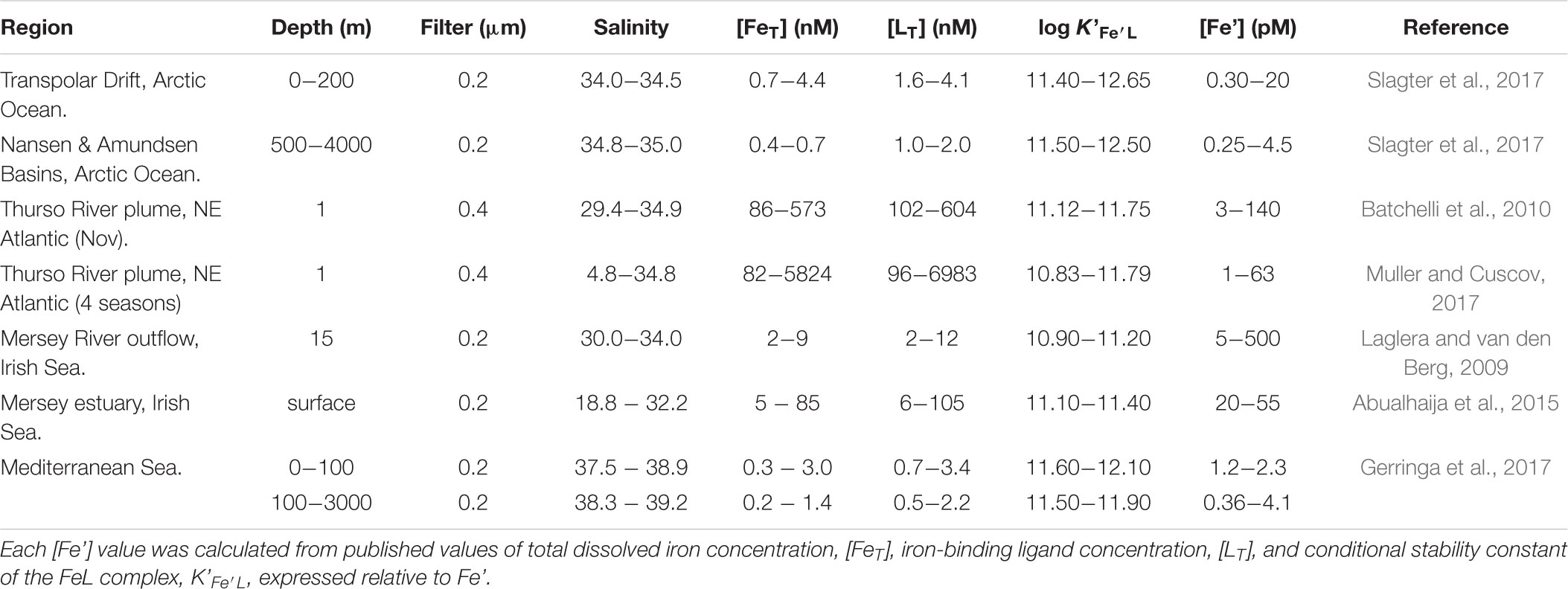
TABLE 1. Compilation of [Fe’] values (unchelated dissolved inorganic iron concentrations) in marine waters where humic ligands are deemed to control the speciation of iron.
What significance the colloidal dimension of humics has when it comes to the supply of bioavailable iron is still an open question. That exposure to UV light makes humic substances more bioavailable to aquatic microorganisms is well recognized (Opsahl and Zepp, 2001; Lalonde et al., 2014). Humic colloids may well facilitate microbial respiration by providing hot spots for (bacteria to congregate as well as a localized source of nutrients. Recent work by Hasegawa et al. (2017) provides some support for this hypothesis. They found that the highest intracellular iron uptake by Prymnesium parvum (haptophyte) occurred with the 10-30 kDa MW fraction of humics in seawater medium rather than with the LMW fraction. This was accompanied by an increase in the iron associated with the LMW fraction, presumably due to the breakup of humic colloids leading to a shift in the MW distribution of the humics (Batchelli et al., 2010; Kuhn et al., 2014).
Recommendations for Future Research Directions
At the present time there is no commercial standard for either marine humics or degraded terrestrial humics, with the result that most biological process studies to date have been conducted with humic substances isolated from soils or freshwater bodies. Future experiments should be conducted under more natural conditions, e.g., with humic substances that have reacted either photochemically or microbially, or with the marine labile organic matter (priming effect) of marine gels (Zielińska et al., 2014). Such experiments will help further our understanding of a host of processes involving iron-humic interactions with algal and bacterial communities. Outstanding questions that need to be addressed are: (1) How available is humic-bound iron to phytoplankton in systems where algae-bacteria interactions are taken into account, and is this bioavailability conditioned by prior photochemical processing of the humic substances? (2) How does microbial and photochemical processing affect the capacity of humics to bind ‘new iron,’ e.g., iron supplied by atmospheric dust deposition?
What Is the Current Status of Measurement Technologies for Iron-Humic Studies in Marine Waters?
Analytical Approaches to Differentiate Terrigenous and Marine DOM
Although the chemically based distinction between humic and fulvic acids is operational, the humic acids are best understood as the colloidal or HMW component while the fulvic acids are the soluble or LMW component (Bleam, 2017). Thus, it is the presence of the humic acid component that endows humic substances with both colloidal and purely chemical properties, both of which being relevant in the context of the iron biogeochemical cycle. Conventionally, XAD-8 resin has been used to isolate humic substances (Thurman and Malcolm, 1981) for detailed characterization in freshwaters (Fimmen et al., 2007) and seawater (Zhang et al., 2017), although it has now been largely superseded by SuperliteTM DAX-8. After passage through the non-ionic, macroporous resin (XAD-8 or DAX-8), humic substances are back eluted, then fractionated on the basis of solubility between humic and fulvic acids. The last step, which subjects the humic substances to extremes of pH, has led a number of authors to argue that these conventional methods of isolation introduce artifacts (De Haan, 1992; Peuravuori and Pihlaja, 1997; Gadmar et al., 2005). These concerns have led to alternative separation methods, as discussed in the next paragraph. The DAX-8 effluent, i.e., the fraction not retained, is sometimes passed through XAD-4 resin to collect a further DOM fraction of higher hydrophilic character. These analytical approaches have allowed Zhang et al. (2017) to proceed with the detailed characterization of the isolated fractions of DOM collected in the Yellow Sea. From FTIR, 13C NMR and Py-GC/MS analyses of the DOM pool they observed that the bulk of the humic substances had a marine signature, indicating that algal and/or microbially derived materials were the predominant precursors for the humics. It is perhaps not surprising that open sea humic material would have characteristics of the marine endmember in terms of stable carbon isotopes and average molecular formulae. Marine compounds are expected to contain molecular formulae of lower MW, fewer carbon atoms, lower O/C than DOM from terrestrial sources (Seidel et al., 2017). They also contain fewer aromatic and unsaturated compounds. Collectively, their δ13C values, which is negatively correlated to aromatic compounds, is higher (less negative) than any δ13C values measured at salinity 0 (Alling et al., 2008). However, it is important to realize that the very small percentage of terrestrial compounds susceptible to be present at the marine endmember would not be detected by the bulk measurement of optical properties, the δ13C isotopic composition or the relative abundance of compounds with different molecular weights and saturation indexes. In theory, lignin biopolymers could represent a powerful tool to detect the presence of terrigenous humic substances in the sea since they are considered to be the most refractory markers for terrigenous OM (Bianchi et al., 1997; Opsahl and Benner, 1998; Stubbins et al., 2010). The evaluation of lignin biomarkers is based on thermally assisted hydrolysis to break down the lignin structure to its constituent monomers and dimers that are then analyzed chromatographically (Hatcher et al., 1995). Non-detection of these monomers is frequently interpreted to mean that lignin has been lost or completely remineralized (Benner et al., 2005; Louchouarn et al., 2010; Hossler and Bauer, 2013). However, recent research has revealed that this is not necessarily the case. Instead, some HPLC fractionated components of lignin extracts have been found to be transformed by hydroxyl radical attack into compounds unrecognizable as having a terrestrial origin (Chen et al., 2014; Koch and Dittmar, 2016). Waggoner and Hatcher (2017) found that the suite of compounds generated under laboratory conditions by the action of hydroxyl radicals no longer carried the lignin biomarker signature. They also found that the lignin derived products included alicyclic and condensed aromatic compounds (Waggoner et al., 2015). They hypothesized that this process, which resulted in oxidation, ring opening and depolymerization (and hence lower average MW), would occur during transit of DOM through sunlit coastal waters. A recent confirmation of their hypothesis can be found in a study by Cao et al. (2018) who examined the changes in the structure of XAD-8 isolated DOM during transit along the Penobscot River, into Penobscot Bay and across the Gulf of Maine. Lignin residues were ‘lost’ through the sequence river-estuary-ocean, while carboxyl-rich alicyclic molecules (CRAMs), which can be clearly differentiated from lignin by NMR spectroscopy, remained major structural components across the whole transect. Whether CRAMs and alteration products of lignin play a significant role in the iron biogeochemical cycle once they have become integrated into (and greatly diluted by) marine DOM is a question that deserves further investigation.
There has been an increasing tendency to use separation methods other than the XAD-8 method over the past 30 years. Methods based on reverse osmosis have allowed unprecedented quantities of aquatic humic substances to be isolated, desalted by cation exchange, freeze dried and homogenized. They are the method of choice for producing reference materials from a range of water bodies and containing the full mix of fulvic and humic components. Methods based on cross-flow (or tangential flow) ultrafiltration (CFF) have allowed the colloidal matter present in different size classes to be concentrated for bulk chemical analysis (Buesseler et al., 1996). Provided careful checks and balances on the permeation/retention behavior of the system are carried out (Schlosser and Croot, 2008; Zhang and Santschi, 2009) and the filter is preconditioned with clean seawater (Fitzsimmons and Boyle, 2014), CFF systems can provide meaningful information on the soluble and colloidal fractions of DOM and iron in the ocean. It is also worth noting that some of this information may be lost when the CFF filter cartridges are used in a single pass-rather than a recirculation-mode (Guo and Santschi, 2007; Schlosser and Croot, 2008). The main disadvantages of CFF are (i) the very long processing times required to minimize artifacts such as the retention of LMW substances and (ii) the very long flushing and cleaning times required to reduce DOC blanks in particular. Its main advantages are (i) the isolation of a large quantity of colloidal matter and (ii) the option of conducting CFF in the diafiltration mode with a view of progressively transferring the colloidal matter into a synthetic solution where the stability, charge and conformation of the humic molecules can be manipulated. The continuous diafiltration mode was selected by Muller and Cuscov (2017) to minimize artifacts associated with potential self-aggregation and to enhance their negative electric charge, thus facilitating their adsorption to positively charged QuantomixTM membranes. This allowed SEM imaging and EDX analysis of individual humic colloids from the River Thurso and its coastal plume (Figure 3). Finally, size-exclusion chromatography in combination with organic carbon detection (SEC-OCD) allows the quantification of humic substances in terms of their organic carbon content. This method was extended by Hubert et al. (2011) to enable the differentiation and quantification of humic substances, biopolymers, LMW acids and neutrals, and hydrophobic compounds. It was recently used by Dulaquais et al. (2018) to map out the vertical distributions of humics in the western Mediterranean Sea.
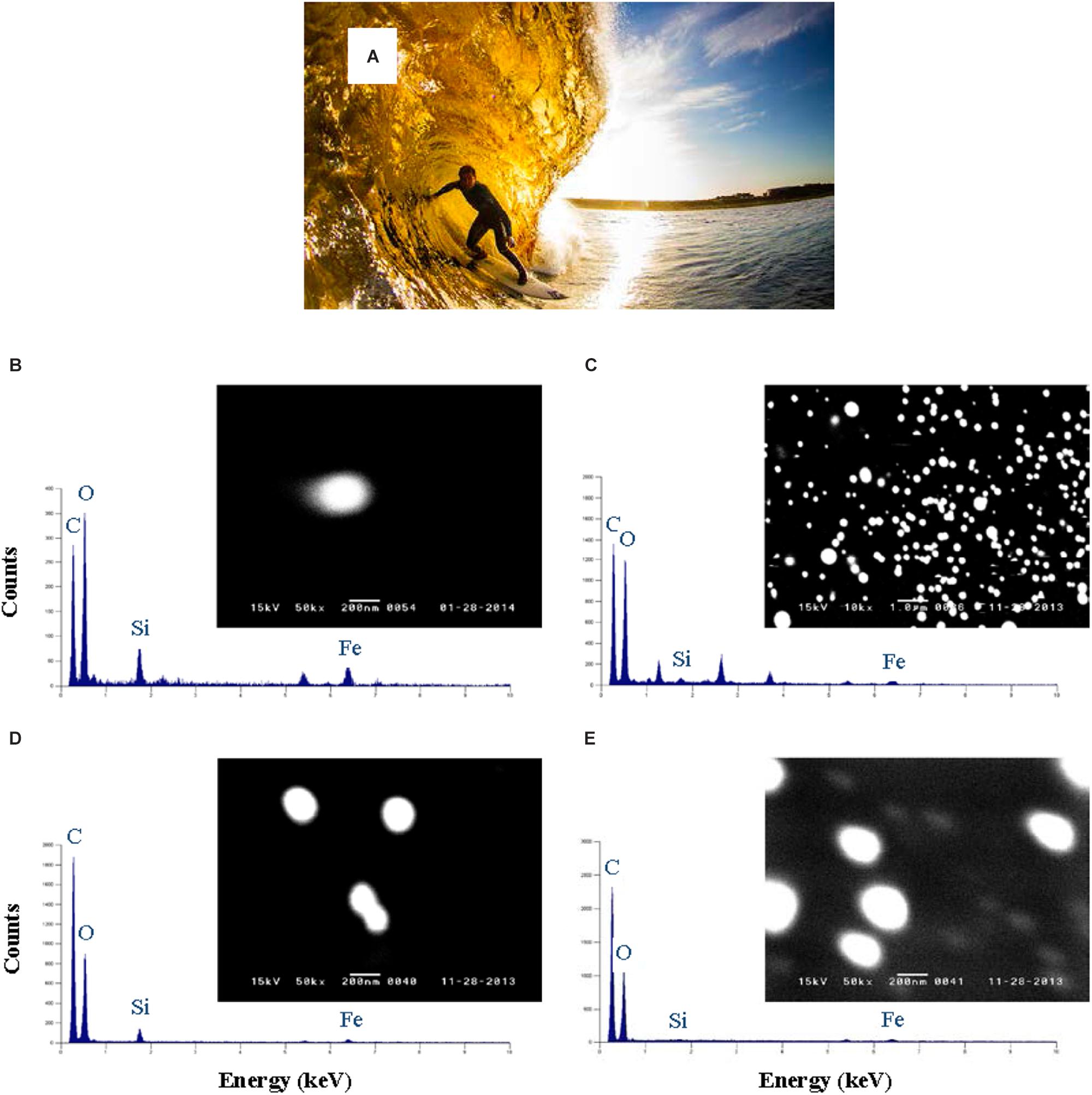
FIGURE 3. (A) Most of the dissolved humic material carried to the sea by River Thurso remains in solution upon mixing with seawater and imparts a yellowish color to coastal plume waters. Photo credit: Roger Sharp. (B–E) WETSEM images and EDXA analyses of colloidal humics collected at salinity 30 on 1 Feb (B) and 3 Nov (D), and at salinity 0 on 1 Feb (C) and 3 Nov (E) 2013; reproduced from Muller and Cuscov (2017) with copyright permission of the American Chemical Society.
Determination of Electroactive Humic and Humic-Like Substances
The direct analysis of humic substances is possible with electrochemical techniques, although the analysis cannot resolve mixed pools of terrestrial and marine humics. The method is based on the addition of Mo(VI) or Fe(III) to the sample, adsorption of the complex formed with humic substances onto a mercury drop electrode and subsequent reduction of the Mo(VI) or Fe(III) complex resulting in a quantifiable cathodic current. The Mo-humic method was originally developed by Quentel et al. (1986) and further tested by Quentel and Filella (2008) while the Fe-humic method was developed by Laglera and van den Berg (2009). The Fe-humic method might be expected to provide a more relevant measure of the iron complexation capacity of humic substances. Yet, Dulaquais et al. (2018) recently made a compelling demonstration of the equivalence of the two methods, using samples from contrasting marine systems. Like others (Quentel and Filella, 2008; Laglera and van den Berg, 2009; Abualhaija et al., 2015), they expressed their results in concentration equivalents of Suwannee River Fulvic Acid (SRFA, IHSS) as there is currently no commercial standard for marine humic-like substances. Developing a marine humics reference material for the analysis of elemental composition, iron binding capacity, etc., would help advance this field of research. It would offer the hope of identifying a property that distinguishes marine from terrestrial humic substances and can be used to quantify both endmembers in a mixture. The available body of evidence suggests that the protonation, redox and complexation properties of both endmembers overlap considerably (Fimmen et al., 2007; Aeschbacher et al., 2011; Gledhill and Buck, 2012), especially after terrestrial humics have suffered photochemical and microbial alterations. One property that might offer some hope of differentiating marine algal/bacterial and terrestrially derived humics within a marine-dominated mixed pool could be their kinetics of adsorption, e.g., onto the mercury drop electrode used in voltammetric work. The differing dependence of adsorption parameters on pH or ionic strength for marine and terrestrial materials might provide a means of quantifying both fractions (Cuscov and Muller, 2015).
Determination of Chromophoric Humic and Humic-Like Substances
Fluorescence techniques have been used a lot more than electrochemical techniques for the characterization of DOM in aquatic environments. They are highly sensitive and versatile techniques which (ideally) are not affected by any blank or background signal. Since humic substances are intrinsically fluorescent, they can be determined directly without using added indicator molecules. As such, humic-like fluorescence has been measured extensively in a wide range of freshwaters and marine waters in the past 20 years (Coble, 1996, 2007; Trubetskoj et al., 2018). Oceanic measurements have mostly relied on the interpretation of excitation-emission matrices of DOM and advanced multivariate analysis including Parallel Factor Analysis (Stedmon and Markarger, 2005; Yamashita et al., 2015; Painter et al., 2018). A number of oceanic and coastal studies have found correlations between dissolved iron concentration and humic-like fluorescence, e.g., in the Arctic Ocean (Slagter et al., 2017), the Atlantic Ocean (Gerringa et al., 2015) and in the Yura River-Tango Bay system, Japan (Watanabe et al., 2018). However, such correlations were not found in all oceanic domains and, in any case, showed a great deal of regional variability (Hassler et al., 2017). Correlations also occur throughout the top 100 m of the global ocean between chromophoric humics and chlorophyll a, suggesting a biologically mediated distribution in surface and epipelagic waters. In deeper waters, the fluorescent signal used as a proxy for humic substances is relatively constant (Yamashita et al., 2010; Heller et al., 2013). By contrast, the electrochemically active component of the humic substances appears to decrease slowly with depth in mesopelagic waters (200-1000 m), with a concomitant increase in apparent oxygen utilisation (AOU), which points to bacterial degradation of this component (Dulaquais et al., 2018). If we consider that the fluorescence signal is essentially a bulk measurement whereas the voltammetric signal is generated by a tiny subset of chelating sites, the two sets of observations may not be mutually incompatible. Also, the fluorescence quenching phenomenon (Yamashita and Jaffé, 2008) could partly explain the difference observed between the vertical distributions of fluorescent and electrochemically active humic substances. The loss of iron from humic chelating sites, e.g., due to bacterial uptake, would tend to increase the humic-like fluorescence and thus mask the process of bacterial degradation. This could then explain why the latter is not reflected in the shape of the fluorescence vertical profiles.
Like electrochemical measurements, direct fluorescence measurements cannot discriminate between marine humics and aged terrestrial humics. Marine humic-like fluorescence is blue shifted compared to terrigenous humics (Coble, 1996, 2007), but a blue shift is also known to occur due to a reduction in the extent of the π electron system, such as a decrease in the number of aromatic rings caused by the breakdown of terrestrial humic substances (Waggoner and Hatcher, 2017; Cao et al., 2018). Therefore, a marine humic-like signature does not rule out the possibility of a small, terrestrial humic component being present, integrated into the DOM pool and playing a role in iron biogeochemistry. In fact, the molecular composition of the DOM in the Baltic Sea outflow to the North Sea (and beyond) clearly reveals the presence of an aged and refractory terrigenous endmember which mixes conservatively with the NE Atlantic Ocean endmember (Seidel et al., 2017) over the 32.0-35.4 salinity range. This terrigenous endmember comprises the refractory products of humic substances that have undergone extensive photochemical and microbial transformations over the 30-year residence time of freshwater in the Baltic Sea. As previously mentioned, these transformations lower the mean molecular weight of DOM and probably make it less reactive (Massicotte et al., 2017). Therefore, stable terrestrial humic products might be expected to appear at least initially associated with LMW DOM as they are exported to the ocean.
What Are the Consequences of Marine and Terrestrial Organic Matter Interacting With Iron Oxides and With Each Other?
It is now recognized that a small but highly stable fraction of terrigenous DOM is exported to the ocean and that humic substances are largely responsible for this transfer (Muller and Batchelli, 2013; Tesi et al., 2014; Krachler et al., 2015; Seidel et al., 2017). From the discussion so far, we have good reasons to expect that these humic substances contain a large portion of the highly specific, strong Fe binding ligands which occur in coastal and shelf waters where they modulate [Fe’] and thus influence the bioavailability of iron. We have also noted that large quantities of terrestrial humics are exported to the Arctic Ocean (Figures 1, 2) by the outflow of large boreal rivers and numerous small blackwater streams. Formation of the North Atlantic Deep Water via winter cooling and evaporation entrains some of this material, and its associated iron, into the deep ocean circulation (Medeiros et al., 2016). While the lifespan of individual compounds in the deep ocean may range from months to millennia, their 14C age reaches thousands of years (Jiao et al., 2010) and the most recalcitrant pool was found to have a 14C age greater than 24,000 years (Lechtenfeld et al., 2014). The stability of terrestrial humic degradation products, as evidenced by recent studies, makes them potential candidates for becoming incorporated in this pool of recalcitrant material trapped in the deep ocean. If remains of terrestrial humics are indeed trapped in the deep-sea, the next question that needs to be addressed is whether this is due to their low susceptibility to bacterial breakdown or to some external stabilization mechanism, perhaps bonding with reactive metal oxide nanoparticles. To date, the latter mechanism has only been documented in sediments (Burdige, 2007; Lalonde et al., 2012; Johnson et al., 2015), not in the water column.
It is now well documented that both refractory and reactive organic matter can be preserved in marine sediments through interactions with, or protection by, reactive iron or manganese phases (Lalonde et al., 2012; Johnson et al., 2015; Barber et al., 2017). In modern sediments this has been described as a ‘rusty carbon sink’ (Raiswell and Canfield, 2012) whereby organic matter is intimately mixed with iron oxide phases. This can lead to the mutual stabilization and preservation of organic carbon and iron, which has significant implications for both geochemical cycles (Lalonde et al., 2012; Schröder et al., 2016). Detailed visualization of the electronic interactions between iron and carbon has recently been made possible through a combination of synchrotron X-ray techniques (XANES and NEXAFS) and scanning transmission X-ray microscopy (STXM) applied to sediments of contrasting depositional regimes (Barber et al., 2017). It revealed that only a small fraction of the reactive iron phase was directly associated with organic carbon. This is consistent with the formation of Fe-OC chelates proposed by Lalonde et al. (2012). These chelates occur within predominantly organic structures that are thought to resemble those depicted by the “onion” model of Mackey and Zirino (1994). According to the “onion” hypothesis, organic compounds of low and high molecular weight are held together by metal ions and gradually coalesce, entrapping the metal in an organic matrix. There is evidence that such chelates may occur in marine colloidal matter, e.g., in poorly flushed sections of the water column and near sediment pore water inputs of Fe or Mn (Muller, 1999). Unlike organic colloids, however, the chelates described by Lalonde et al. (2012) have an average Fe/OC ratio which greatly exceeds the ratios reported for humic complexation with iron (Laglera and van den Berg, 2009; Batchelli et al., 2010; Krachler et al., 2010; Kritzberg et al., 2014; Muller and Cuscov, 2017). This suggests that organic matter preserved in sediments must be associated with nanoparticulate iron oxides (Barber et al., 2017), probably formed during diagenetic recycling of Fe(III)/Fe(II). Little is known about the nature and origin of this organic matter, although we do know some of the factors that promote its bonding with reactive iron oxides. One of the most important factors is the relative abundance of carbonyl and hydroxyl-containing aromatic acids which bind iron (oxyhydr)oxides via a ligand exchange mechanism (Gu et al., 1995). The mixed σ and π bond characteristics of these organic acids allow for the formation of strong OC-Fe bonds (Barber et al., 2017) similar to those occurring in siderophores (Hocking et al., 2010; Orlowska et al., 2017b) and in iron encapsulated carbon nanotubes (Chen et al., 2015). Interestingly, we have seen that these very same organic acids are also responsible for binding iron in terrigenous humic substances (Fujii et al., 2014; Orlowska et al., 2017b). Therefore, the preservation of humic substances in coastal and shelf sediments receiving large inputs of iron and humics would seem worth investigating. The origin of the stabilized organic matter (algal/bacterial decomposition products, biopolymers or humics) could be explored by investigating how different types of organic matter are taken up by freshly precipitated nanophases of iron (oxyhydr)oxides.
One issue that has received little attention, despite its relevance to the functioning of the biological pump, is the direct interactions between terrestrial humic substances and marine gels. Two types of interactions have recently been documented. These interactions lower and increase the mean MW of marine DOM, respectively, so this is bound to complicate efforts to establish their net effect on the biological pump and the global carbon and iron cycles. The first type of interaction, as documented by Shiu et al. (2018), occurs when river-borne humic substances are transported to coastal waters. Through electrostatic interactions and the binding of Ca2+ ions holding the gel together, the terrigenous humics can destabilize marine microgels and impede the formation of new gel. The net effect is to shift the coupled equilibria monomer ↔ polymer ↔ gel toward lower MW. This might reduce the loss of particulate gels from surface waters via sinking, and thus act to maintain essential nutrients (and iron) in surface waters. The second type of interactions is the phase partitioning of terrestrial humic substances between aqueous solution and gel (Zielińska et al., 2014; Shiu et al., 2018). Zielińska et al. (2014) found that the first step in phase partitioning is the formation of a surface accumulation film, which facilitates the associative tendencies of the humic compounds, resulting in the formation of larger aggregates. This type of interaction might be anticipated to hasten the removal of humics and gel from surface waters, thus negatively impacting the marine phytoplankton community (Wells, 2002). On the other hand, the absorption of humics by marine gels could create hot spots for microbial activity and thus contribute to the degradation of humics with ageng of deep waters recorded by Dulaquais et al. (2018) in the western Mediterranean Sea.
Figure 4 is a schematic diagram representing the marine cycle of humic substances and its connections to iron biogeochemistry. The abovementioned marine gel is represented in the top center of the diagram by a tangled network of fibrils stabilized by cations such as Fe3+ (red dots) and Ca2+ or Mg2+ (purple dots).
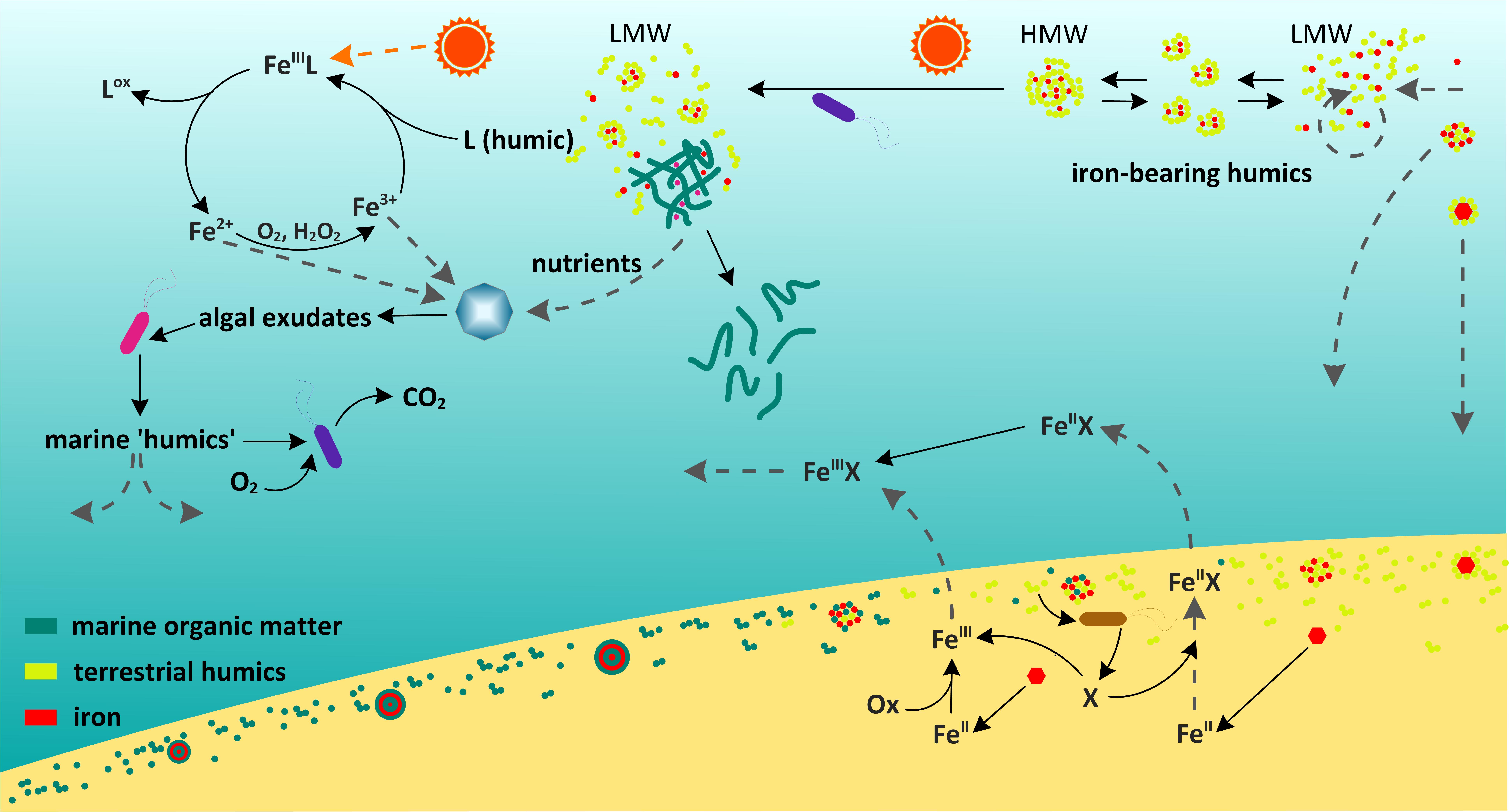
FIGURE 4. Schematic diagram representing the involvement of humics in the stabilization of Fe in solution, the enhancement of Fe uptake by phytoplankton via humic mediated reactions in surface waters (section “How Bioavailable Is Humic-Bound Iron in the Marine Environment?”), the synthesis and preservation of humics in sediments (section “What Are the Consequences of Marine and Terrestrial Organic Matter Interacting With Iron Oxides and With Each Other?”) and the sediment flux of Fe-humic complexes back into the water column.
Conclusion
The importance of the role played by iron in the marine biogeochemistry of carbon is undisputed. Recently, there has been a shift in our understanding of the major controls on the iron inventory of the ocean (Tagliabue et al., 2017). The notion that fluvial iron inputs can be neglected is being challenged by the realization that some dissolved and colloidal constituents of aquatic humic substances are not subject to coagulation across the estuarine salinity gradient. As a result, these humic constituents and their associated iron load can be transported to the coastal ocean. Here we have discussed some of the coordination, steric and electrostatic properties of the terrestrial humic substances that are likely to make them excellent iron carriers and thus enhance iron export to the coastal ocean. Catchment science tells us how seasonal and hydrological factors influence the quality and quantity of humic substances exported to the coast (Lumsdon et al., 2005; Billett et al., 2006; Koehler et al., 2009; Muller and Tankéré-Muller, 2012; Muller and Cuscov, 2017). Photochemical and microbial degradation experiments show us how aromatic and phenolic groups are degraded and what new products are formed (Boyd and Carlucci, 1996; Hernes and Benner, 2003; Helms et al., 2008, 2013; Chen et al., 2014; Sharpless et al., 2014; Waggoner and Hatcher, 2017). Yet the effects of these alterations on iron speciation are still largely unknown. In short, although the main drivers of the fluvial inputs, transport and transformations of terrestrial humics have been identified, their net effects on the speciation and the immediate fate of iron still elude us. One of the major obstacles to revealing these effects, and more generally the interconnections and relationships between terrestrial humics and iron, is the lack of analytical methods to selectively quantify terrestrial humic substances at a marine site. Once this has been overcome, it will be possible to address in a direct way the unresolved issue concerning the fate of terrestrial humic matter and its associated metals, including iron. It may then be possible to quantify how much of the total humic and iron riverine input is exported to the open sea and how much is removed by aggregation during passage through estuaries or by biological uptake.
Laboratory and mesocosm studies will be key to providing the detailed knowledge needed to understand these processes. For example, we have seen how humic stability and transport properties are governed by their colloidal dimension and the nature of their association with iron within the colloidal matrix. At the moment, biogeochemical models either ignore colloids (Tagliabue et al., 2016) or assume that aggregated ligands are immediately lost from the water column (Völker and Tagliabue, 2015). Biological processing of terrestrial humics will also need to be better represented. Recent evidence indicates that the first step may be a stimulation of bacterial metabolism in coastal waters (Vähätalo et al., 2011; Le Fouest et al., 2018), resulting in the transfer of iron into the biogenic phase and disrupting the coupling between terrestrial humics and iron. As such, biological processes may complicate efforts to model the fate of humic substances beyond coastal influences. Nevertheless, understanding the above processes will be critical for generating accurate models of oceanic iron cycling. First, such models will enable us, for the first time, to integrate terrestrial humic inputs into coupled biogeochemical models of iron and carbon. Secondly, model improvements will enable us to test the impacts of climate-driven changes occurring on land on marine systems (Goberville et al., 2010).
Author Contributions
FM fully contributed to the ideas, writing and preparation of this review.
Funding
Support from the Royal Society of Edinburgh, which I received through their Scotland-Taiwan International Exchange Program, helped me develop some of the ideas presented here. Funding from the Marine Alliance for Science and Technology Scotland (MASTS Small Grant SG88) is also gratefully acknowledged.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
FM particularly grateful to Andrea Lazenby for inviting me to produce a critical review of this subject, as well as to Halldór Jóhannsson for kindly providing the map reproduced in Figure 1.
References
Abualhaija, M. M., Whitby, H., and van den Berg, C. M. G. (2015). Competition between copper and iron for humic ligands in estuarine waters. Mar. Chem. 172, 46–56. doi: 10.1016/j.marchem.2015.03.010
Aeschbacher, M., Vergari, D., Schwarzenbach, R. P., and Sander, M. (2011). Electrochemical analysis of proton and electron transfer equilibria of the reducible moieties in humic acids. Environ. Sci. Technol. 45, 8385–8394. doi: 10.1021/es201981g
Aiken, G. R. (1985). “Isolation and concentration techniques for aquatic humic substances,” in Humic Substances in Soil, Sediment and Water, eds G. R. Aiken, D. M. McKnight, R. L. Wershaw, and P. MacCarthy (New York, NY: Wiley), 363–385.
Aiken, G. R., Hsu-Kim, H., and Ryan, J. N. (2011). Influence of dissolved organic matter on the environmental fate of metals, nanoparticles, and colloids. Environ. Sci. Technol. 45, 3196–3201. doi: 10.1021/es103992s
Aiken, G. R., and Malcolm, R. L. (1987). Molecular weight of aquatic fulvic acids by vapor pressure osmometry. Geochim. Cosmochim. Acta 51, 2177–2184. doi: 10.1016/0016-7037(87)90267-5
Alling, V., Humborg, C., Mörth, C.-M., Rahm, L., and Pollehne, F. (2008). Tracing terrestrial organic matter by (14C and (13C signatures in a subarctic estuary. Limnol. Oceanogr. 53, 2594–2602. doi: 10.4319/lo.2008.53.6.2594
Asmala, E., Autio, R., Kaartokallio, H., Stedmon, C. A., and Thomas, D. N. (2014a). Processing of humic-rich riverine dissolved organic matter by estuarine bacteria: effects of predegradation and inorganic nutrients. Aquat. Sci. 76, 451–463. doi: 10.1007/s00027-014-0346-7
Asmala, E., Bowers, D. G., Autio, R., Kaartokallio, H., and Thomas, D. N. (2014b). Qualitative changes of riverine dissolved organic matter at low salinities due to flocculation. J. Geophys. Res. Biogeosci. 119, 1919–1933. doi: 10.1002/2014JG002722
Barbeau, K. A., Kujawinski, E. B., and Moffett, J. W. (2001). Remineralization and recycling of iron, thorium and organic carbon by heterotrophic marine protists in culture. Aquat. Microb. Ecol. 24, 69–81. doi: 10.3354/ame024069
Barber, A., Brandes, J., Leri, A., Lalonde, K., Balind, K., Wirick, S., et al. (2017). Preservation of organic matter in marine sediments by inner-sphere interactions with reactive iron. Sci. Rep. 7:366. doi: 10.1038/nrmicro2386
Batchelli, S., Muller, F. L. L., Baalousha, M., and Lead, J. R. (2009). Size fractionation and optical properties of colloids in an organic-rich estuary (Thurso, UK). Mar. Chem. 113, 227–237. doi: 10.1016/j.marchem.2009.02.006
Batchelli, S., Muller, F. L. L., Chang, K.-C., and Lee, C.-L. (2010). Evidence for strong but dynamic iron(humic colloidal associations in humic-rich coastal waters. Environ. Sci. Technol. 44, 8485–8490. doi: 10.1021/es101081c
Belzile, C., and Guo, L. (2006). Optical properties of low molecular weight and colloidal organic matter: application of the ultrafiltration permeation model to DOM absorption and fluorescence. Mar. Chem. 98, 183–196. doi: 10.1016/j.marchem.2005.08.009
Benner, R., Louchouarn, P., and Amon, R. M. W. (2005). Terrigenous dissolved organic matter in the Arctic Ocean and its transport to surface and deep waters of the North Atlantic. Glob. Biogeochem. Cycles 19:GB2025. doi: 10.1029/2004GB002398
Bianchi, T. S., Rolff, C., and Lambert, C. D. (1997). Sources and composition of particulate organic carbon in the Baltic Sea: the use of plant pigments and lignin-phenols as biomarkers. Mar. Ecol. Prog. Ser. 156, 25–31. doi: 10.3354/mepsl56025
Billett, M. F., Deacon, C. M., Palmer, S. M., Dawson, J. J. C., and Hope, D. (2006). Connecting organic carbon in stream water and soils in a peatland catchment. J. Geophys. Res. 111:G02010. doi: 10.1029/2005JG000065
Blazevic, A., Orlowska, E., Kandioller, W., Jirsa, F., Keppler, B. K., Tafili-Kryeziu, M., et al. (2016). Photoreduction of terrigenous Fe-humic substances leads to bioavailable iron in oceans. Angew. Chem. Int. Ed. 55, 6417–6422. doi: 10.1002/anie.201600852
Bleam, W. F. (ed.) (2017). “Natural organic matter,” in Soil and Environmental Chemistry, Vol. 7, (Cambridge, MA: Academic Press), 333–384. doi: 10.1016/B978-0-12-804178-9-00007-0
Boehme, J., and Wells, M. L. (2006). Fluorescence variability of marine and terrestrial colloids: examining size fractions of chromophoric dissolved organic matter in the Damariscotta River estuary. Mar. Chem. 101, 95–103. doi: 10.1016/j.marchem.2006.02.001
Bonnain, C., Breitbart, M., and Buck, K. N. (2016). The ferrojan horse hypothesis: iron-virus interactions in the ocean. Front. Mar. Sci. 3:82. doi: 10.3389/fmars.2016.00082
Boyd, T. J., and Carlucci, A. F. (1996). Rapid microbial degradation of phenolic materials in California (USA) coastal environments. Aquat. Microb. Ecol. 11, 171–179. doi: 10.3354/ame011171
Boye, M., Nishioka, J., Croot, P., Laan, P., Timmermans, K. R., Strass, V. H., et al. (2010). Significant portion of dissolved organic Fe complexes in fact is Fe colloids. Mar. Chem. 122, 20–27. doi: 10.1016/j.marchem.2010.09.001
Breitbarth, E., Achterberg, E. P., Ardelan, M. V., Baker, A. R., Bucciarelli, E., Chever, F., et al. (2010). Iron biogeochemistry across marine systems progress from the past decade. Biogeosciences 7, 1075–1097. doi: 10.5194/bg-7-1075-2010
Buesseler, K. O., Bauer, J. E., Chen, R. F., Eglinton, T. I., Gustafsson, O., Landing, W., et al. (1996). An intercomparison of cross-flow filtration techniques used for sampling marine colloids: overview and organic carbon results. Mar. Chem. 55, 1–31. doi: 10.1016/S0304-4203(96)00046-1
Buffle, J., and Altmann, R. S. (1987). “Interpretation of metal complexation by heterogeneous complexants,” in Aquatic Surface Chemistry, ed. W. Stumm (New York, NY: John Wiley & Sons), 351–383. doi: 10.1017/S0025315400050335
Buffle, J., Wilkinson, K., Stoll, S., Filella, M., and Zhang, J. (1998). A generalized description of aquatic colloidal interactions: the three-colloidal component approach. Environ. Sci. Technol. 32, 2887–2899. doi: 10.1021/es980217h
Bundy, R. M., Abdulla, H. A., Hatcher, P. G., Biller, D. V., Buck, K. N., and Barbeau, K. A. (2015). Iron-binding ligands and humic substances in the San Francisco Bay estuary and estuarine-influenced shelf regions of coastal California. Mar. Chem. 173, 183–194. doi: 10.1016/j.marchem.2014.11.005
Burdige, D. J. (2007). Preservation of organic matter in marine sediments: controls, mechanisms, and an imbalance in sediment organic carbon budgets? Chem. Rev. 107, 467–485. doi: 10.1021/cr050347q
Cao, X., Aiken, G. R., Butler, K. D., Huntington, T. G., Balch, W. M., Mao, J., et al. (2018). Evidence for major input of riverine organic matter into the ocean. Org. Geochem. 116, 62–76. doi: 10.1016/j.orggeochem.2017.11.001
Chen, H., Abdulla, H. A. N., Sanders, R. L., Myneni, S. C. B., Mopper, K., and Hatcher, P. G. (2014). Production of black carbon-like and aliphatic molecules from terrestrial dissolved organic matter in the presence of sunlight and iron. Environ. Sci. Technol. Lett. 1, 399–404. doi: 10.1021/ez5002598
Chen, M., and Wang, W.-X. (2008). Accelerated uptake by phytoplankton of iron bound to humic acids. Aquat. Biol. 3, 155–166. doi: 10.3354/ab00064
Chen, X., Xiao, J., Wang, J., Deng, D., Hu, Y., Zhou, J., et al. (2015). Visualising electronic interactions between iron and carbon by X-ray chemical imaging and spectroscopy. Chem. Sci. 6, 3262–3267. doi: 10.1039/c5sc00353a
Chin, W.-C., Orellana, M. V., and Verdugo, P. (1998). Spontaneous assembly of marine dissolved organic matter into polymer gels. Nature 391, 568–572. doi: 10.1038/35345
Coble, P. G. (1996). Characterization of marine and terrestrial DOM in seawater using excitation emission matrix spectroscopy. Mar. Chem. 51, 325–346. doi: 10.1016/0304-4203(95)00062-3
Coble, P. G. (2007). Marine optical biogeochemistry: the chemistry of ocean color. Chem. Rev. 107, 402–418. doi: 10.1021/cr050350
Colatriano, D., Tran, P. Q., Guéguen, C., Williams, W. J., Lovejoy, C., and Walsh, D. A. (2018). Genomic evidence for the degradation of terrestrial organic matter by pelagic Arctic Ocean Chloroflexi bacteria. Commun. Biol. 1:90. doi: 10.1038/s42003-018-0086-7
Conway, T. M., and John, S. G. (2014). Quantification of dissolved iron sources to the North Atlantic Ocean. Nature 511, 212–215. doi: 10.1038/nature13482
Ćosović, B., Vojvodić, V., Bošcović, N., Plavšić, M., and Lee, C. (2010). Characterization of natural and synthetic humic substances (melanoidins) by chemical composition and adsorption measurements. Org. Geochem. 41, 200–205. doi: 10.1016/j.orggeochem.2009.10.002
Crocket, K., Abell, R., and Brand, T. (2015). Iron BREW: Iron Beyond River Etive Water. Small Grant Scheme Report CPDSG3. Scotland: Marine Alliance for Science and Technology,
Croot, P. L., Bowie, A. R., Frew, R. D., Maldonado, M. T., Hall, J. A., Safi, K. A., et al. (2001). Retention of dissolved iron and Fe(II) in an iron induced Southern Ocean phytoplankton bloom. Geophys. Res. Lett. 28, 3425–3428. doi: 10.1021/2001GL013023
Cuscov, M., and Muller, F. L. L. (2015). Differentiating humic and algal surface-active substances in coastal waters by their pH-dependent adsorption behaviour. Mar. Chem. 174, 35–45. doi: 10.1016/j.marchem.2015.05.002
Cuss, C. W., and Guéguen, C. (2015). Relationships between molecular weight and fluorescence properties for size-fractionated dissolved organic matter from fresh and aged sources. Water Res. 68, 487–497. doi: 10.1016/j.watres.2014.10.013
Dai, M.-H., and Martin, J.-M. (1995). First data on trace metal level and behaviour in two major Arctic river-estuarine systems (Ob and Yenisey) and in the adjacent Kara Sea, Russia. Earth Planet. Sci. Lett. 131, 127–141. doi: 10.1016/0012-821X(95)00021-4
De Haan, H. (1992). Impacts of environmental changes on the biogeochemistry of aquatic humic substances. Hydrobiologia 229, 59–71. doi: 10.1007/s11356-009-0176-7
Du, Y., Ramirez, C. E., and Jaffé, R. (2018). Fractionation of dissolved organic matter by co-precipitation with iron: effects of composition. Environ. Process. 5, 5–21. doi: 10.1007/s40710-017-0281-4
Dulaquais, G., Waeles, M., Gerringa, L. J. A., Middag, R., Rijkenberg, M., and Riso, R. (2018). The biogeochemistry of electroactive humic substances and its connection to iron chemistry in the North East Atlantic and the Western Mediterranean Sea. J. Geophys. Res. 123, 5481–5499. doi: 10.1029/2018JC014211
Elliott, S., Burrows, S. M., Deal, C., Liu, X., Long, M., Ogunro, O., et al. (2014). Prospects for simulating macromolecular surfactant chemistry at the ocean-atmosphere boundary. Environ. Res. Lett. 9:064012. doi: 10.1088/1748-9326/9/6/064012
Falkowski, P. G. (1997). Evolution of the nitrogen cycle and its influence on the biological sequestration of CO2 in the ocean. Nature 387, 272–275. doi: 10.1038/387272a0
Falkowski, P. G. (2012). Ocean Science: the power of plankton. Nature 483, S17–S20. doi: 10.1038/483S17a
Fichot, C. G., and Benner, R. (2012). The spectral slope coefficient of chromophoric dissolved organic matter S275-295) as a tracer of terrigenous dissolved organic carbon in river-influenced ocean margins. Limnol. Oceanogr. 57, 1453–1466. doi: 10.4319/lo.2012.57.5.1453
Fimmen, R. L., Cory, R. M., Chin, Y.-P., Trouts, T. D., and McKnight, D. M. (2007). Probing the oxidation-reduction properties of terrestrially and microbially derived dissolved organic matter. Geochim. Cosmochim. Acta 71, 3003–3015. doi: 10.1016/j.gca.2007.04.009
Fitzsimmons, J. N., and Boyle, E. A. (2014). Assessment and comparison of Anopore and cross flow filtration methods for the determination of dissolved iron size fractionation into soluble and colloidal phases in seawater. Limnol. Oceanogr. Methods 12, 244–261. doi: 10.4319/lom.2014.12.244
Fitzsimmons, J. N., Bundy, R., Al-Subiai, S. N., Barbeau, K. A., and Boyle, E. A. (2015). The composition of dissolved iron in the dusty surface ocean: an exploration using size-fractionated iron-binding ligands. Mar. Chem. 173, 125–135. doi: 10.1016/j.marchem.2014.09.002
Fritzsche, A., Schröder, C., Wiekzorek, A., Händel, M., Ritschel, T., and Totsche, K. U. (2015). Structure and composition of Fe-OM coprecipitates that form in soil-derived solutions. Geochim. Cosmochim. Acta 169, 167–183. doi: 10.1016/j.gca.2015.07.041
Fujii, M., Imaoka, A., Yoshimura, C., and Waite, T. D. (2014). Effects of molecular composition of natural organic matter on ferric iron complexation at circumneutral pH. Environ. Sci. Technol. 48, 4414–4424. doi: 10.1021/es405496b
Gadmar, T. C., Vogt, R. D., and Evje, L. (2005). Artefacts in XAD-8 NOM fractionation. Int. J. Environ. Anal. Chem. 85, 365–376. doi: 10.1080/03067310500053910
Gerringa, L. J. A., Rijkenberg, M. J. A., Schoemann, V., Laan, P., and de Baar, H. J. W. (2015). Organic complexation of iron in the West Atlantic Ocean. Mar. Chem. 177, 434–446. doi: 10.1016/j.marchem.2015.04.007
Gerringa, L. J. A., Slagter, H. A., Bown, J., van Haren, H., Laan, P., de Baar, H. J. W., et al. (2017). Dissolved fe and fe-binding organic ligands in the mediterranean sea – GEOTRACES G04. Mar. Chem. 194, 100–113. doi: 10.1016/j.marchem.2017.05.012
Gilliam, A. H., and Riley, J. P. (1981). Correction of osmometric number-average molecular weights of humic substances for dissociation. Chem. Geol. 33, 355–366. doi: 10.1016/0009-2541(81)90108-X
Gledhill, M., and Buck, K. N. (2012). The organic complexation of iron in the marine environment: a review. Front. Microbiol. 3:69. doi: 10.3389/fmicb.2012.00069
Goberville, E., Beaugrand, G., Sautour, B., Tréguer, P., and Somlit, T. (2010). Climate-driven changes in coastal marine systems of western Europe. Mar. Ecol. Prog. Ser. 408, 129–148. doi: 10.3354/meps08564
Green, S. A., and Blough, N. V. (1994). Optical absorption and fluorescence properties of chromophoric dissolved organic matter in natural waters. Limnol. Oceanogr. 39, 1903–1916. doi: 10.4319/lo.1994.39.8.1903
Gu, B., Schmitt, J., Chen, Z., Liang, L., and McCarthy, J. F. (1995). Adsorption and desorption of different organic matter fractions on iron oxide. Geochim. Cosmochim. Acta 59, 219–229. doi: 10.1016/0016-7037(94)00282-Q
Guo, L., and Santschi, P. H. (2007). “Ultrafiltration and its applications to sampling and characterisation of aquatic colloids,” in Environmental Colloids and Particles, eds K. J. Wilkinson and J. R. Lead (Hoboken, NJ: Wiley), 159–221. doi: 10.1002/9780470024539
Guo, L., Wen, L.-S., Tang, D., and Santschi, P. H. (2000). Re-examination of cross-flow ultrafiltration for sampling aquatic colloids: evidence from molecular probes. Mar. Chem. 69, 75–90. doi: 10.1016/S0304-4203(99)000097-3
Hansell, D. A. (2002). “DOC in the global ocean carbon cycle,” in Biogeochemistry of Marine Dissolved Organic Matter, eds D. A. Hansell and C. A. Carlson (Orlando, FA: Academic Press), 685–715. doi: 10.1016/B978-012323841-2/50017-8
Hasegawa, H., Tate, Y., Ogino, M., Maki, T., Begum, Z. A., Ichijo, T., et al. (2017). Laboratory culture experiments to study the effect of lignite humic acid fractions on iron solubility and iron uptake rates in phytoplankton. J. Appl. Phycol. 2, 903–915. doi: 10.1007/s10811-016-0982-5
Hassler, C. S., van den Berg, C. M. G., and Boyd, P. W. (2017). Toward a regional classification to provide a more inclusive examination of the ocean biogeochemistry of iron-binding ligands. Front. Mar. Sci. 4:19. doi: 10.3389/fmars.2017.00019
Hatcher, P. G., Nanny, M. A., Minard, R. D., Dible, S. D., and Carson, D. M. (1995). Comparison of two thermochemolytic methods for the analysis of lignin in decomposing gymnosperm wood: the CuO oxidation method and the method of thermochemolysis with tetramethylammonium hydroxide (TMAH). Org. Geochem. 23, 881–888. doi: 10.1016/0146-6380(95)00087-9
Heller, M. I., Gaiero, S. M., and Croot, P. L. (2013). Basin scale survey of marine humic fluorescence in the Atlantic: relationship to iron solubility and H2O2. Glob. Biogeochem. Cycles 27, 88–100. doi: 10.1029/2012GB004427
Helms, J. R., Mao, J., Schmidt-Rohr, K., Abdulla, H., and Mopper, K. (2013). Photochemical flocculation of terrestrial dissolved organic matter and iron. Geochim. Cosmochim. Acta 121, 398–413. doi: 10.1016/j.gca.2013.07.025
Helms, J. R., Stubbins, A., Ritchie, J. D., Minor, E. C., Kieber, D. J., and Mopper, K. (2008). Absorption spectral slopes and slope ratios as indicators of molecular weight, source, and photobleaching of chromophoric dissolved organic matter. Limnol. Oceanogr. 53, 955–969. doi: 10.4319/lo.2008.53.3.0955
Hernes, P. J., and Benner, R. (2003). Photochemical and microbial degradation of dissolved lignin phenols: implications for the fate of terrigenous dissolved organic matter in marine environments. J. Geophys. Res. 108:3291. doi: 10.1029/2002JC001421
Herzog, S. D., Persson, P., and Kritzberg, E. S. (2017). Salinity effects on iron speciation in boreal river waters. Environ. Sci. Technol. 51, 9747–9755. doi: 10.1021/acs.est/7b02309
Hocking, R. K., George, S. D., Raymond, K. N., Hodgson, K. O., Hedman, B., and Solomon, E. L. (2010). FeL-edge X-ray absorption spectroscopy determination of different orbital covalency of siderophore model compounds: electronic structure contributions to high stability constants. J. Am. Chem. Soc. 132, 4006–4015. doi: 10.1021/ja9090098
Hossler, K., and Bauer, J. E. (2013). Amounts, isotopic character, and ages of organic and inorganic carbon exported from rivers to ocean margins. 1: estimates of terrestrial losses and inputs to the Middle Atlantic Bight. Glob. Biogeochem. Cycles 27, 331–346. doi: 10.1002/gbc.20033
Hubert, S. A., Balz, A., Abert, M., and Pronk, W. (2011). Characterisation of aquatic humic and non-humic matter with size exclusion chromatography organic carbon detection organic nitrogen detection (LC-OCD-OND). Water Res. 45, 879–885. doi: 10.1016/j.watres.2010.09.023
Hutchins, D. A., and Boyd, P. W. (2016). Marine phytoplankton and the changing ocean iron cycle. Nat. Clim. Change 6, 1071–1079. doi: 10.1038/nclimate/3147
Hutchins, D. A., DiTullio, G. R., Zhang, Y., and Bruland, K. W. (1998). An iron limitation mosaic in the California upwelling regime. Limnol. Oceanogr. 43, 1037–1054. doi: 10.4319/lo.1998.43.6.1037
Hutchins, D. A., Witter, A. E., Butler, A., and Luther, G. W. (1999). Competition among marine phytoplankton for different chelated iron species. Nature 400, 858–861. doi: 10.1038/23680
Jiao, N., Herndl, G. J., Hansell, D. A., Benner, R., Kattner, G., Wilhelm, S. W., et al. (2010). Microbial production of recalcitrant dissolved organic matter: long-term carbon storage in the global ocean. Nat. Rev. Microbiol. 8, 593–599. doi: 10.1038/nrmicro2386
Jickells, T. D., An, Z. S., Andersen, K. K., Baker, A. R., Bergametti, G., Brooks, N., et al. (2005). Global iron connections between desert dust, ocean biogeochemistry, and climate. Science 308, 67–71. doi: 10.1126/science.1105959
Johnson, K., Purvis, G., Lopez-Capel, E., Peacock, C., Gray, N., Wagner, T., et al. (2015). Towards a mechanistic understanding of carbon stabilization in manganese oxides. Nat. Commun. 6:7628. doi: 10.1038/ncomms8628
Jones, T. G., Evans, C. D., Jones, D. L., Hill, P. W., and Freeman, C. (2016). Transformation in DOC along a source to sea continuum; impact of photo-degradation, biological processes and mixing. Aquat. Sci. 78, 433–446. doi: 10.1007/s00027-015-0461-0
Karlsson, T., and Persson, P. (2010). Coordination chemistry and hydrolysis of Fe(III) in a peat humic acid studied by X-ray absorption spectroscopy. Geochim. Cosmochim. Acta 74, 30–40. doi: 10.1016/j.gca.2009.09.023
Kisand, V., Rocker, D., and Simon, M. (2008). Significant decomposition of riverine humic-rich DOC by marine but not estuarine bacteria assessed in sequential chemostat experiments. Aquat. Microb. Ecol. 53, 151–160. doi: 10.3354/ame01240
Klunder, M. B., Bauch, D., Laan, P., de Baar, H. J. W., van Heuven, S., and Ober, S. (2012). Dissolved iron in the arctic shelf seas and surface waters of the central arctic ocean: impact of arctic river water and ice-melt. J. Geophys. Res. 117:C01027. doi: 10.1029/2011JC007133
Koch, B. P., and Dittmar, T. (2016). From mass to structure: an aromaticity index for high-resolution mass data of natural organic matter. Rapid Commun. Mass Spectrom. 30:250. doi: 10.1002/rcm.7433
Koch, B. P., Witt, M., Engbrodt, R., Dittmar, T., and Kattner, G. (2005). Molecular formulae of marine and terrigenous dissolved organic matter detected by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Geochim. Cosmochim. Acta 69, 3299–3308. doi: 10.1016/j.gca.2005.02.027
Koehler, A. K., Murphy, K., Kiely, G., and Sottocornola, M. (2009). Seasonal variation of DOC concentration and annual loss of DOC from an Atlantic blanket bog in Southwestern Ireland. Biogeochemistry 95, 231–242. doi: 10.1007/s10533-009-933-9
Krachler, R., Krachler, R. F., von der Kammer, F., Süphandag, A., Jirsa, F., Ayromlou, S., et al. (2010). Relevance of peat-draining rivers for the riverine input of dissolved iron into the ocean. Sci. Total Environ. 408, 2402–2408. doi: 10.1016/j.scitotenv.2010.02.018
Krachler, R., Krachler, R. F., Wallner, G., Hahn, S., Laux, M., Cervantez Recalde, M. F., et al. (2015). River-derived humic substances as iron chelators in seawater. Mar. Chem. 174, 85–93. doi: 10.1016/j.marchem.2015.05.009
Krachler, R., Krachler, R. F., Wallner, G., Steier, P., El Abiead, Y., Wiesinger, H., et al. (2016). Sphagnum-dominated bog systems are highly effective yet variable sources of bio-available iron to marine waters. Sci. Total Environ. 556, 53–62. doi: 10.1016/j.scitotenv.2016.03.012
Krachler, R., von der Kammer, F., Jirsa, F., Süphandag, A., Krachler, R. F., Plessl, C., et al. (2012). Nanoscale lignin particles as sources of dissolved iron to the ocean. Glob. Biogeochem. Cycles 26:GB3024. doi: 10.1029/2012GB004294
Kritzberg, E. S., Bedmar Villanueva, A., Jung, M., and Reader, H. E. (2014). Importance of boreal rivers in providing iron to marine waters. PLoS One 9:e107500. doi: 10.1371/journal.pone.0107500
Kuhn, K. M., Maurice, P. A., Neubauer, E., Hofmann, T., and von der Kammer, F. (2014). Accessibility of humic-associated Fe to a microbial siderophore: implications for bioavailability. Environ. Sci. Technol. 48, 1015–1022. doi: 10.1021/es404186v
Kuma, K., Nishioka, J., and Matsugaga, K. (1996). Controls on Fe(III) hydroxide solubility in seawater: the influence of pH and natural organic chelators. Limnol. Oceanogr. 41, 396–407. doi: 10.4319/lo.1996.41.3.0396
Laglera, L. M., and van den Berg, C. M. G. (2009). Evidence for geochemical control of iron by humic substances in seawater. Limnol. Oceanogr. 54, 610–619. doi: 10.4319/lo.2009.54.2.0610
Lalonde, K., Mucci, A., Ouellet, A., and Gélinas, Y. (2012). Preservation of organic matter in sediments promoted by iron. Nature 483, 198–200. doi: 10.1038/nature10855
Lalonde, K., Vähätalo, A. V., and Gélinas, Y. (2014). Revisiting the disappearance of terrestrial dissolved organic matter in the ocean: a 13C study. Biogeosciences 11, 3707–3719. doi: 10.5194/bg-11-3707-2014
Le Fouest, V., Matsuoka, A., Manizza, M., Shernetsky, M., Tremblay, B., and Babin, M. (2018). Towards an assessment of riverine dissolved organic carbon in surface waters of the western Arctic Ocean based on remote sensing and biogeochemical modelling. Biogeosciences 15, 1335–1346. doi: 10.5194/bg-15-1335-2018
Lechtenfeld, O. J., Kattner, G., Flerus, R., McCallister, S. L., Schmitt-Koplin, P., and Koch, B. P. (2014). Molecular transformation and degradation of refractory dissolved organic matter in the Atlantic and Southern Ocean. Geochim. Cosmochim. Acta 126, 321–337. doi: 10.1016/j.gca.2013.11.009
Lis, H., Kranzler, C., Keren, N., and Shaked, Y. (2015). A comparative study of iron uptake rates and mechanisms amongst marine and freshwater cyanobacteria: prevalence of reductive iron uptake. Life 5, 841–860. doi: 10.3390/life5010841
Lis, H., Shaked, Y., Kranztler, C., Keren, N., and Morel, F. M. M. (2014). Iron bioavailability to phytoplankton: an empirical approach. ISME J. 9, 1003–1013. doi: 10.1038/ismej.2014.199
Louchouarn, P., Amon, R. M. W., Duan, S., Pondell, C., Seward, S. M., and White, N. (2010). Analysis of lignin-derived phenols in standard reference materials and ocean dissolved organic matter by gas chromatography/tandem mass spectrometry. Mar. Chem. 118, 85–97. doi: 10.1016/j.marchem.2009.11.003
Lumsdon, D. G., Stutter, M. I., Cooper, R. J., and Manson, J. R. (2005). Model assessment of biogeochemical controls on dissolved organic carbon partitioning in an acid organic soil. Environ. Sci. Technol. 39, 8057–8063. doi: 10.1021/es050266b
Luther, G. W. III (2016). Inorganic Chemistry for Geochemistry and Environmental Sciences: Fundamentals and Applications. New York, NY: Wiley.
Mackey, D. J., and Zirino, A. (1994). Comments on trace metal speciation in seawater or do “onions” grow in the sea? Anal. Chim. Acta 284, 635–647. doi: 10.1016/0003-2670(94)85068-2
Maldonado, M. T., Allen, A. E., Chong, J. S., Lin, K., Leus, D., Karpenko, N., et al. (2006). Copper-dependent iron transport in coastal and oceanic diatoms. Limnol. Oceanogr. 51, 1729–1743. doi: 10.4319/lo.2006.51.4.1729
Masini, J. C., Godinho, O. E. S., and Aleixo, L. M. (1998). Determination of ionizable groups of proteins by potentiometric titration in concentrated solutions of guanidine hydrochloride. Fresenius J. Anal. Chem. 360, 104–111. doi: 10.1007/s002160050651
Massicotte, P., Asmala, E., Stedmon, C., and Markager, S. (2017). Global distribution of dissolved organic matter along the aquatic continuum: across rivers, lakes and oceans. Sci. Total Environ. 609, 180–191. doi: 10.1016/j.scitotenv.2017.07.076
Medeiros, P. M., Seidel, M., and Niggemann, J. (2016). A novel molecular approach for tracing terrigenous dissolved organic matter into the deep ocean. Glob. Biogeochem. Cycles 30, 689–699. doi: 10.1002/2015GB005320
Moran, S. B., Yeats, P. A., and Balls, P. W. (1996). On the role of colloids in trace metal solid-solution partitioning in continental shelf waters: a comparison of model results and field data. Cont. Shelf Res. 16, 397–408. doi: 10.1016/0278-4343(95)98840-7
Morel, F. M. M., and Hering, J. G. (1993). Principles and Applications of Aquatic Chemistry. New York, NY: John Wiley & Sons Inc.
Muller, F. L. L. (1996). Measurement of electrokinetic and size characteristics of estuarine colloids by dynamic light scattering spectroscopy. Anal. Chim. Acta 331, 1–15. doi: 10.1016/0003-2670(96)00190-0
Muller, F. L. L. (1998). Colloid/solution partitioning of metal-selective organic ligands and its relevance to Cu, Pb and Cd cycling in the Firth of Clyde. Estuar. Coast. Shelf Sci. 46, 419–437. doi: 10.1006/ecss.1997.0267
Muller, F. L. L. (1999). Evaluation of the effects of natural dissolved and colloidal organic ligands on the electrochemical lability of Cd, Pb and Cd in the Arran Deep, Scotland. Mar. Chem. 67, 43–60. doi: 10.1016/S0304-4203(99)00048-1
Muller, F. L. L., and Batchelli, S. (2013). Copper binding by terrestrial versus marine organic ligands in the coastal plume of River Thurso, North Scotland. Estuar. Coast. Shelf Sci. 133, 137–146. doi: 10.1016/j.ecss.2013.08.024
Muller, F. L. L., and Cuscov, M. (2017). Alteration of the copper-binding capacity of iron-rich humic colloids during transport from peatland to marine waters. Environ. Sci. Technol. 51, 3214–3222. doi: 10.1021/acs.est.6b05303
Muller, F. L. L., and Tankéré-Muller, S. P. C. (2012). Seasonal variations in surface water chemistry at disturbed and pristine peatland sites in the Flow Country of northern Scotland. Sci. Total Environ. 43, 351–362. doi: 10.1016/j.scitotenv.2012.06.048
Murphy, K. R., Stedmon, C. A., Waite, T. D., and Ruiz, G. M. (2008). Distinguishing between terrestrial and autochthonous organic matter sources in marine environments using fluorescence spectroscopy. Mar. Chem. 108, 40–58. doi: 10.1016/j.marchem.2007.10.003
Neal, C., Lofts, S., Evans, C. D., Reynolds, B., Tipping, E., and Neal, M. (2008). Increasing iron concentrations in UK upland waters. Aquat. Geochem. 14, 263–288. doi: 10.1007/s10498-008-9036-1
Nowostawska, U., Kim, J. P., and Hunter, K. A. (2008). Aggregation of riverine colloidal iron in estuaries: a new kinetic study using stopped-flow mixing. Mar. Chem. 110, 205–210. doi: 10.1016/j.marchem.2008.03.001
Nuzzo, A., Sánchez, A., Fontaine, B., and Piccolo, A. (2013). Conformational changes of dissolved humic and fulvic superstructures with progressive iron complexation. J. Geochem. Explor. 129, 1–5. doi: 10.1016/j.gexplo.2013.01.010
Opsahl, S., and Benner, R. (1998). Photochemical reactivity of dissolved lignin in river and ocean waters. Limnol. Oceanogr. 43, 1297–1304. doi: 10.4319/lo.1998.43.6.1297
Opsahl, S. P., and Zepp, R. G. (2001). Photochemically-induced alteration of stable carbon isotope ratios (13C) in terrigenous dissolved organic carbon. Geophys. Res. Lett. 28, 2417–2420. doi: 10.1029/2000GL012686
Orlowska, E., Enyedy, E. A., Pignitter, M., Jirsa, F., Krachler, R., Kandioller, W., et al. (2017a). β-O-4 type dilignol compounds and their iron complexes for modeling of iron binding to humic acids: synthesis, characterization, electrochemical studies and algal growth experiments. New J. Chem. 41, 11546–11555. doi: 10.1039/c7nj02328f
Orlowska, E., Roller, A., Pignitter, M., Jirsa, F., Krachler, R., Kandioller, W., et al. (2017b). Synthetic iron complexes as models for natural iron-humic compounds: synthesis, characterization and algal growth experiments. Sci. Total Environ. 577, 94–104. doi: 10.1016/j.scitotenv.2016.10.109
Painter, S. C., Lapworth, D. J., Woodward, E. M. S., Kroeger, S., Evans, C. D., Mayor, D. J., et al. (2018). Terrestrial dissolved organic matter distribution in the North Sea. Sci. Total Environ. 630, 630–647. doi: 10.1016/j.scitotenv.2018.02.237
Pasquier, B., and Holzer, M. (2017). Inverse-model estimates of the ocean’s coupled phosphorus, silicon and iron cycles. Biogeosciences 14, 4125–4159. doi: 10.5194/bg-14-4125-2017
Peuravuori, J., and Pihlaja, K. (1997). Molecular size distribution and spectroscopic properties of aquatic humic substances. Anal. Chim. Acta 337, 133–149. doi: 10.1016/S0003-2670(96)00412-6
Piccolo, A., Conte, P., and Cozzolino, A. (1999). Conformational association of dissolved humic substances as affected by interactions with mineral and monocarboxylic acids. Eur. J. Soil Sci. 50, 687–694. doi: 10.1046/j.1365-2389.1999.00276.x
Pokrovsky, O. S., Shirokova, L. S., Viers, J., Gordeev, V. V., Shevchenko, V. P., Chupakov, A. V., et al. (2014). Fate of colloids during estuarine mixing in the Arctic. Ocean Sci. 10, 107–125. doi: 10.5194/os-10-107-2014
Quentel, F., and Filella, M. (2008). Quantification of refractory organic substances in freshwaters: further insight into the response of the voltammetric method. Anal. Bioanal. Chem. 392, 1225–1230. doi: 10.1007/s00216-008-2366-4
Quentel, F., Madec, C., Bihan, A. L., and Courtot-Coupez, J. (1986). Détermination des substances humiques en milieu marin par redissolution cathodique à l’électrode à goutte pendante de mercure. Anal. Lett. 19, 325–344. doi: 10.1080/00032718608064499
Raiswell, R., and Canfield, D. E. (2012). The iron biogeochemical cycle past and present. Geochem. Perspect. 1, 1–220. doi: 10.7185/geochempersp.1.1
Rathgeb, A., Causon, T., Krachler, R., and Hann, S. (2017). From the peat bog to the estuarine mixing zone: common features and variances in riverine dissolved organic matter determined by non-targeted analysis. Mar. Chem. 192, 158–167. doi: 10.1016/j.marchem.2017.06.012
Reid, P. M., Wilkinson, A. E., Tipping, E., and Jones, M. N. (1990). Determination of molecular weights of humic substances by analytical (UV scanning) ultracentrifugation. Geochim. Cosmochim. Acta 54, 131–138. doi: 10.1016/0016-7037(90)90201-U
Rossel, P. E., Vähätalo, A. V., Witt, M., and Dittmar, T. (2013). Molecular composition of dissolved organic matter from a wetland plant (Juncus effusus) after photochemical and microbial decomposition (1.25 yr): common features with deep sea dissolved organic matter. Org. Geochem. 60, 62–71. doi: 10.1016/j.orggeochem.2013.04.013
Santschi, P. H., Balnois, E., Wilkinson, K. J., Zhang, J., and Buffle, J. (1998). Fibrillar polysaccharides in marine macromolecular organic matter as imaged by atomic force microscopy and transmission electron microscopy. Limnol. Oceanogr. 43, 896–908. doi: 10.4319/lo.1998.43.5.0896
Schellekens, J., Buurman, P., Kalbitz, K., van Zomeren, A., Vidal-Torrado, P., Cerli, C., et al. (2017). Molecular features of humic acids and fulvic acids from contrasting environments. Environ. Sci. Technol. 51, 1330–1339. doi: 10.1021/acs.est.6b03925
Schlosser, C., and Croot, P. L. (2008). Application of cross-flow filtration for determining the solubility of iron species in open ocean seawater. Limnol. Oceanogr. Methods 6, 630–642. doi: 10.4319/lom.2008.6.630
Schröder, C., Köhler, I., Muller, F. L. L., Chumakov, A. I., Kupenko, I., Rüffer, R., et al. (2016). The biogeochemical iron cycle and astrobiology. Hyperfine Interact. 237:85. doi: 10.1007/s10751-016-1289-2
Seidel, M., Manecki, M., Herlemann, D. P. R., Deutsch, B., Schulz-Bull, D., Jürgens, K., et al. (2017). Composition and transformation of dissolved organic matter in the Baltic Sea. Front. Earth Sci. 5:31. doi: 10.3389/feart.2017.00031
Sharpless, C. M., Aeschbacher, M., Page, S. E., Wenk, J., Sander, M., and McNeill, K. (2014). Photooxidation-induced changes in optical, electrochemical, and photochemical properties of humic substances. Environ. Sci. Technol. 48, 2688–2696. doi: 10.1021/es403925g
Shiu, R.-F., Lee, C.-L., and Chin, W.-C. (2018). Reduction in the exchange of coastal dissolved organic matter and microgels by inputs of extra riverine organic matter. Water Res. 131, 161–166. doi: 10.1016/j.watres.2017.12.030
Sholkovitz, E. R., and Copland, D. (1981). The coagulation, solubility and adsorption properties of Fe, Mn, Cu, Ni, Cd, Co and humic acids in a river water. Geochim. Cosmochim. Acta 45, 181–189. doi: 10.1016/0016-7037(81)90161-7
Sigman, D. M., and Boyle, E. A. (2000). Glacial/interglacial variations in atmospheric carbon dioxide. Nature 407, 859–869. doi: 10.1038/35038000
Slagter, H. A., Reader, H. E., Rijkenberg, M. J. A., van der Loeff, M. R., de Baar, H. J. W., and Gerringa, L. J. A. (2017). Organic Fe speciation in the Eurasian basins of the Arctic Ocean and its relation to terrestrial DOM. Mar. Chem. 197, 11–25. doi: 10.1016/j.marchem.2017.10.005
Stedmon, C. A., and Markarger, S. (2005). Resolving the variability in dissolved organic matter fluorescence in a temperate estuary and its catchment using PARAFAC analysis. Limnol. Oceanogr. 50, 686–697. doi: 10.4319/lo.2005.50.2.0686
Stevenson, F. J. (1994). Humus Chemistry: Genesis, Composition, Reactions, 2nd Edn. New York, NY: Wiley, 512.
Stolpe, B., Guo, L., Shiller, A. M., and Aiken, G. R. (2013). Abundance, size distribution and trace-element binding of organic and iron-rich nanocolloids in Alaskan rivers, as revealed by field-flow fractionation and ICP-MS. Geochim. Cosmochim. Acta 105, 221–239. doi: 10.1016/j.gca.2012.11.018
Stolpe, B., and Hassellöv, M. (2010). Nanofibrils and other colloidal biopolymers binding trace elements in coastal seawater: significance for variations in element size distribution. Limnol. Oceanogr. 55, 187–202. doi: 10.4319/lo.2010.55.1.0187
Stubbins, A., Spencer, R. G. M., Chen, H. M., Hatcher, P. G., Mopper, K., Hernes, P. J., et al. (2010). Illuminated darkness: molecular signatures of Congo River dissolved organic matter and its photochemical alteration as revealed by ultrahigh precision mass spectrometry. Limnol. Oceanogr. 55, 1467–1477. doi: 10.4319/lo.2010.55.4.1467
Stumm, W., and Morgan, J. J. (1996). Aquatic Chemistry, 3rd Edn. New York, NY: John Wiley & Sons, Inc.
Sun, L., Xu, C., Zhang, S., Lin, P., Schwehr, K. A., Quigg, A., et al. (2017). Light-induced aggregation of microbial exopolymeric substances. Chemosphere 181, 675–681. doi: 10.1016/j.chemosphere.2017.04.099
Sunda, W. G. (2001). “Bioavailability and bioaccumulation of iron in the sea,” in The Biogeochemistry of Iron in Seawater, eds D. R. Turner and K. A. Hunter (Chichester: John Wiley & Sons, Ltd), 41–84.
Sunda, W. G., and Huntsman, S. A. (1995). Iron uptake and growth limitation in oceanic and coastal phytoplankton. Mar. Chem. 50, 189–208. doi: 10.1016/0304-4203(95)00035-P
Sutton, R., and Sposito, G. (2005). Molecular structure in soil humic substances: the new view. Environ. Sci. Technol. 39, 9009–9015. doi: 10.1021/es050778q
Tagliabue, A., Aumont, O., DeAth, R., Dunne, J. P., Dutkiewicz, S., Galbraith, E., et al. (2016). How well do global ocean biogeochemistry models simulate dissolved iron distributions? Glob. Biogeochem. Cycles 30, 149–174. doi: 10.1002/2015GB005289
Tagliabue, A., Bowie, A. R., Boyd, K. W., Buck, K. N., Johnson, K. S., and Saito, M. A. (2017). The integral role of iron in ocean biogeochemistry. Nature 543, 51–59. doi: 10.1038/nature21058
Tesi, T., Semiletov, I., Hugelius, G., Dudarev, O., Kuhry, P., and Gustafsson, Ö. (2014). Composition and fate of terrigenous organic matter along the Arctic land ocean continuum in East Siberia: insights from biomarkers and carbon isotopes. Geochim. Cosmochim. Acta 133, 235–256. doi: 10.1016/j.gca.2014.02.045
Thurman, E. M., and Malcolm, R. L. (1981). Preparative isolation of aquatic humic substances. Environ. Sci. Technol. 15, 463–466. doi: 10.1021/es00086a012
Trubetskoj, O. A., Richard, C., Voyard, G., Marchenkov, V. V., and Trubetskaya, O. E. (2018). Molecular size distribution of fluorophores in aquatic natural organic matter: application of HPSEC with multi-wavelength absorption and fluorescence detection following LPSEC-PAGE fractionation. Environ. Sci. Technol. 52, 5287–5295. doi: 10.1021/acs.est.7b03924
Vähätalo, A. V., Haarnos, H., Hoikkala, L., and Lignell, R. (2011). Photochemical transformation of terrestrial dissolved organic matter supports hetero- and autotrophic production in coastal waters. Mar. Ecol. Prog. Ser. 423, 1–14. doi: 10.3354/meps09010
Völker, C., and Tagliabue, A. (2015). Modeling organic iron-binding ligands in a three-dimensional biogeochemical ocean model. Mar. Chem. 173, 67–77. doi: 10.1016/j.marchem.2014.11.008
Waggoner, D. C., Chen, H., Willoughby, A. S., and Hatcher, P. G. (2015). Formation of black carbon-like and alicyclic compounds by hydroxyl radical initiated degradation of lignin. Org. Geochem. 82, 69–76. doi: 10.1016/j.orggeochem.2015.02.007
Waggoner, D. C., and Hatcher, P. G. (2017). Hydroxyl radical alteration of HPLC fractionated lignin: formation of new compounds from terrestrial organic matter. Org. Geochem. 113, 315–325. doi: 10.1016/j.orggeochem.2017.07.011
Walker, B. D., Beaupré, S. R., Guilderson, T. P., McCarthy, M. D., and Druffel, E. R. (2016). Pacific carbon cycling constrained by organic matter size, age and composition relationships. Nat. Geosci. 9, 888–891. doi: 10.1038/ngeo2830
Ward, N. D. (2017). Editorial: integrative research on organic matter cycling across aquatic gradients. Front. Mar. Sci. 4:131. doi: 10.3389/fmars.2017.00131
Watanabe, K., Fukuzaki, K., Fukushima, K., Aimoto, M., Yoshioka, T., and Yamashita, Y. (2018). Iron and fluorescent dissolved organic matter in an estuarine and coastal system in Japan. Limnology 19, 229–240. doi: 10.1007/s10201-017-0536-9
Wells, M. L. (2002). “Marine colloids and trace metals,” in Biogeochemistry of Marine Dissolved Organic Matter, eds D. A. Hansell and C. A. Carlson (San Diego, CA: Academic Press), 367–404. doi: 10.1016/B978-012323841-2/50009-9
Willey, J. D., Kieber, R. J., Seaton, P. J., and Miller, C. (2008). Rainwater as a source of Fe(II)-stabilizing ligands to seawater. Limnol. Oceanogr. 53, 1678–1684. doi: 10.4319/lo.2008.53.4.1678
Yamashita, Y., Cory, R. M., Nishioka, J., Kuma, K., Tanoue, E., and Jaffé, R. (2010). Fluorescence characteristics of dissolved organic matter in the deep waters of the Okhotsk Sea and the northwestern North Pacific Ocean. Deep Sea Res. Part 2 Top. Stud. Oceanogr. 57, 1478–1485. doi: 10.1016/j.dsr2.2010.02.016
Yamashita, Y., Fichot, C. G., Shen, Y., Jaffé, R., and Benner, R. (2015). Linkages among fluorescent dissolved organic matter, dissolved amino acids and lignin-derived phenols in a river-influenced ocean margin. Front. Mar. Sci. 2:92. doi: 10.3389/fmars.2015.00092
Yamashita, Y., and Jaffé, R. (2008). Characterizing the interactions between trace metals and dissolved organic matter using excitation-emission matrix and parallel factor analysis. Environ. Sci. Technol. 42, 7374–7379. doi: 10.1021/es801357h
Zhang, S., and Santschi, P. H. (2009). Application of cross-flow ultrafiltration for isolating exopolymeric substances from a marine diatom (Amphora sp.). Limnol. Oceanogr. Methods 7, 419–429. doi: 10.4319/lom.2009.7.419
Zhang, Y., Yang, K., Du, J., Zhang, F., Dong, Y., and Wu, L. (2017). Chemical characterization of fractions of dissolved humic substances from a marginal sea a case from the Southern Yellow Sea. Chin. J. Oceanol. Limnol. 36, 238–248. doi: 10.1007/s00343-017-6202-6
Keywords: colloids, iron, humic substances, land-ocean transfer, organic ligands, seawater, terrigenous DOM
Citation: Muller FLL (2018) Exploring the Potential Role of Terrestrially Derived Humic Substances in the Marine Biogeochemistry of Iron. Front. Earth Sci. 6:159. doi: 10.3389/feart.2018.00159
Received: 29 July 2018; Accepted: 24 September 2018;
Published: 15 October 2018.
Edited by:
Selvaraj Kandasamy, Xiamen University, ChinaReviewed by:
Peter H. Santschi, Texas A&M University, United StatesRegina Krachler, Universität Wien, Austria
Copyright © 2018 Muller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: François L. L. Muller, fmuller@mail.nsysu.edu.tw