 Ting Yuan
Ting Yuan Izhar Hyder Qazi
Izhar Hyder Qazi Jinhao Li
Jinhao Li Peijia Yang
Peijia Yang- South China Agriculture University, College of Animal Science, Regional Sericulture Training Center for Asia-Pacific, Guangzhou, Guangdong, China
Introduction: Mulberry bacterial wilt is a serious destructive soil-borne disease caused by a complex and diverse group of pathogenic bacteria. Given that the bacterial wilt has been reported to cause a serious damage to the yield and quality of mulberry, therefore, elucidation of its main pathogenic groups is essential in improving our understanding of this disease and for the development of its potential control measures.
Methods: In this study, combined metagenomic sequencing and culture-dependent approaches were used to investigate the microbiome of healthy and bacterial wilt mulberry samples.
Results: The results showed that the healthy samples had higher bacterial diversity compared to the diseased samples. Meanwhile, the proportion of opportunistic pathogenic and drug-resistant bacterial flora represented by Acinetobacter in the diseased samples was increased, while the proportion of beneficial bacterial flora represented by Proteobacteria was decreased. Ralstonia solanacearum species complex (RSSC), Enterobacter cloacae complex (ECC), Klebsiella pneumoniae, K. quasipneumoniae, K. michiganensis, K. oxytoca, and P. ananatis emerged as the main pathogens of the mulberry bacterial wilt.
Discussion: In conclusion, this study provides a valuable reference for further focused research on the bacterial wilt of mulberry and other plants.
Introduction
Mulberry is a perennial dicotyledonous tree or shrub (Dai et al., 2020) that is widely cultivated throughout subtropical and temperate regions, and has a significant economic value (Xie et al., 2020). Mulberry leaves are exclusively used as a food source for the domesticated silkworm Bombyx mori L. (Ji et al., 2008; Chan et al., 2016). Besides its use as silkworm forage, mulberry is now used as a raw material in animal feed (Jiang et al., 2022), medicine (Meng et al., 2020) and food industry (Maqsood et al., 2022). However, the occurrence of mulberry diseases has seriously affected the healthy and stable development of the sericulture industry (Dong et al., 2021). For instance, mulberry bacterial wilt is a destructive disease that seriously affects the yield and quality of mulberry (Dong et al., 2021). Mulberry bacterial wilt was first reported in 1969 in Shunde City, Guangdong province of China, and has spread to most mulberry planting areas in Guangdong (Lai et al., 1979). Mulberry bacterial wilt is still prevalent in the main sericulture-producing areas of Guangdong, Guangxi and other places in China (Dai et al., 2016), and has been reported in many other mulberry planting areas in the country.
Mulberry bacterial wilt is a vascular disease which is difficult to diagnose with the naked eye at the initial stage of infection. However, in the middle stage of the disease, the leaves lose moisture and then curl or wilt, turning black or brown. In the late stage of the disease, the leaves of the whole plant are withered until they fall off, the xylem turns brown streaked or dark brown, and white pus-like bacteria overflow from the cross-section of the diseased root (Wang et al., 2008; Zhu et al., 2010; Zhou et al., 2021; Luo et al., 2022; Yuan et al., 2023a).
The pathogen of mulberry bacterial wilt has complex and diverse characteristics. Lai et al. (1979) isolated and identified the pathogen of mulberry bacterial wilt for the first time. Initially, Pseudomonas solanacearum was considered as a pathogen causing mulberry bacterial wilt, which was later renamed as Ralstonia solanacearum, and now classified as R. pseudosolanacearum. Wang et al. (2008) reported for the first time that the mulberry wilt was also caused by Enterobacter cloacae complex (ECC). Subsequently, Zhu et al. (2010) isolated E. mori from mulberry wilt disease samples. Zhou et al. (2021) isolated E. roggenkampii strain KQ-01 from the bacterial wilt-resistant mulberry cultivar YS283, which can cause mulberry wilt. Luo et al. (2022) isolated Klebsiella michiganensis AKKL-001 from mulberry bacterial disease samples, which can also cause mulberry wilt. Recently, Pantoea ananatis strain LCFJ-001 was isolated from mulberry bacterial wilt disease samples and was reported to cause mulberry wilt (Yuan et al., 2023a).
Currently reported pathogens of mulberry bacterial wilt can be divided into four categories: Ralstonia, Enterobacter, Klebsiella, and Pantoea (Supplementary Figure 1). The gradual increase in sericulture production and exchange activities in the recent times has also led to an increased occurrence of bacterial wilt in mulberry fields in China, leading to significant challenges to the healthy development of the sericulture industry in the country. As this complex disease is caused by a number of pathogens, it still remains to be known which pathogen is the main pathogen, making its prevention and control difficult. It has been reported that the occurrence of plant diseases is related to changes in their crop microbiome, and that the study of changes in their microbiome can further reveal their pathogenesis (Li et al., 2022). Therefore, in order to further understand the basis of pathogenesis and provide a valuable reference for prevention and control, this study was carried out to explore the changes in mulberry microbiome in bacterial wilt and healthy samples of mulberry. In the present study, we collected (2017 to 2022) 35 mulberry bacterial wilt disease samples from Guangdong, Guangxi, and other regions of China. The diseased mulberry samples were isolated and tested for pathogenicity of pathogenic bacteria. At the same time, due to the limitations of traditional culture-dependent method, we also used the metagenomic sequencing to further explore the main pathogenic groups in the diseased and healthy mulberry samples.
Materials and methods
Metagenomic sequencing of mulberry samples
Collection of mulberry samples
A survey of mulberry fields where mulberry wilt was prevalent in Liucheng (109.24°, 24.65°) and Rong’an (109.35°, 25.15°) counties of Guangxi, China was conducted (see Supplementary Figures 2A–I for description). The mulberry samples with typical disease symptoms in the field were processed for laboratory verification.
Metagenomic sequencing of mulberry samples
The pH values of the diseased (wilted) plants and rhizosphere soil of typical mulberry in Liucheng and Rong’an were tested. Eight samples were collected (Supplementary Table 1) and sent to the Science Corporation of Gene Co., Ltd. for metagenomic sequencing to analyze the types, and abundance of pathogens in the samples.
Extraction of genomic DNA
The genome DNA was extracted from samples using the Ezup Column Bacteria genomic DNA purification kit (Sangon Biotech (Shanghai) Co., Ltd., China). DNA purity and concentration were measured by gel electrophoresis and NanoDrop 2000 (Thermo Scientific) spectrophotometer (Yuan et al., 2023b).
Amplification of the target region
The 16S rRNA gene consists of nine hypervariable regions flanked by regions of more conserved sequence. To maximize the effective length of PE 250 sequencing reads of Illumina HiSeq2500, the region encompassing the V3 and V4 hypervariable regions of the 16S rRNA gene was targeted for sequencing. The V3-V4 hypervariable region was amplified using a specific primer with the barcode (Supplementary Table 2). All PCRs were carried out in 40 μL reactions with 20 μL of 2×Taq MasterMix, 0.5 μM forwards and reverse primers, and approximately 10 ng of template DNA. Temperature cycling consisted of denaturation at 94°C for 2 min, followed by 30 cycles of denaturation at 94°C for 30 s, annealing at 60°C for 30 s, and elongation at 72°C for 20 s., and finally, 72°C for 7 min. The purity and concentration of all amplicons were characterized by gel electrophoresis and Qubit@ 2.0 Fluorometer (Thermo Scientific). The amplicons with bright main strips and the right length were chosen for the subsequent experiments (Yuan et al., 2023b).
Library preparation and metagenomic sequencing
PCR products with different barcodes were mixed in equidensity ratios. Then, a mixture of PCR products was used to prepare pair-end sequencing libraries. The libraries were generated following the Illumina HiSeq 2500 standard protocol (Illumina, Inc., San Diego, CA). Paired-end reads (250 bp) were generated on the Illumina HiSeq2500 platform. Three replicates of each sample were used for metagenomic sequencing (Yuan et al., 2023b). The metagenomic sequencing data have been uploaded to the NCBI (National Center for Biotechnology Information) with accession number PRJNA911049. Finally, based on taxonomy, the abundance of each bacterial genus was counted, and Origin 2019b software (OriginLab Corporation, Northampton, MA, USA) was used to make bacterial abundance maps of different samples and the Shannon, Chao-1 and Simpson values of each sample were calculated.
Analysis of mulberry samples using culture-dependent approach
Collection of mulberry samples
During 2019 to 2022, a total of 35 samples of diseased plants were collected from Guangxi, Guangdong and Hainan in China (Supplementary Table 3). M. atropurpurea varieties Lun40 and Kangqing10 were used as the healthy group (Supplementary Table 3), and 20 copies of each variety were collected in mulberry field of the South China Agricultural University, Guangzhou, Guangdong, China (113.35°, 23.17°).
Isolation of bacteria from mulberry samples
The experimental design is depicted in Supplementary Figure 3. Firstly, the collected diseased or healthy roots were rinsed under the faucet, and the surface stains were washed with soapy water and the samples were wiped with a clean gauze. The roots were cut into small sections of three centimeters in length using clean scissors. Then, the sections were soaked in 75% ethanol for 1 min, rinsed with sterile water three times, soaked in 0.1% mercuric chloride for 5 min, and rinsed with sterile water five times. Then, the surface-sterilized small section was placed in a sterile glass petri dish, the xylem in the center was removed with sterile tweezers and scissors, cut into pieces and ground in a sterile mortar. The ground xylem was placed in 10 mL of sterile saline and in a shaker at 28°C and 140 r/min for 10 minutes to form a liquid containing xylem bacteria. The liquid was then removed and diluted eight times according to the 10-fold dilution method (Yuan et al., 2023a).
A total of 0.1 mL of each gradient was spread evenly on Lysogeny Borth (LB) agar plates (Guangdong Huankai Co., Ltd., China) and nutrient agar plates (Guangdong Huankai Co., Ltd., China). Then, the plates were placed in a biochemical incubator at 28°C for two days for cultivation. Finally, single colonies were picked from LB agar and nutrient agar media and drawn on new nutrient agar plates, and each colony was purified for seven generations (Yuan et al., 2023a).
Classification of bacteria
Classification of the bacteria was based on analysis of 16S rRNA gene using universal primer 27F/1492R (Bredow et al., 2015). All strains were inoculated in nutrient broth medium (Guangdong Huan Kai Co., Ltd., China) and placed in a shaker at 28°C and 140 r/min for 12 h. Bacterial genomic DNA was extracted using the Ezup Column Bacteria Genomic DNA purification kit (Sangon Biotech (Shanghai) Co., Ltd., China). DNA from all purified isolates was used for PCR amplification of the 16S rRNA gene, which was performed in a 25 μL volume under the following conditions: one cycle of 98°C for 4 min, followed by 30 cycles of 98°C for 30 seconds, 55°C for 30 seconds, 72°C for 1 minute, and final an extension at 72°C for 10 minutes. The PCR-amplified products were transferred to a laboratory in Shanghai, China, at Sangon Biotechnology Co. Ltd. in Shanghai, China, and then sequenced by the Sanger method (Yuan et al., 2023a).
The generated sequences were aligned using BioEdit software version 7.0 and then subjected to analysis by the Basic Local Alignment Search Tool (BLAST) search program of the NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi) to determine the sequence homology with closely related organisms (Altschul et al., 1997). Microorganisms with high homology (97-100%) were selected as the closest matches. All bacterial isolates were assigned to the genus level separately based on information from the closest microorganisms. In addition, the NCBI taxonomic database was used to classify all bacterial strains at the phylum, class, order, and family levels (Yuan et al., 2023b). All bacterial 16S rDNA sequences generated in this study have been submitted to the NCBI. The accession numbers OP990608-OP990981 are bacterial 16S rDNA sequences derived from healthy samples; OP989957-OP990607 are bacterial 16S rDNA sequences derived from diseased samples. Finally, based on the 16S rRNA gene bacterial identification results, the abundance of each bacterial genus was counted, and the bacterial abundance maps of different samples were made using Origin 2019b software (OriginLab Corporation, Northampton, MA, USA) and the Shannon, Chao-1, and Simpson values of healthy and diseased samples were calculated.
16S rRNA gene phylogenetic tree construction
The 16S rDNA sequences of typical strains of Ralstonia, Enterobacter, Klebsiella, and Pantoea were downloaded from the List of Prokaryotic names with Standing in Nomenclature (https://lpsn.dsmz.de/). The 16S rDNA of six strains of Ralstonia, 30 strains of Enterobacter, 12 strains of Klebsiella, and 12 strains of Pantoea were selected for construction of phylogenetic trees (Supplementary Table 4). At the same time, the 16S rDNA sequences of strains identified in our laboratory (Supplementary Table 4) and the 16S rDNA sequences downloaded from the NCBI were used as references. The sequences were compared using MUSCLEv.3.8.31 software. The phylogenetic trees were constructed using the maximum likelihood tree with MEGA-X software, and the bootstrap value was set to 1000 (Yuan et al., 2023a).
Cultivation of mulberry branches
Healthy 15-year-old healthy M. atropurpurea cultivar Lun40 (susceptible to bacterial wilt) obtained from the South China Agricultural University mulberry field (Guangzhou, Guangdong, China), (113.35°, 23.17°) was selected as plant material for this study. The samples were collected in December 2021. Firstly, old branches of Lun40 with a diameter of 0.5-0.75 centimeter were selected and cut into stem segments of 10-12 centimeters in length and containing three lateral shoots. The stems were washed with soapy water to remove surface dust and soaked in 0.5% sodium hypochlorite solution for five hours. The stems were then inserted into the sterile MS liquid medium and placed in an artificial climate incubator at 25°C, 12 h/d light, and 85% humidity. The culture was incubated for 25 days until the lateral shoots sprouted and exhibited 2-3 leaves. During this period, the sterile MS liquid medium was changed every day (Yuan et al., 2023b).
Pathogenicity test
To investigate the pathogenicity of Ralstonia, Enterobacter, Klebsiella, and Pantoea, the following procedures were adopted: 1) the pure cultures of all the bacteria (Supplementary Table 4) were placed in nutrient broth medium overnight. The overnight cultured bacteria solution was adjusted to OD600 nm=0.1 with sterile MS liquid medium. 2) After cultivation, the Lun40 mulberry branch with 2~3 leaves was placed into the diluted bacterial solution. The sterile MS liquid medium was set as the healthy group. 3) The treated Lun40 mulberry branches were cultured for 12 days in an artificial climate incubator at 28°C, 12 h/d light, and 85% humidity, and the disease incidence in plants was observed. 4) Morbidity rate= (A-B)/C×100%. A: The total number of diseased mulberry branches in the experimental group; B: The total number of diseased mulberry branches in the control group; C: The total number of mulberry branches (Yuan et al., 2023b).
Analysis of variance (ANOVA) was performed using Excel software. Each set of experiments for each pathogen species was tested using six healthy mulberry branches. With the sterile MS liquid medium as a control, each group had three replicates (Yuan et al., 2023b).
Data statistics
Data analysis was performed using a one-way analysis of variance (Levene’s test was used to evaluate the equality of variance before analysis), and the least significant difference test was used to determine the significant difference between the means as a post hoc analysis. P<0.05 was considered significant. Excel 2016 software (Microsoft, Redmond, WA, USA) and Origin 2019b 64Bit were used to analyze and map the data.
Results
Analysis of metagenomic sequencing quality
The description of sequencing data of the bacterial 16S rDNA V3-V4 regions collected from mulberry xylem and rhizosphere soil are shown in Table 1. Briefly, the GC was greater than 53%, Q20 was greater than 96%, and Q30 was greater than 94%. This indicates a low sequencing error rate and high quality and reliability of the data.
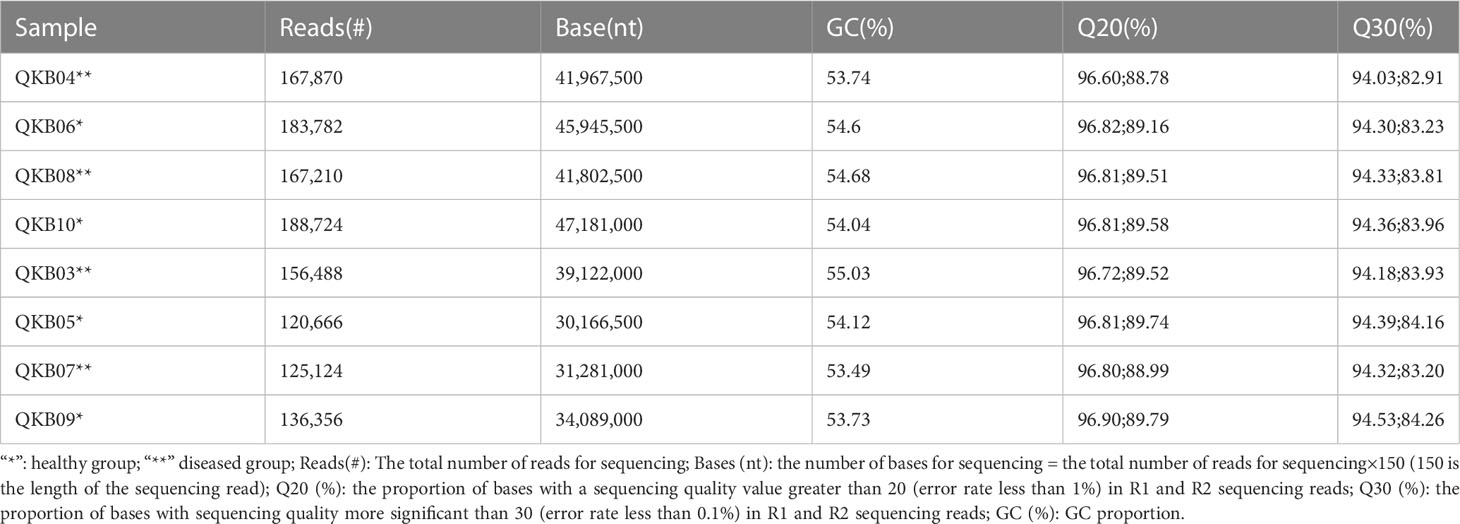
Table 1 Description of metagenomic sequencing data.
Analysis of diversity of bacterial community in mulberries based on metagenomic sequencing
As shown in Table 2, the bacterial community OTU numbers, Shannon index, Chao-1 index and Simpson index of the diseased rhizosphere soil (QKB04 and QKB08) and xylem (QKB03 and QKB07) were lower compared to the healthy rhizosphere soil (KB06 and QKB10) and xylem (QKB05 and QKB09). From these, the bacterial diversity of the rhizosphere soil and xylem of the diseased mulberry was lower compared to the healthy mulberry.
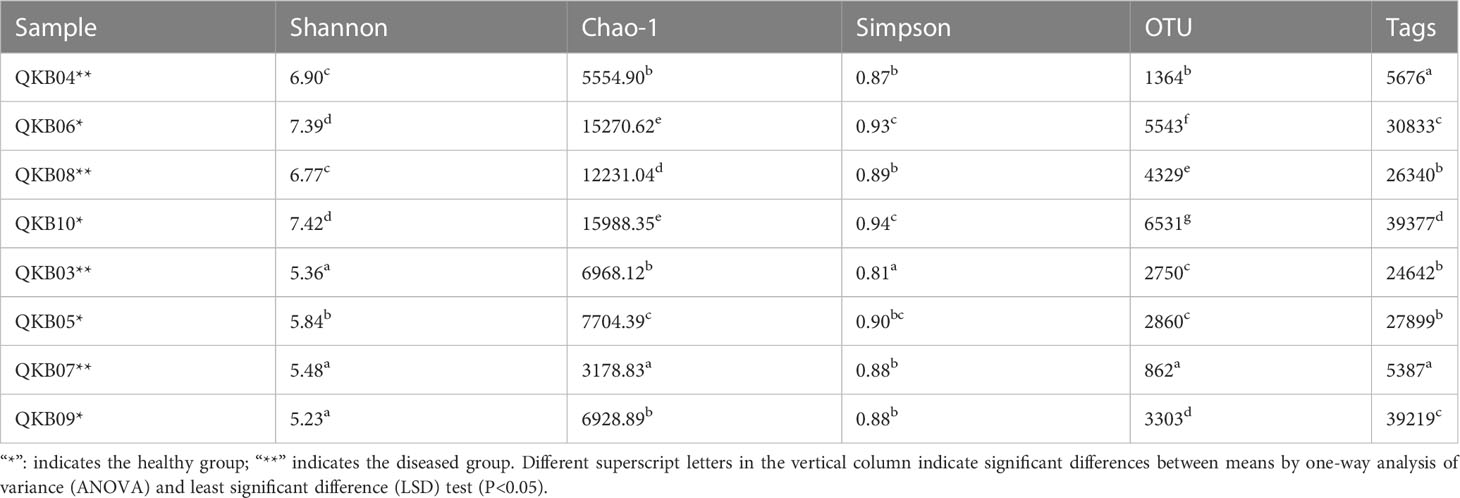
Table 2 The number of OTUs and diversity index of the read sequence (Tags) bacterial community of the sequenced branch samples.
Analysis of bacterial community composition of mulberries based on metagenomic sequencing
The phylum-level abundance distribution of bacterial populations in the mulberry rhizosphere soil was associated with 17 phyla (Figure 1A). From these, Proteobacteria had the highest abundance in diseased and healthy samples, followed by Actinomycetes and unclassified bacteria. At the genus level, the taxonomic sequence of the mulberry rhizosphere soil was associated with 49 genera (Figure 1B). The abundance distribution of the dominant flora are shown in Table 3. Pseudomonas accounted for the largest proportion, followed by Mycobacteria, Erwinia and Ralstonia, respectively. The Pseudomonas disease samples showed a significant downward trend (P<0.05), whereas the Erwinia disease samples showed a significant upward trend (P<0.05). Interestingly, there was no significant difference in the abundance of Ralstonia between healthy and diseased samples (P<0.05).
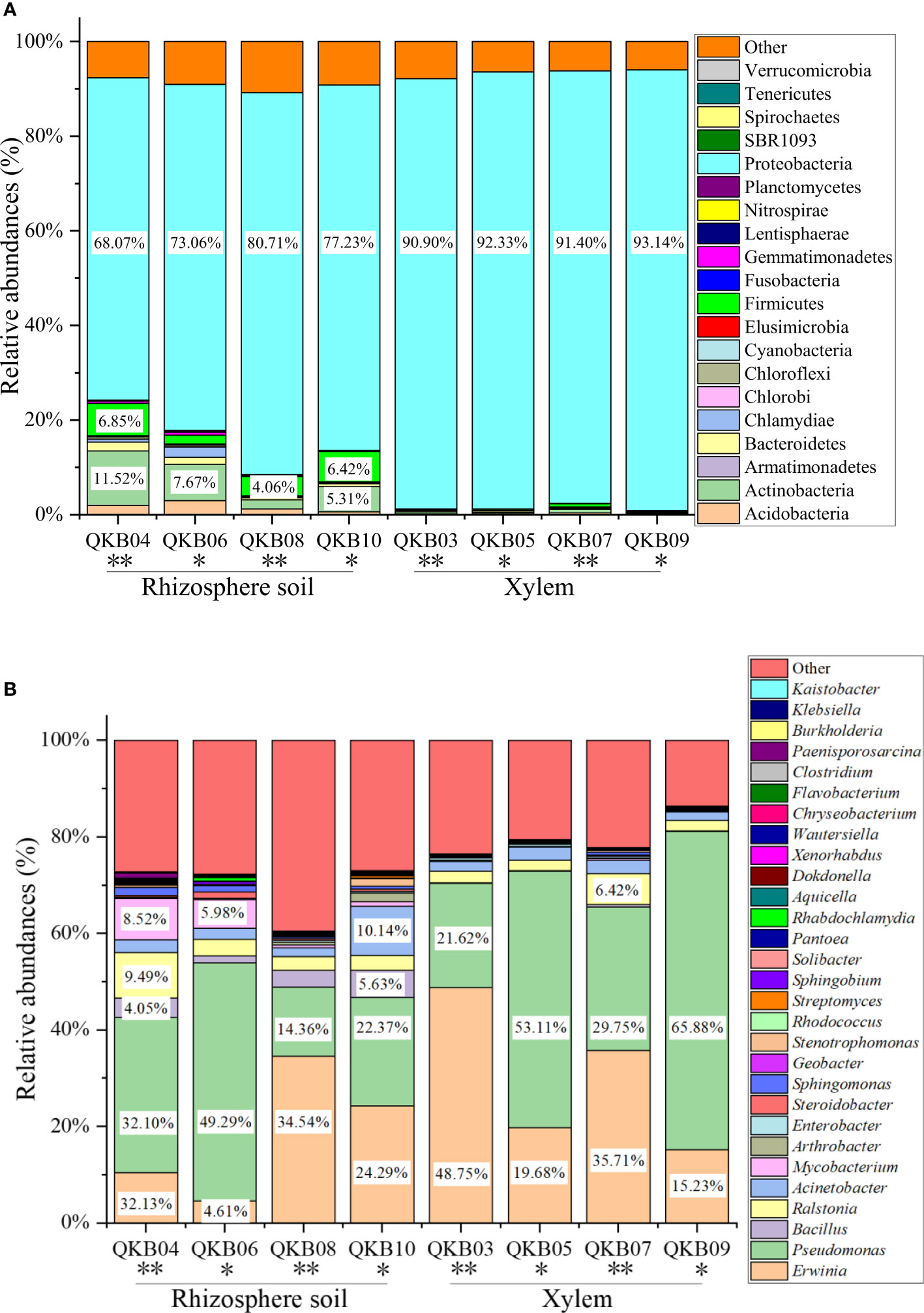
Figure 1 Abundance distribution of bacterial populations based on metagenomic sequencing at the phylum (A) and genus (B) levels. QKB04: Liucheng (109.24°, 24.65°) diseased sample rhizosphere soil; QKB06: Liucheng (109.24°, 24.65°) healthy sample rhizosphere soil; QKB08: Rong’an (109.35°, 25.15°) diseased sample rhizosphere soil; QKB10: Rong’an (109.35°, 25.15°) healthy sample rhizosphere soil; QKB03: Liucheng (109.24°, 24.65°) diseased sample xylem; QKB05: Liucheng (109.24°, 24.65°) healthy sample xylem; QKB07: Rong’an (109.35°, 25.15°) diseased sample xylem; QKB09: Rong’an (109.35°, 25.15°) diseased sample xylem; “*”: indicates the healthy group; “**” indicates the diseased group.
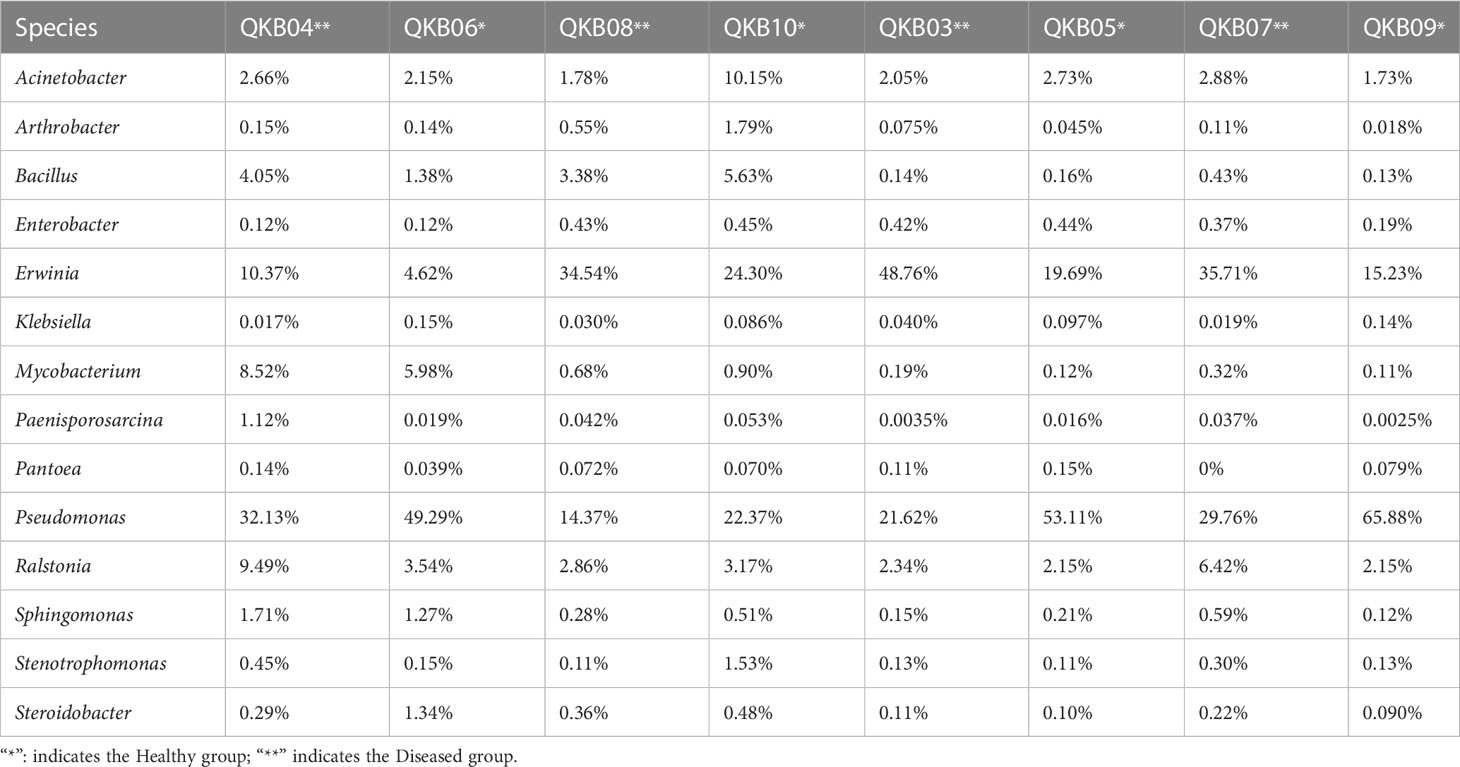
Table 3 Richness of important bacterial genera in different samples based on metagenomic sequencing data.
The phylum-level abundance distribution of bacterial populations in mulberry xylem was associated with seven phyla (Figure 1A). Proteus was the first dominant bacterial group in both healthy and diseased mulberry samples and its abundance accounted for more than 90% in both healthy and diseased groups. Many sequences in xylem could not be classified (7.8% richness), indicating the diversity of the xylem bacteria. At the genus level, the bacteria in the mulberry xylem part were related to 23 genera (Figure 1B). The abundance distribution of the dominant bacterial taxa is shown in Table 3. Pseudomonas was found to be the most abundant, followed by Erwinia and Ralstonia, respectively. Interestingly, the abundance of Pseudomonas was lower in the diseased group compared to the healthy mulberry group, whereas abundance of Erwinia and Ralstonia showed a reverse trend.
Bacterial composition and diversity in mulberries based on a culture-dependent approach
A total of 1052 strains of the xylem bacteria were isolated from all samples. From these, 389 strains were from the healthy mulberry samples (CKS) (Supplementary Table 5), 663 strains were from the diseased (bacterial wilt) mulberry samples (MBWS) (Supplementary Table 6). Based on the results of 16S rRNA gene, CKS culturable strains were divided into 58 genera, distributed in 4 phyla, 6 classes, 18 orders and 28 families (Supplementary Table 5). The Shannon, Simpson and Pielou values were 3.03, 0.90 and 0.75, respectively (Table 4). The culturable strains of MBWS were divided into 69 genera, distributed in 4 phyla, 9 classes, 17 orders and 31 families (Supplementary Table 6). The values of Shannon, Simpson and Pielou were 3.17, 0.92 and 0.75, respectively (Table 4). This finding indicated that the diversity of the xylem bacteria in the MBWS samples was slightly higher compared to the CKS samples (P>0.05).
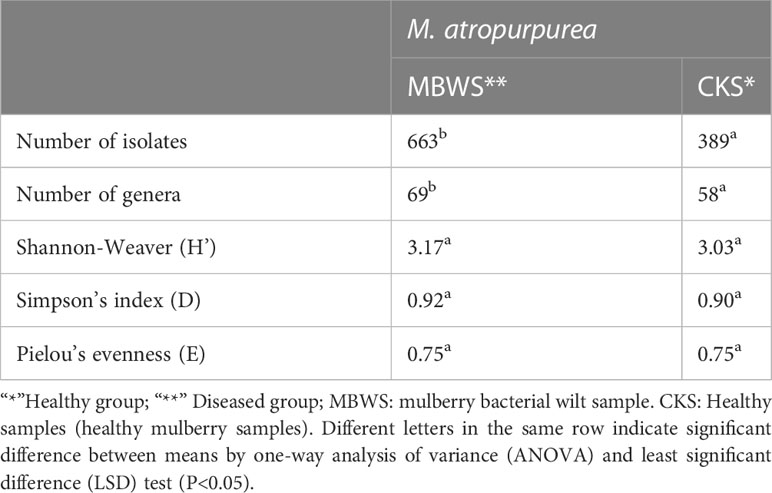
Table 4 Profiles of bacterial community diversity in the biomass of diseased and healthy mulberry samples based on culture-dependent approach.
All isolates belonged to Actinobacteria, Bacteroidetes, Firmicutes and Proteobacteria phyla. From these, Proteobacteria was found to the dominant phylum in the bacterial community of mulberry xylem (Figure 2A). The most abundant Proteobacteria (CKS 70.95%, MBWS 88.98%) mainly contained the Alphaproteobacteria, Betaproteobacteria, and Gammaproteobacteria-like bacteria. Firmicutes was the second most dominant phylum (CKS 19.53%, MBWS 4.67%) and contained only bacilli, consisting of Bacillus (Figure 2A). Actinomycetes was the third most dominant bacterial phylum (CKS 8.99%, MBWS 2.71%) and was represented by Microbacteria (Figure 2A). At the genus level (Figure 2B), Pseudomonas, Enterobacter, and Acinetobacter were found to be the main groups of the xylem bacteria.
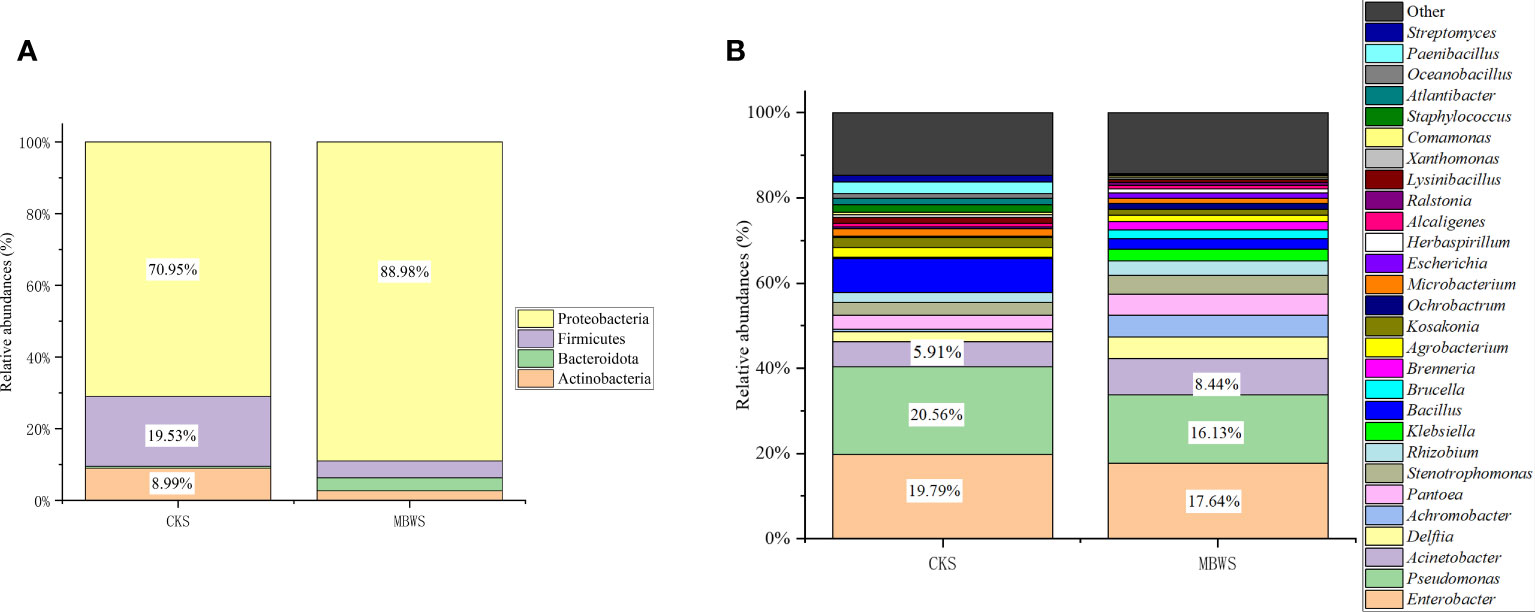
Figure 2 Relative abundance (%) of cultivable bacteria in different communities isolated from healthy and diseased mulberry at the phylum (A) and genus (B) levels. MBWS, mulberry bacterial wilt sample; CKS, Healthy samples (healthy mulberry samples).
Analysis of bacterial community of mulberries based on culture-dependent approach
Unique and shared bacterial genera between healthy and diseased mulberry groups are shown in the Venn diagram (Figure 3). The number of shared attributes across all groupings was 33 (Figure 3). In addition, the number of unique genera in the MBWS was higher than the number of unique and shared genera in the CKS group. Genera including Enterobacter, Pseudomonas, Acinetobacter, Delftia, Pantoea, Stenotrophomonas, Rhizobium, Bacillus, Agrobacterium, Kosakonia, and Microbacterium, with an average segregation rate of >1% in MBWS and CKS, were the 11 core genera of mulberry xylem bacteria (Figure 2B).
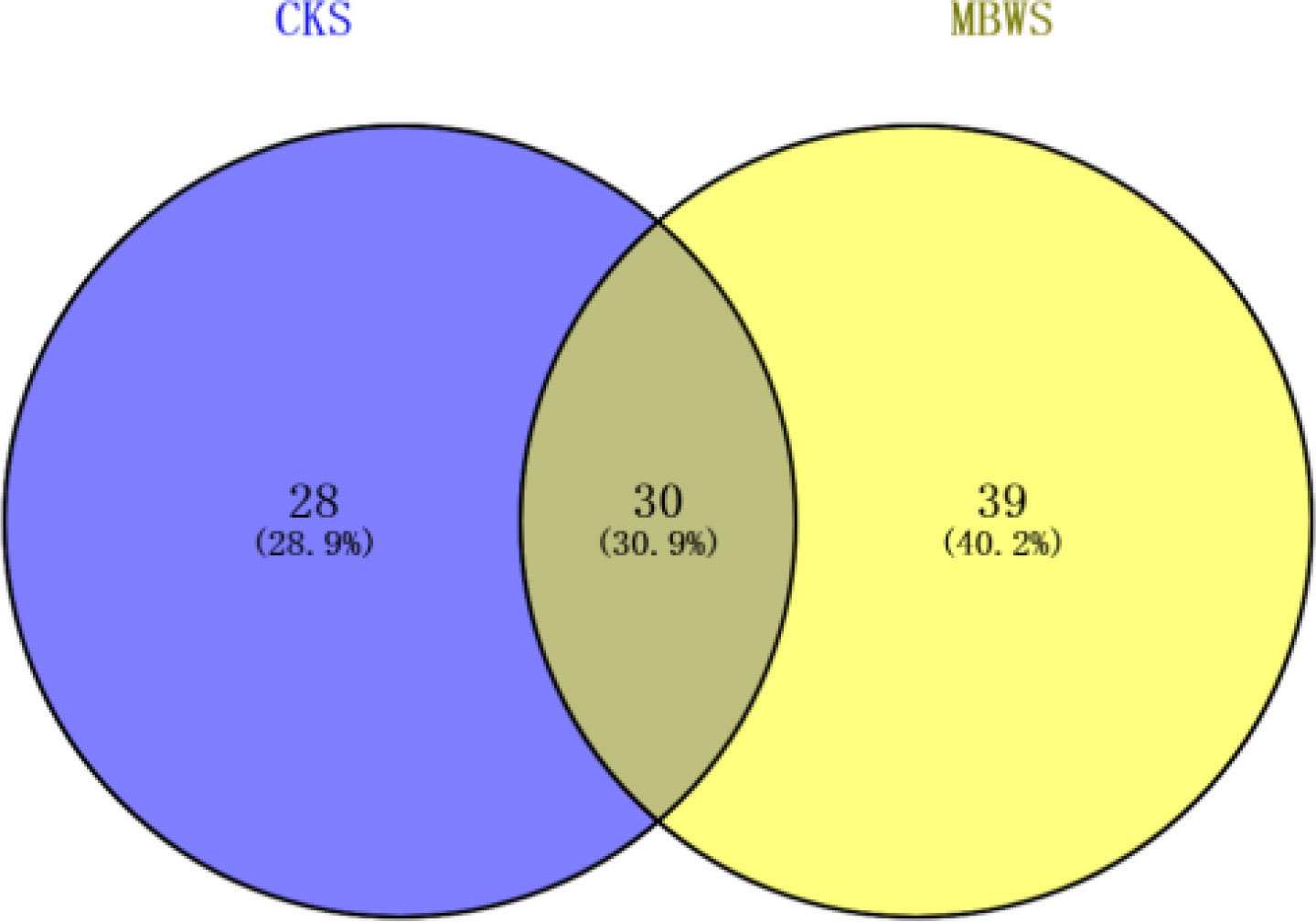
Figure 3 Bacterial Venn diagrams of healthy and diseased mulberry samples. MBWS, mulberry bacterial wilt sample; CKS, healthy mulberry samples.
The separation frequencies of Achromobacter, Acinetobacter, Brenneria, Brucella, Delftia, Escherichia, Herbaspirillum, Klebsiella, Ochrobactrum, Pantoea, Ralstonia, Rhizobium and Stenotrophomonas in the CKS were significantly lower (P<0.05) compared to the MBWS group. Additionally, Herbaspirillum, Brenneria, Klebsiella and Ralstonia were isolated only in the diseased samples (Figure 4A).
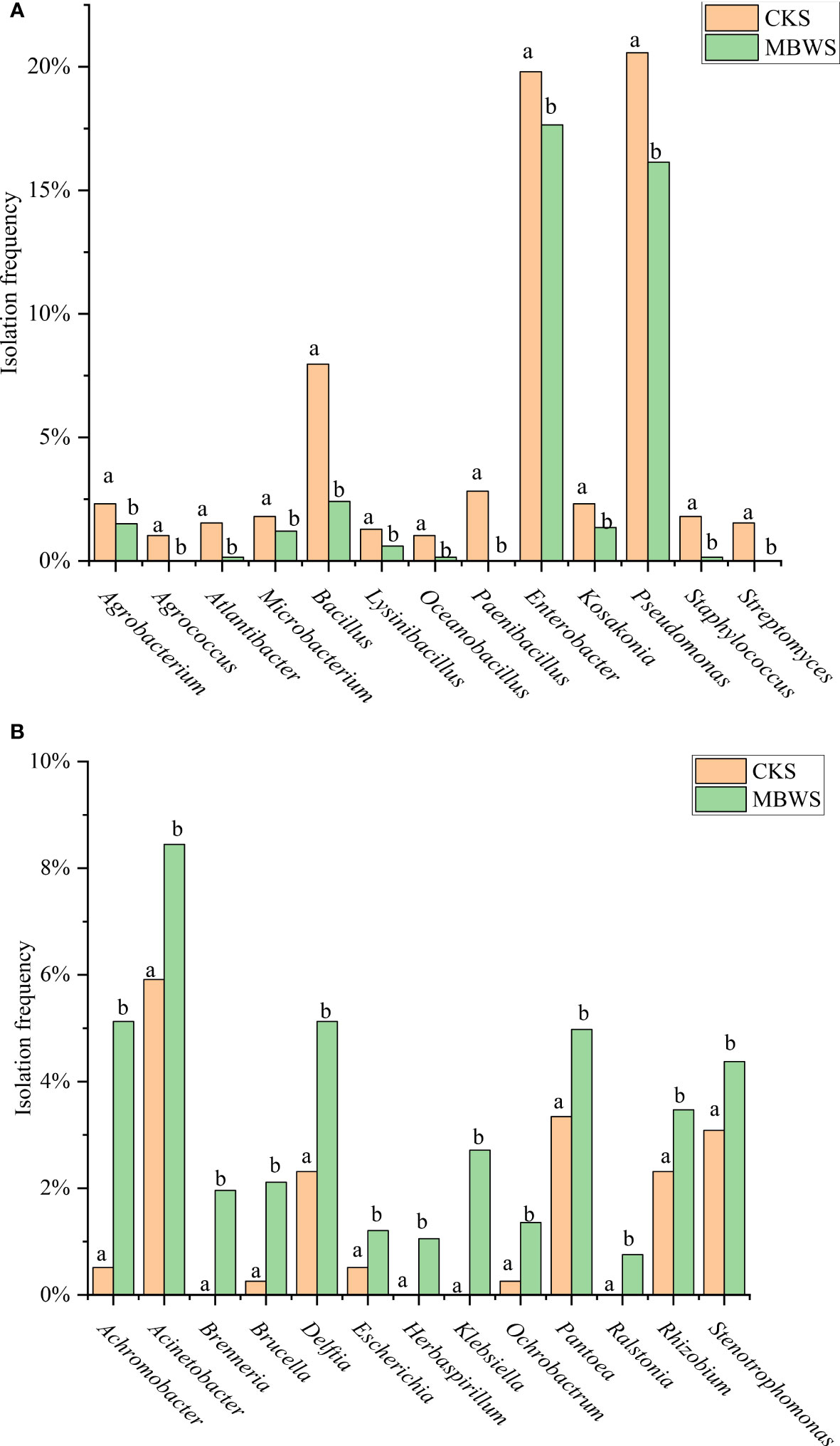
Figure 4 Isolation rates of abundant bacteria isolated from healthy and diseased mulberry. (A) Bacterial species and classification rates greater than CK in MBWS; (B) Bacterial species and classification rates greater than MBWS in CK; MBWS: mulberry bacterial wilt sample. CKS: Healthy samples (healthy mulberry samples). Bars with different letters indicate a significant difference between means by one-way analysis of variance (ANOVA) and least significant difference (LSD) tests (P< 0.05).
The isolation frequencies of Agrobacterium, Agrococcus, Atlantibacter, Microbacterium, Bacillus, Lysinibacillus, Oceanobacillus, Paenibacillus, Enterobacter, Kosakonia, Pseudomonas, Staphylococcus and Streptomyces were significantly higher (P<0.05) in the CKS compared to the MBWS group. Additionally, Agrococcus, Paenibacillus, and Streptomyces were not isolated in the MBWS group (Figure 4B).
The distribution of four main types of pathogenic bacteria in mulberries
In order to explore the main group of pathogenic bacteria causing bacterial wilt of mulberry, the distribution of Ralstonia, Enterobacter, Klebsiella, and Pantoea in 35 diseased samples was analyzed (Figure 5). From these diseased samples, Ralstonia, Enterobacter, Klebsiella, and Pantoea were isolated from 6 (17.14%), 30 (85.71%), 12 (34.28%) and 12 (34.28%) diseased samples, respectively. From the 30 diseased samples in which Enterobacter was isolated, Klebsiella, Pantoea, and Ralstonia were isolated from 10, 9, and 5 samples, respectively.

Figure 5 Isolation of Ralstonia, Enterobacter, Klebsiella, and Pantoea in different mulberry bacterial wilt samples.
Phylogenetic analysis of four main types of pathogenic bacteria
Classification was based on 16S rDNA sequences of Ralstonia, Enterobacter, Klebsiella, and Pantoea (Figure 6). Ralstonia was mainly concentrated in the RSSC and R. pickettii (Figure 6A). There were two main groups of Enterobacter: the ECC (E. kobei, E. chengduensis, E. chuandaensis, E. hormaechei, E. cloacae, E. sichuanensis, E. roggenkampii, E. ludwigii, and E. cancerogenerus.) and E. lignolyticus (Figure 6B). Klebsiella species were mainly divided into K. michiganensis and K. oxytoca (Figure 6C). Pantoea species were mainly clustered into two groups i.e., P. dispersa and P. anthophila (Figure 6D).
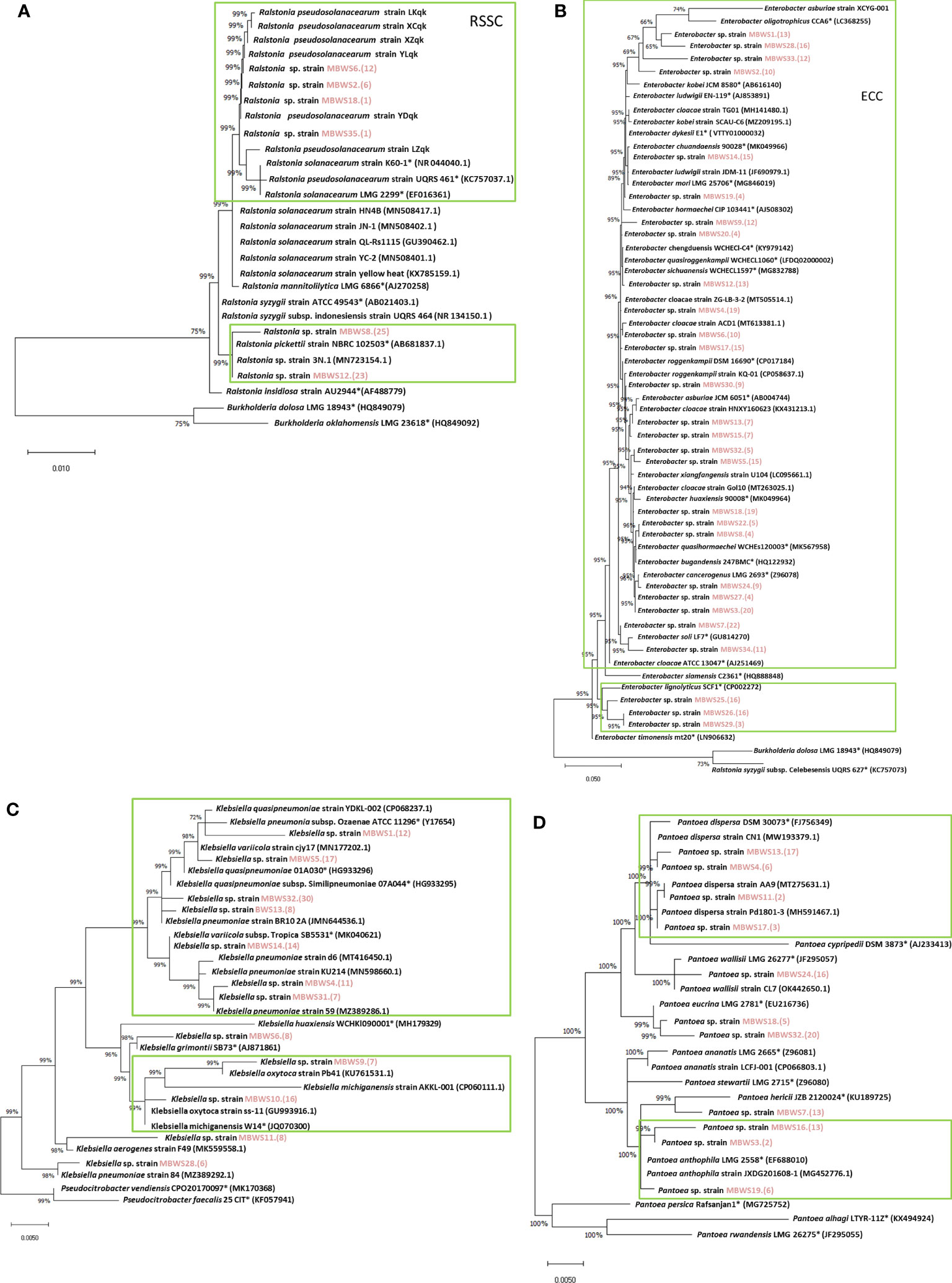
Figure 6 Phylogenetic trees of Ralstonia (A), Enterobacter (B), Klebsiella (C), and Pantoea (D) based on 16S rRNA genes. “*” indicates the representative species; the red marks are the isolates of this study.
Pathogenicity test of four main types of pathogenic bacteria
To further understand the role of Ralstonia, Enterobacter, Klebsiella and Pantoea in mulberry wilt, the pathogenicity test was conducted. As shown in Table 5, it was found that the average pathogenicity rate of Ralstonia derived from the MBWS was found to be 60.13%. The pathogenicity rate of Ralstonia with 16S rRNA accumulated in the RSSC (R. solanacearum species complex) clade was higher than 43.33%, while the pathogenicity rate of Ralstonia aggregated in R. pickettii was 0% (Figure 6A). The average pathogenicity rate of Enterobacter derived from the MBWS was found to be 44.89%. From these, the main pathogenic group was concentrated in the ECC (E. cloacae complex) (Figure 6B), with greatly varying pathogenicity rates between them. The average pathogenicity rate of Klebsiella derived from the MBWS was found to be 44.76%. From these, the main pathogenic groups were K. michiganensis, K. quasipneumoniae, K. oxytoca and K. pneumoniae (Figure 6C), with greatly varying pathogenicity rates between them. The average pathogenicity rate of Pantoea derived from the MBWS was found to be 6.79%. From these, P. ananatis strain LCFJ-001 had the highest pathogenicity rate of 38.33%, while the others showed 0% pathogenicity rate (Figure 6D).
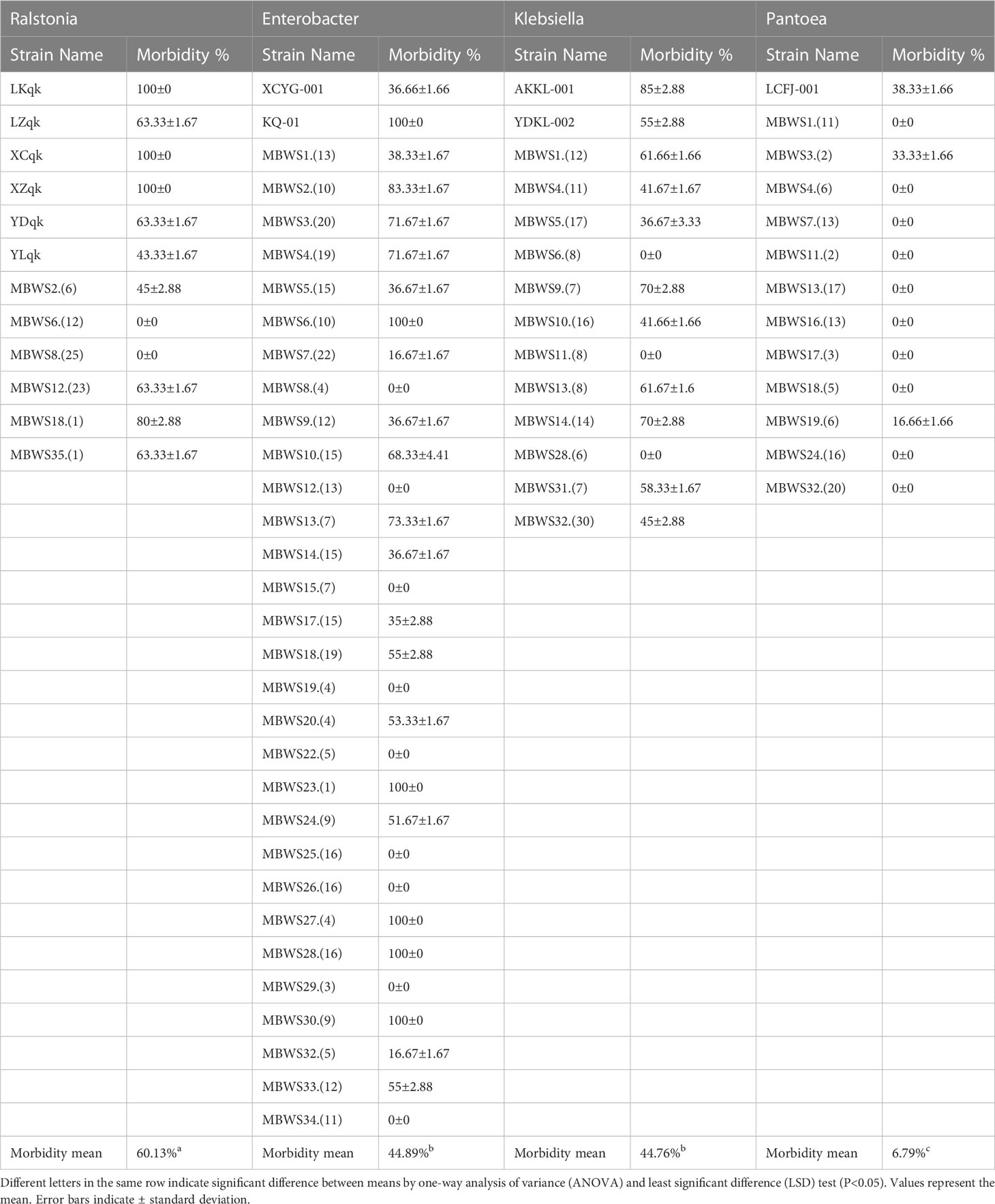
Table 5 Pathogenicity tests of Ralstonia, Enterobacter, Klebsiella, and Pantoea.
Discussion
Plant bacterial wilt is generally considered a highly destructive xylem disease caused by the R. solanacearum complex (RSSC). However, the advancement of bacterial wilt research shows that, in addition to other pathogens, the E. cloacae complex (ECC) can also cause bacterial wilt in African marigoldx (Jeevan et al., 2022), ginger (Cosmas et al., 2016) and mulberry plants (Wang et al., 2008; Zhu et al., 2011). Although the pathogenic bacteria of mulberry bacterial wilt are said to be complex and diverse, they mainly include Ralstonia (Pan et al., 2013), Enterobacter (Wang et al., 2008; Wang et al., 2010; Zhu et al., 2010; Zhu et al., 2011; Zhou et al., 2021), Klebsiella (Luo et al., 2022) and Pantoea (Yuan et al., 2023a). In order to better elucidate the interaction between the microbiome and mulberry, we used combined metagenomic sequencing and a culture-dependent approaches to explore the composition and diversity of bacterial communities in mulberry bacterial wilt samples.
We found 19 phyla and 112 genera in the diseased and healthy mulberry rhizosphere soil and xylem using Illumina HiSeq2500 sequencing. In contrast, four phyla and 97 genera were isolated and characterized using a culture-dependent approach. This discrepancy in the result infers that this phenomenon maybe linked to the inherent limitation of the culture-dependent method, as it is not entirely possible to isolate all xylem bacteria due to the limitation of the medium. On the contrary, it has been said that the metagenomic sequencing method can compensate for this limitation of the culture-dependent method (Zhang et al., 2020). Based on the results of metagenomic sequencing and Shannon, Chao-1, Simpson, and OTU, it was observed that the number and diversity of microbial flora in rhizosphere soil and xylem of healthy mulberry were higher than those of the diseased mulberry samples. Suhaimi et al. (2017) and Tao et al. (2022) also found that the diseased samples had a lower microbial diversity compared to the healthy samples. However, Kaushal et al. (2020) found contrasting result and reported higher OTU richness and diversity in the symptomatic roots. It is generally believed that a low microbial diversity in microbial communities favors pathogen invasion (Locey and Lennon, 2016). This argument is supported by finding of our recent report in which we found that the diversity of endophytes in highly resistant or moderately resistant varieties of mulberry bacterial wilt was significantly higher compared to the weakly resistant or susceptible varieties (Yuan et al., 2023b). This evidence also supports finding of the present study and demonstrate a potential link between diversity of microbial species and susceptibility.
Interestingly, both metagenomic sequencing and culture-dependent approaches revealed that Proteobacteria was a main phylum in both diseased and healthy mulberry rhizosphere soils and xylem, followed by Firmicutes and Actinobacteria. Proteobacteria, Firmicutes, and Actinobacteria were essential components of bacteria in healthy and diseased mulberry xylem. This finding is in line with the evidence reported by Yuan et al. (2023b); Xu et al. (2019) and Ou et al. (2019). The culture-dependent method also revealed that Proteobacteria and Bacteroidetes had greater (P<0.05) abundance in the diseased mulberry xylem compared to the healthy samples. In contrast, Actinobacteria and Firmicutes had greater (P<0.05) abundance in the healthy xylem compared to the diseased samples. Interestingly, Kaushal et al. (2020) have reported that the abundance of Proteobacteria and Actinobacteria showed a similar trend in the banana Mchare cultivar. Suhaimi et al. (2017) have also reported that the healthy samples had higher richness of Proteobacteria than the diseased samples.
At the subordination level, metagenomic sequencing revealed that Erwinia, Pseudomonas, Ralstonia, and Acinetobacter were the dominant genera, accounting for more than 1% of the eight samples tested. On the other hand, the culture-dependent approach revealed that Enterobacter, Pseudomonas, Acinetobacter, Delftia, Pantoea, Stenotrophomonas, Rhizobium, Bacillus, Agrobacterium, Kosakonia, and Microbacterium accounted for more than 1% of the microbial populations in healthy and diseased mulberry xylem. Overall, Pseudomonas and Acinetobacter were found to be the main constituent groups of the mulberry microbiome. This finding is supported by similar evidence reported by previous studies of Xu et al. (2019) and Ou et al. (2019), who reported that Pseudomonas was indeed an essential endophytic flora of mulberry. This evidence is further reinforced by reports of Suhaimi et al. (2017) and Kaushal et al. (2020) who also showed that Pseudomonas was an essential component of the banana bacterial flora. In our previous study, we found that Pseudomonas was one of the component of the endophytic flora in mulberry, but had no obvious control effect on the bacterial wilt of mulberry trees caused by E. roggenkampii strain KQ-01 (Yuan et al., 2023b). Acinetobacter was found to be an endophyte in mulberry and its proportion was significantly higher in mulberry varieties susceptible to bacterial wilt compared to the resistant varieties. In addition, the control rate of Acinetobacter against bacteria wilt caused by E. roggenkampii strain KQ-01 was higher than 80% (Yuan et al., 2023b). Seemingly, these results are contrasting and highlight that the precise roles played by Pseudomonas and Acinetobacter in plants need to be elucidated in future focused research.
Intriguingly, both metagenomic sequencing and culture-dependent methods employed in the present study showed that the proportion of Pseudomonas in the rhizosphere soil and xylem of healthy mulberry was higher compared to the diseased mulberry samples. Similarly, Suhaimi et al. (2017) showed that the abundance of Pseudomonas in the healthy banana samples was higher compared to the diseased samples. Using metagenomic sequencing, we found that the proportion of Erwinia bacteria in the rhizosphere soil and xylem of the diseased mulberry was higher compared to the healthy mulberry samples. However, this result was not supported by finding of the culture-dependent method. This finding is reinforced by evidence of our previous study in which a similar phenomenon was observed in mulberry samples (Yuan et al., 2023b).
In addition, the culture-dependent method revealed that the abundance of many opportunistic pathogens and drug-resistant bacteria was significantly higher in the xylem of the diseased samples compared to their healthy counterparts. Infections in humans have been reported mostly with opportunistic pathogens, including Achromobacter (Menetrey et al., 2021), Acinetobacter (Amorim and Nascimento, 2017), Brucella (Roop et al., 2021), Delftia (Deb et al., 2020), Escherichia (Bhatt et al., 2019), Herbaspirillum (Bloise et al., 2021), Klebsiella (Rodríguez–Medina et al., 2019), Ochrobactrum (Bratschi et al., 2020), Pantoea (Cobo et al., 2021), Ralstonia (Ryan and Adley, 2014) and Stenotrophomonas (Menetrey et al., 2021). Moreover, Acinetobacter (Shin et al., 2020), Escherichia (Tang et al., 2022), Klebsiella (Dong et al., 2022), Pantoea (Yoshimura et al., 2022) and Stenotrophomonas (Ferreira et al., 2020) have been shown to have multidrug resistance.
In the present study, Herbaspirillum, Klebsiella, and Ralstonia were not isolated in the healthy mulberry xylem, indicating that these bacteria might have invaded after infection. Similar results were obtained by Hu et al. (2020), who found an increase in the relative abundance of Ralstonia, Stenotrophomonas and Achromobacter in the infected samples compared to the healthy samples. Although we suspect that the overuse of agricultural antibiotics and untreated farmyard manure exacerbates this situation, the precise underlying basis of this phenomenon remains to be explored. In addition, Brenneria was only isolated in the diseased mulberry samples but not in the healthy xylem. Brenneria has been reported to be a pathogen of woody plants that can cause cankers in plants including walnut (Poret–Peterson et al., 2019), oak (Denman et al., 2012), willow (Maes et al., 2009), alder (Maes et al., 2009) and poplar (Li et al., 2015). Currently, Brenneria is rarely reported in mulberry, and whether this pathogen is emerging as a new pathogen of mulberry still needs further investigation.
The culture-dependent method showed that many bacteria that have been reported to promote plant growth or control bacterial wilt were present in the mulberry samples. These included: Agrobacterium (Soares et al., 2020), Microbacterium (Singh and Singh, 2019), Bacillus (Im et al., 2020), Lysinibacillus (Lelapalli et al., 2021), Oceanobacillus (Alhindi and Albdaiwi, 2022), Paenibacillus (Abdallah et al., 2019), Enterobacter (Anand et al., 2021), Kosakonia (Brock et al., 2018), Pseudomonas (Zhuo et al., 2022) and Streptomyces (Olanrewaju and Babalola, 2019). In agreement with our finding, Hu et al. (2020) also reported similar results. They found that the relative abundance of Pseudomonas, Bacillus, and Falsibacillus, which are generally considered beneficial to plants, was significantly higher in the healthy mulberry samples compared to the diseased samples. This group of bacteria can be considered as a bank of beneficial microbial flora of mulberry. Interestingly, in the present study, the abundance of these bacteria was lower in the diseased samples compared to the healthy mulberry samples.
We further revealed that Enterobacter was the most widely distributed among the four types of pathogenic bacteria, accounting for 85.71%, followed by Klebsiella and Pantoea, which accounted for 34.28%. In contrast, Ralstonia accounted for the lowest (17.14%) proportion. This result indicated that Enterobacter might be the primary pathogen group causing bacterial wilt of mulberry, however, further focused research is needed to reinforce this evidence and gain more insights in this domain. Based on the 16S rDNA sequence and its pathogenicity, Ralstonia was mainly clustered into two clades, the RSSC and R. pickettii. The pathogenicity of Ralstonia clustered in the same clade as the RSSC was greater than 45%, while clustered in the other clade, R. pickettii showed no pathogenicity. Meanwhile, Enterobacter was mainly clustered into the ECC and E. lignolyticus. A total of 73.91% of Enterobacter bacteria clustering in the ECC showed pathogenicity. E. lignolyticus clustered in one clade and showed no pathogenicity. Klebsiella was mainly clustered into two clades centered on K. pneumoniae, K. quasipneumoniae, K. oxytoca, and K. michiganensis, and both showed pathogenicity. Pantoea mainly clustered into two clades centered on P. dispersa and P. anthophila and did not show strong pathogenicity. However, P. ananatis strain LCFJ-001 (CP066803.1) which was discovered earlier (Yuan et al., 2023a) by our laboratory was shown to be pathogenic, with a pathogenicity rate of 38.33%. The RSSC, ECC, K. pneumoniae, K. quasipneumoniae, K. oxytoca, K. michiganensis, and P. ananatis were found to be the main components of the pathogenic bacteria of mulberry bacterial wilt.
During the RSSC infection, the Sol system can be regulated to produce an acylated homoserine lactone (AHL) quorum signaling factor, which is ubiquitous in various gram-negative bacteria, but it is poorly studied in the RSSC (Flavier et al., 1997). When AHL reaches a critical concentration, it diffuses into the cell to bind transcriptional regulators and activates other virulence regulators (Baltenneck et al., 2021). Density-dependent signaling systems centered on AHL are standard in gram-negative bacteria and have been reported in Enterobacter (Shastry et al., 2018), Klebsiella (Hosny and Fadel, 2021), and Pantoea (Jiang et al., 2015). It remains to be explored if there is a possibility that the RSSC can secrete enough AHL through the Sol regulation system to cooperate with other pathogenic bacteria and to infect together.
Conclusion
The llumina HiSeq2500 sequencing and traditional culture medium approaches employed in the present study revealed that the bacterial diversity of healthy mulberry was higher compared to the diseased mulberry. The phyla Proteobacteria, Firmicutes and Actinobacteria constituted an important component of bacteria in the healthy and diseased mulberry. In addition, the abundance of many opportunistic pathogens and drug-resistant bacteria was significantly higher in the diseased samples compared to the healthy counterparts. It was found that the RSSC, ECC, K. pneumoniae, K. quasipneumoniae, K. oxytoca, K. michiganensis, and P. ananatis were the main components of the pathogenic bacteria of mulberry wilt. From these, the ECC was found to be the most widely distributed in the diseased samples. This study provides reference data for further focused research on the bacterial wilt of mulberry and other plants.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA911049; OP990608-OP990981;OP989957-OP990607.
Author contributions
TY wrote the initial draft of manuscript, conceived experiment design, performed experiments, data analysis and implementation. IHQ participated in data analysis, interpretation of the results and revised and edited the draft. JHL, HY, PY, XZ, WL, and YQ collected materials, and assisted in the experiment and data analysis. JPL provided experimental platform and support, project supervision, and funding. All authors contributed to the article and approved the submitted version.
Funding
The research was financially supported by the China Agriculture Research System (CARS-18-ZJ0304).
Acknowledgments
We thank all lab mates who supported during this project.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1206691/full#supplementary-material
References
Abdallah, Y., Yang, M., Zhang, M., Masum, M. M., Ogunyemi, S. O., Hossain, A., et al. (2019). Plant growth promotion and suppression of bacterial leaf blight in rice by Paenibacillus polymyxa Sx3. Lett. Appl. Microbiol. 68 (5), 423–429. doi: 10.1111/lam.13117
Alhindi, T., Albdaiwi, R. (2022). Draft genome sequence of Oceanobacillus jordanicus strain GSFE11, a halotolerant plant growth–promoting bacterial endophyte isolated from the Jordan valley. Evol. Bioinform. 18, 1–6. doi: 10.1177/11769343211071114
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI–BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 (17), 3389–3402. doi: 10.1093/nar/25.17.3389
Amorim, A. M., Nascimento, J. D. (2017). Acinetobacter: an underrated foodborne pathogen? J. Infect. Dev. Countr. 11 (02), 111–114. doi: 10.3855/jidc.8418
Anand, G., Bhattacharjee, A., Shrivas, V. L., Dubey, S., Sharma, S. (2021). ACC deaminase positive Enterobacter–mediated mitigation of salinity stress, and plant growth promotion of Cajanus cajan: A lab to field study. Physiol. Mol. Biol. Plants 27 (7), 1547–1557. doi: 10.1007/s12298–021–01031–0
Baltenneck, J., Reverchon, S., Hommais, F. (2021). Quorum sensing regulation in phytopathogenic bacteria. Microorganisms 9 (2), 239. doi: 10.3390/microorganisms9020239
Bhatt, S., Egan, M., Critelli, B., Kouse, A., Kalman, D., Upreti, C. (2019). The evasive enemy: Insights into the virulence and epidemiology of the emerging attaching and effacing pathogen Escherichia albertii. Infect. Immun. 87 (1), e00254–e00218. doi: 10.1128/IAI.00254–18
Bloise, I., Guedez–López, G. V., Tejedor–Rodríguez, M., Romero–Gómez, M. P., García–Rodríguez, J., Mingorance, J., et al. (2021). Bloodstream infection due to Herbaspirillum sp.: Case series and review of literature. Eur. J. Clin. Microbiol. 40, 779–785. doi: 10.1007/s10096–020–04075–4
Bratschi, C., Ly, T., Weber, A., Meuli–Simmen, C., Conen, A., Mauler, F. (2020). Ochrobactrum anthropi Infection of the hand. J. Handb. Surg. Glob. 2, 365–367. doi: 10.1016/j.jhsg.2020.08.006
Bredow, C., Azevedo, J. L., Pamphile, J. A., Mangolin, C. A., Rhoden, S. A. (2015). In silico analysis of the 16S rRNA gene of endophytic bacteria, isolated from the aerial parts and seeds of important agricultural crops. Genet. Mol. Res. 14 (3), 9703–9721. doi: 10.4238/2015.August.19.3
Brock, A. K., Berger, B., Schreiner, M., Ruppel, S., Mewis, I. (2018). Plant growth–promoting bacteria Kosakonia radicincitans mediate anti–herbivore defense in Arabidopsis thaliana. Planta 248, 1383–1392. doi: 10.1007/s00425–018–2964–0
Chan, E. W., Lye, P. Y., Wong, S. K. (2016). Phytochemistry, pharmacology, and clinical trials of Morus alba. Chin. J. Natural Med. 14 (1), 17–30. doi: 10.3724/SP.J.1009.2016.00017
Cobo, F., González, A., Pérez–Carrasco, V., García–Salcedo, J. A. (2021). Pantoea stewartii: A new pathogen as a cause of bacteremia? Enferm. Infecc. Microbiol. Clin. 40 (5), 278–280. doi: 10.1016/j.eimce.2021.03.005
Cosmas, L. L., Atong, M., Poili, E. (2016). Preliminary studies towards identification of ginger wilt disease in Sabah, Malaysia. Pertanika J. Trop. Agr. Sci. 39 (3), 373–380. Available at: https://web.s.ebscohost.com/abstract?direct=true&profile=ehost&scope=site&authtype=crawler&jrnl=15113701&asa=Y&AN=119631060&h=2pCKvL4kTEhP6Ep%2fbJiooj3w79p98h2NcEDL63gvm2pwmEIpvRgUfj4v2VQeAItpTSqVVIrT7JJGikFZxyCZjw%3d%3d&crl=c&resultNs=AdminWebAuth&resultLocal=ErrCrlNotAuth&crlhashurl=login.aspx%3fdirect%3dtrue%26profile%3dehost%26scope%3dsite%26authtype%3dcrawler%26jrnl%3d15113701%26asa%3dY%26AN%3d119631060.
Dai, F., Luo, G., Li, Z., Wei, X., Wang, Z., Lin, S., et al. (2020). Physiological and transcriptomic analyses of mulberry (Morus atropurpurea) response to cadmium stress. Ecotox. Environ. Safe. 205, 111298. doi: 10.1016/j.ecoenv.2020.111298
Dai, F., Wang, Z., Luo, G., Tang, C. (2016). Transcriptional analysis of different mulberry cultivars in response to Ralstonia solanacearum. Can. J. Forest. Res. 46 (2), 152–162. doi: 10.1139/cjfr–2015–0235
Deb, A. K., Chavhan, P., Chowdhury, S. S., Sistla, S., Sugumaran, R., Panicker, G. (2020). Endophthalmitis due to Delftia acidovorans: An unusual ocular pathogen. Indian. J. Ophthalmol. 68 (11), 2591–2594. doi: 10.4103/ijo.IJO_373_20
Denman, S., Brady, C., Kirk, S., Cleenwerck, I., Venter, S., Coutinho, T., et al. (2012). Brenneria goodwinii sp. nov., associated with acute oak decline in the UK. Int. J. Syst. Evol. Micr. 62, 2451–2456. doi: 10.1099/ijs.0.037879–0
Dong, Z., Guo, Y., Yu, C., Zhixian, Z., Rongli, M., Deng, W., et al. (2021). The dynamics in rhizosphere microbial communities under bacterial wilt resistance by mulberry genotypes. Arch. Microbiol. 203 (3), 1107–1121. doi: 10.1007/s00203-020-02098-1
Dong, N., Yang, X., Chan, E. W., Zhang, R., Chen, S. (2022). Klebsiella species: Taxonomy, hypervirulence and multidrug resistance. EBioMedicine 79, 103998. doi: 10.1016/j.ebiom.2022.103998
Ferreira, M. A., Pereira, M. L., Dos–Santos, K. V. (2020). Drug–induced tolerance: the effects of antibiotic pre–exposure in Stenotrophomonas maltophilia. Future Microbiol. 15 (7), 497–508. doi: 10.2217/fmb–2019–0253
Flavier, A. B., Ganova–Raeva, L. M., Schell, M. A., Denny, T. P. (1997). Hierarchical autoinduction in Ralstonia solanacearum: Control of acyl–homoserine lactone production by a novel autoregulatory system responsive to 3–hydroxypalmitic acid methyl ester. J. Bacteriol. 179 (22), 7089–7097. doi: 10.4049/jimmunol.175.7.4433
Hosny, R. A., Fadel, M. A. (2021). Detection of quorum sensing n–acyl–homoserine lactone molecules produced by different resistant Klebsiella pneumoniae isolates recovered from poultry and different environmental niches. Appl. Biochem. Biotechnol. 193 (10), 3351–3370. doi: 10.1007/s12010–021–03605–w
Hu, Q., Tan, L., Gu, S., Xiao, Y., Xiong, X., Zeng, W., et al. (2020). Network analysis infers the wilt pathogen invasion associated with non–detrimental bacteria. NPJ Biofilms Microbi. 6 (1), 8. doi: 10.1038/s41522–020–0117–2
Im, S. M., Yu, N. H., Joen, H. W., Kim, S. O., Park, H. W., Park, A. R., et al. (2020). Biological control of tomato bacterial wilt by oxydifficidin and difficidin–producing Bacillus methylotrophicus DR–08. Pestic. Biochem. Phys. 163, 130–137. doi: 10.1016/j.pestbp.2019.11.007
Jeevan, U., Kurian, P. S., Sreelatha, U., Mathew, D., Narayanankutty, C. (2022). Morphological, symptomatological and molecular characterization of Enterobacter cloacae causing bacterial wilt in African marigold (Tagetes erecta L.). Indian Phytopathol. 75 (1), 279–285. doi: 10.1016/j.pestbp.2019.11.007
Ji, X., Lu, G., Gai, Y., Zheng, C., Mu, Z. (2008). Biological control against bacterial wilt and colonization of mulberry by an endophytic Bacillus subtilis strain. FEMS Microbiol. Ecol. 65 (3), 565–573. doi: 10.1111/j.1574-6941.2008.00543.x
Jiang, W., Lin, Y., Qian, L., Miao, L., Liu, B., Ge, X., et al. (2022). Mulberry leaf meal: A potential feed supplement for juvenile Megalobrama amblycephala “Huahai No. 1”. Fish Shellfish Immunol. 128, 279–287. doi: 10.1016/j.fsi.2022.07.022
Jiang, J., Wu, S., Wang, J., Feng, Y. (2015). AHL–type quorum sensing and its regulation on symplasmata formation in Pantoea agglomerans YS19. J. Basic Microbiol. 55 (5), 607–616. doi: 10.1002/jobm.201400472
Kaushal, M., Mahuku, G., Swennen, R. (2020). Metagenomic insights of the root colonizing microbiome associated with symptomatic and non–symptomatic bananas in Fusarium wilt infected fields. Plants 9 (2), 263. doi: 10.3390/plants9020263
Lai, W., Zeng, X., Tan, B., Wu, G., Chen, J., Guan, W., et al. (1979). Preliminary identification of pathogenic bacteria of mulberry bacterial wilt. Guangdong Silk Newslett. 2, 21–24. Available at: https://kns.cnki.net/KCMS/detail/detail.aspx?dbname=cjfd1979&filename=gdcy197902006&dbcode=cjfq.
Lelapalli, S., Baskar, S., Jacob, S. M., Paranthaman, S. (2021). Characterization of phosphate solubilizing plant growth promoting rhizobacterium Lysinibacillus pakistanensis strain PCPSMR15 isolated from Oryza sativa. Curr. Res. Microbial Sci. 2, 100080. doi: 10.1016/j.crmicr.2021.100080
Li, Y., Fang, W., Xue, H., Liang, W. X., Wang, L. F., Tian, G. Z., et al. (2015). Brenneria populi sp. nov., isolated from symptomatic bark of Populus×euramericana canker. Int. J. Syst. Evol. Micr. 65 (2), 432–437. doi: 10.1099/ijs.0.066068–0
Li, M., Pommier, T., Yin, Y., Wang, J., Gu, S., Jousset, A., et al. (2022). Indirect reduction of Ralstonia solanacearum via pathogen helper inhibition. ISME J. 16 (3), 868–875. doi: 10.1038/s41396–021–01126–2
Locey, K. J., Lennon, J. T. (2016). Scaling laws predict global microbial diversity. PNAS. 113 (21), 5970–5975. doi: 10.7287/peerj.preprints.1451v1
Luo, L., Huang, Y., Liu, J. (2022). Genome sequence resources of Klebsiella michiganensis AKKL–001, Which causes bacterial blight of mulberry. Mol. Plant Microbe In. 35 (4), 349–351. doi: 10.1094/MPMI–09–21–0222–A
Maes, M., Huvenne, H., Messens, E. (2009). Brenneria salicis, the bacterium causing watermark disease in willow, resides as an endophyte in wood. Environ. Microbiol. 11 (6), 1453–1462. doi: 10.1111/j.1462–2920.2009.01873.x
Maqsood, M., Anam Saeed, R., Sahar, A., Khan, M. I. (2022). Mulberry plant as a source of functional food with therapeutic and nutritional applications: A review. J. Food Biochem. 46 (11), e14263. doi: 10.1111/jfbc.14263
Menetrey, Q., Sorlin, P., Jumas–Bilak, E., Chiron, R., Dupont, C., Marchandin, H. (2021). Achromobacter xylosoxidans and Stenotrophomonas maltophilia: Emerging pathogens well–armed for life in the cystic fibrosis patients’ lung. Genes 12 (5), 610. doi: 10.3390/genes12050610
Meng, Q., Qi, X., Fu, Y., Chen, Q., Cheng, P., Yu, X., et al. (2020). Flavonoids extracted from mulberry (Morus alba L.) leaf improve skeletal muscle mitochondrial function by activating AMPK in type 2 diabetes. J. Ethnopharmacol. 248, 112326. doi: 10.1016/j.jep.2019.112326
Olanrewaju, O. S., Babalola, O. O. (2019). Streptomyces: implications and interactions in plant growth promotion. Appl. Microbiol. Biotechnol. 103 (3), 1179–1188. doi: 10.1007/s00253–018–09577–y
Ou, T., Xu, W. F., Wang, F., Strobel, G., Zhou, Z. Y., Xiang, Z. H., et al. (2019). A microbiome study reveals seasonal variation in endophytic bacteria among different mulberry cultivars. Comput. Struct. Biotec. 17, 1091–1100. doi: 10.1016/j.csbj.2019.07.018
Pan, Z. C., Xu, J., Prior, P., Xu, J. S., Zhang, H., Chen, K. Y., et al. (2013). Development of a specific molecular tool for the detection of epidemiologically active mulberry causing–disease strains of Ralstonia solanacearum phylotype I (historically race 5–biovar 5) in China. Eur. J. Plant Pathol. 137 (2), 377–391. doi: 10.1007/s10658–013–0249–9
Poret–Peterson, A. T., McClean, A. E., Chen, L., Kluepfel, D. A. (2019). Complete genome sequences of Brenneria rubrifaciens strain 6D370 and Brenneria nigrifluens strain ATCC 13028, causative agents of bark cankers in walnut. Microbiol. Resour. Ann. 8 (37), e00597–e00519. doi: 10.1128/MRA.00597–19
Rodríguez–Medina, N., Barrios–Camacho, H., Duran–Bedolla, J., Garza–Ramos, U. (2019). Klebsiella variicola: An emerging pathogen in humans. Emerg. Microbes Infect. 8 (1), 973–988. doi: 10.1080/22221751.2019.1634981
Roop, R. M., Barton, I. S., Hopersberger, D., Martin, D. W. (2021). Uncovering the hidden credentials of Brucella Virulence. Microbiol. Mol. Biol. R. 85 (1), e00021–e00019. doi: 10.1128/MMBR.00021–19
Ryan, M. P., Adley, C. C. (2014). Ralstonia spp.: Emerging global opportunistic pathogens. Eur. J. Clin. Microbiol. 33 (3), 291–304. doi: 10.1007/s10096–013–1975–9
Shastry, R. P., Dolan, S. K., Abdelhamid, Y., Vittal, R. R., Welch, M. (2018). Purification and characterisation of a quorum quenching AHL–lactonase from the endophytic bacterium Enterobacter sp. CS66. FEMS Microbiol. Lett. 365 (9), fny054. doi: 10.1093/femsle/fny054
Shin, B., Park, C., Park, W. (2020). Stress responses linked to antimicrobial resistance in Acinetobacter species. Appl. Microbiol. Biot. 104 (4), 1423–1435. doi: 10.1007/s00253–019–10317–z
Singh, T., Singh, D. K. (2019). Rhizospheric Microbacterium sp. P27 showing potential of lindane degradation and plant growth promoting traits. Curr. Microbiol. 76 (7), 888–895. doi: 10.1007/s00284–019–01703–x
Soares, R., Trejo, J., Lorite, M. J., Figueira, E., Sanjuán, J., Videira, E., et al. (2020). Diversity, phylogeny and plant growth promotion traits of nodule associated bacteria isolated from lotus parviflorus. Microorganisms 8 (4), 499. doi: 10.3390/microorganisms8040499
Suhaimi, N. S. M., Goh, S. Y., Ajam, N., Othman, R. Y., Chan, K. G., Thong, K. L. (2017). Diversity of microbiota associated with symptomatic and non–symptomatic bacterial wilt–diseased banana plants determined using 16S rRNA metagenome sequencing. World J. Microb. Biot. 33 (9), 1–10. doi: 10.1007/s11274–017–2336–0
Tang, B., Chang, J., Chen, Y., Lin, J., Xiao, X., Xia, X., et al. (2022). Escherichia fergusonii, an underrated repository for antimicrobial resistance in food animals. Microbiol. Spect. 10 (1), e0161721. doi: 10.1128/spectrum.01617–21
Tao, J., Yu, S., Jin, J., Lu, P., Yang, Z., Xu, Y., et al. (2022). The wilt pathogen induces different variations of root–associated microbiomes of plant. Front. Plant Sci. 13, 1023837. doi: 10.3389/fpls.2022.1023837
Wang, G. F., Praphat, K., Xie, G. L., Zhu, B., Li, B., Liu, B., et al. (2008). Bacterial wilt of mulberry (Morus alba) caused by Enterobacter cloacae in China. Plant Dis. 92 (3), 483. doi: 10.1094/pdis–92–3–0483b
Wang, G., Xie, G., Zhu, B., Huang, J., Liu, B., Kawicha, P., et al. (2010). Identification and characterization of the Enterobacter complex causing mulberry (Morus alba) wilt disease in China. Eur. J. Plant Pathol. 126 (4), 465–478. doi: 10.1007/s10658–009–9552–x
Xie, S., Vallet, M., Sun, C., Kunert, M., David, A., Zhang, X., et al. (2020). Biocontrol potential of a novel endophytic bacterium from mulberry (Morus) tree. Front. Bioeng. Biotechnol. 7. doi: 10.3389/fbioe.2019.00488
Xu, W., Wang, F., Zhang, M., Ou, T., Wang, R., Strobel, G., et al. (2019). Diversity of cultivable endophytic bacteria in mulberry and their potential for antimicrobial and plant growth–promoting activities. Microbiol. Res. 229, 126328. doi: 10.1016/j.micres.2019.126328
Yoshimura, M., Tokushige, C., Maruyama, J. I., Kawano, Y., Ishikura, H., Matsunaga, A., et al. (2022). Emerging resistance to beta–lactams in Pantoea ananatis isolated from an immunocompetent patient with bacteremia. Diagn. Micr. Infect. Dis. 102 (4), 115633. doi: 10.1016/j.diagmicrobio.2022.115633
Yuan, T., Huang, Y., Luo, L., Wang, J., Li, J., Chen, J., et al. (2023a). Complete genome sequence of Pantoea ananatis strain LCFJ–001, isolated from bacterial wilt mulberry. Plant Dis. 1, 1–22. doi: 10.1094/PDIS–10–22–2473–A
Yuan, T., Qazi, I. H., Yang, P., Zhang, X., Li, J., Liu, J. (2023b). Analysis of endophytic bacterial flora of mulberry cultivars susceptible and resistant to bacterial wilt using metagenomic sequencing and culture-dependent approach. World J. Microbiol. Biotechnol. 39 (6), 163. doi: 10.1007/s11274-023-03599-z
Zhang, C. B., Ren, C. H., Wang, Y. L., Wang, Q. Q., Wang, Y. S., Weng, Q. B. (2020). Uncovering fungal community composition in natural habitat of Ophiocordyceps sinensis using metagenomic sequencing and culture–dependent approaches. BMC Microbiol. 20 (1), 331. doi: 10.21203/rs.3.rs–28393/v2
Zhou, Y., Yang, H., Liu, J. (2021). Complete genome sequence of Enterobacter roggenkampii strain KQ–01, isolated from bacterial wilt–resistant mulberry cultivar YS283. Plant Dis. 105 (3), 688–690. doi: 10.1094/PDIS–07–20–1468–A
Zhu, B., Lou, M. M., Xie, G. L., Wang, G. F., Zhou, Q., Wang, F., et al. (2011). Enterobacter mori sp. nov., associated with bacterial wilt on Morus alba L. Int. J. Syst. Evol. Micr. 61 (11), 2769–2774. doi: 10.1099/ijs.0.028613–0
Zhu, B., Wang, G., Xie, G., Zhou, Q., Zhao, M., Praphat, K., et al. (2010). Enterobacter spp.: A new evidence causing bacterial wilt on mulberry. Sci. China Life Sci. 53 (2), 292–300. doi: 10.1007/s11427–010–0048–x
Keywords: bacterial wilt, drug-resistant bacteria, Enterobacter cloacae complex, Klebsiella, mulberry, opportunistic pathogens, RSSC
Citation: Yuan T, Qazi IH, Li J, Yang P, Yang H, Zhang X, Liu W and Liu J (2023) Analysis of changes in bacterial diversity in healthy and bacterial wilt mulberry samples using metagenomic sequencing and culture-dependent approaches. Front. Plant Sci. 14:1206691. doi: 10.3389/fpls.2023.1206691
Received: 16 April 2023; Accepted: 21 July 2023;
Published: 23 August 2023.
Edited by:
Mamoona Rauf, Abdul Wali Khan University Mardan, PakistanReviewed by:
Vijay K. Sharma, Agricultural Research Organization (ARO), IsraelMahananda Chutia, Central Muga Eri Research and Training Institute (CMERTI), India
Copyright © 2023 Yuan, Qazi, Li, Yang, Yang, Zhang, Liu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiping Liu, liujiping@scau.edu.cn
†These authors share first authorship