 Jianxia Jiang1†
Jianxia Jiang1† Pengfei Xu2†
Pengfei Xu2† Junying Zhang1Yanli Li1
Junying Zhang1Yanli Li1 Xirong Zhou1Meiyan Jiang1Jifeng Zhu1Weirong Wang1
Xirong Zhou1Meiyan Jiang1Jifeng Zhu1Weirong Wang1 Liyong Yang1*
Liyong Yang1*- 1Crop Breeding and Cultivation Research Institute, Shanghai Academy of Agricultural Sciences, Shanghai, China
- 2Joint International Research Laboratory of Metabolic and Developmental Sciences, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai, China
Rapeseed is the third leading source of edible oil in the world. Genic male sterility (GMS) lines provide crucial material for harnessing heterosis for rapeseed. GMS lines have been widely used successfully for rapeseed hybrid production. The physiological and molecular mechanism of pollen development in GMS lines of rapeseed (Brassica napus L.) need to be determined for the creation of hybrids and cultivation of new varieties. However, limited studies have focused on systematically mining genes that regulate the pollen development of GMS lines in B. napus. In the present study, to determine the stage at which pollen development begins to show abnormality in the GMS lines, we performed semi-thin section analysis of the anthers with five pollen development stages. The results indicated that the abnormal pollen development in DGMS lines might start at the meiotic stage, and abnormal pollen development in RGMS lines probably occurred before the tetrad stage. To investigate the critical genes and pathways involved in pollen development in GMS lines, we constructed and sequenced 24 transcriptome libraries for the flower buds from the fertile and sterile lines of two recessive GMS (RGMS) lines (6251AB and 6284AB) and two dominant GMS (DGMS) lines (4001AB and 4006AB). A total of 23,554 redundant DEGs with over two-fold change between sterile and fertile lines were obtained. A total of 346 DEGs were specifically related to DGMS, while 1,553 DEGs were specifically related to RGMS. A total of 1,545 DEGs were shared between DGMS and RGMS. And 253 transcription factors were found to be differentially expressed between the sterile and fertile lines of GMS. In addition, 6,099 DEGs possibly related to anther, pollen, and microspore development processes were identified. Many of these genes have been reported to be involved in anther and microspore developmental processes. Several DEGs were speculated to be key genes involved in the regulation of fertility. Three differentially expressed genes were randomly selected and their expression levels were verified by quantitative PCR (qRT-PCR). The results of qRT-PCR largely agreed with the transcriptome sequencing results. Our findings provide a global view of genes that are potentially involved in GMS occurrence. The expression profiles and function analysis of these DEGs were provided to expand our understanding of the complex molecular mechanism in pollen and sterility development in B. napus.
Introduction
Brassica napus L. is an evolutionarily young allopolyploid (AACC) brassicaceous species, which was derived from Brassica rapa and Brassica oleracea ~7,500 years ago by natural hybridization and chromosome doubling (Wang et al., 2011; Chalhoub et al., 2014). Rapeseed is the third leading source of edible oil in the world. Rapeseed oil has high oleic acid, linoleic acid, and linolenic acid content and with low erucic acid and glucosinolate contents. Through natural and artificial selection, rapeseed has become a multipurpose crop (i.e., vegetable and oil for human; forage crop for livestock) (Allender and King, 2010). China is a supplier of rapeseed and is the oldest country that is involved in the cultivation of rapeseed in the world. The annual rapeseed plantation area and total output of oilseed yield in China have always been at the forefront of the world (Wang et al., 2007). The improvement of yield is mainly attributed to the large-scale application of hybrid rapeseed. More than 60% of rapeseed production involves hybrid rapeseed. The main resource used in heterosis in rapeseed contains genic male sterility (GMS), cytoplasmic male sterility (CMS), self-incompatibility, and chemical male sterility (Kaul, 1988).
GMS contains dominant GMS (DGMS) and recessive GMS (RGMS), which retain complete and stable male sterility. In cabbage (B. oleracea var. capitata), a male-sterile line (79-399-3) that carries a dominant QTL (Ms-cd-1) for male sterility was identified from line 79-399. This QTL causes abnormal callose degeneration and arrest of microspore cell division at the tetrad stage (Lou et al., 2007). Eventually, a novel gene (Bol357N3) was identified as a candidate gene for Ms-cd1 (Liang et al., 2017). In rapeseed, DGMS and RGMS are widely used for hybrid rapeseed production (Fu, 2000). In 2005, Song et al. found multiple alleles in one locus inheritance in the DGMS line 609AB. In this model, Mf, Ms, and ms are three alleles at the same locus, with a relationship of Mf > Ms > ms. The recessive allele is associated with normal fertility. Multiple-allele DGMS is widely used for hybrid rapeseed seed production through the construction of a three-line hybrid system (Song et al., 2006; Liu et al., 2008). In the RGMS system, most inbred lines can restore their fertility and hybrids with strong heterosis are easily bred. In 1998, 9012AB was used in a three-line hybrid production system, and its male sterility was thought to be controlled by three independent genes, namely, BnMs3, BnMs4, and BnRf (Chen et al., 1998). In 2012, Dong et al. demonstrated that the BnMs4 locus is an actual allele of BnRf, which was designated as BnRfa. The allele from 9012A was designated as BnRfb, and the allele from temporary maintainer was designated as BnRfc. BnRfa was dominant over BnRfb, and BnRfb was dominant over BnRfc (Dong et al., 2012). In total, the DGMS and RGMS lines are valuable resource, and they have been used successfully for rapeseed hybrid production in the world.
As a male gametophyte, pollen participates in sexual reproduction in flowering plants and directly influences seed generation (McCormick, 2004). Pollen development is a complex process that involves the formation of microsporocyte in the anther, male meiosis, and microspores being released from the tetrads, pollen wall development, and pollen maturation. Then, the mature pollen grains are released after anther dehiscence (Gómez et al., 2015). Fertility and pollen development involve many regulatory pathways and related genes. In Arabidopsis, 13,977 male gametophyte-expressed mRNAs were identified using Affymetrix ATH1 genome arrays. Among these mRNAs, 1,355 specific mRNAs were identified as male gametophyte-specific transcripts, including AtPAB3 (At1g22760), AtPAB6 (At3g16380), AtPAB7 (At2g36660), AteIF2-B3 (At3g07920), AteIF4G-like (At4g30680), and AteIF6-2 (At2g39820) (Honys and Twell, 2004). Two transcriptome factors AMS and AtBZIP34 control pollen wall formation during Arabidopsis anther or pollen development (Sorensen et al., 2003; Gibalova et al., 2009; Xu et al., 2010). Five transcription factors (MYB80, MS1, AMS, DYT1, and TDF1) regulate tapetum development and further influence the development of microspores by controlling callose dissolution, pollen extine formation, and tapetal programmed cell death (Zhang et al., 2006; Yang et al., 2007; Zhang et al., 2007; Zhu et al., 2008; Gu et al., 2014). In Brassica campestris, through comparative transcriptome analysis and ChIP-sequencing, 8,288 genes were differentially expressed in at least one stage of sterile floral buds compared with the fertile buds of B. campestris. Among these DEGs, Bra016531 was associated with tapetum development and function during pollen development (Shen et al., 2019). Moreover, various genes in B. campestris are involved in pollen development, such as BcMF2 (Huang et al., 2009), BcMF3 (Liu et al., 2006), BcMF4 (Liu et al., 2006), up to BcMF23 (Lin et al., 2017). Among these genes, BcMF6 (Zhang et al., 2008), BcMF2 (Huang et al., 2009), and BcMF9 (Huang et al., 2009) are polygalacturonase genes, which participate in pollen extine or intine development. BcMF21 is involved in pollen extine development and germination (Jiang et al., 2014). In B. napus, a global dynamic transcriptome programming of rapeseed anther was performed at different development stages. The results indicated that 35,470 transcripts were expressed in at least one of the anther development stages from the pollen mother cell stage to the mature pollen stage, suggesting that much more genes that are involved in pollen development need to be studied (Li et al., 2016). These findings provide important information for understanding the gene regulatory networks of pollen development. However, the mechanisms of pollen development are still incomplete, and more studies are needed.
In the present study, two RGMS lines (6251AB and 6284AB) and two DGMS lines (4001AB and 4006AB) were used to systematically explore the genes involved in pollen development in GMS lines. First, morphological differences were observed in the flower organs of the fertile and sterile lines. Semi-thin section was conducted to determine the stage at which pollen development begins to show abnormality in the sterile lines. Thereafter, 24 transcriptome libraries were constructed and sequenced for the flower buds from the fertile and sterile lines. DEGs between the fertile and sterile line were screened, and their expression differences were analyzed by transcriptome sequencing. Then, the gene function and related special pathway of the DEGs were analyzed using GO classification and KEGG annotation analysis. Additionally, DEGs that are specifically related to DGMS or RGMS and the shared DEGs involved in both DGMS and RGMS were identified. The differentially expressed TFs were investigated. Screening of DEGs that may be involved in anther and microspore development was conducted and the potential gene functions were analyzed according to the functions of their orthologous Arabidopsis genes. Several potential key DEGs involved in RGMS and DGMS were speculated. These findings would provide a foundation for evaluating the complex molecular mechanisms in pollen development and GMS occurrence in Brassica crops.
Materials and methods
Plant materials
The two recessive genic male sterile lines (6251AB and 6284AB) and two dominant genic male sterile lines (4001AB and 4006AB) of B. napus were used in this study and planted in the experimental farm of Zhuanghang comprehensive experimental station of Shanghai Academy of Agricultural Sciences. The 6251A and 6284A lines are male sterile lines, and 6251B and 6284B are fertile lines. The 4001A and 4006A lines are male sterile lines, and 4001B and 4006B are fertile lines (Jiang et al., 2021). During flowering stage, floral buds at five developing stages were sampled, which were corresponding five pollen development stages (stage I, pollen mother cell stage; stage II, tetrad stage; stage III, uninucleate microspore stage; stage IV, binucleate microspore stage; and stage V, mature pollen stage) based on the bud size (Huang et al., 2008). The floral buds at five developing stages were harvested from more than 10 plants of the eight lines, including 6251A, 6251B, 6284A, 6284B, 4001A, 4001B, 4006A, and 4006B. Then, the samples were quickly frozen in liquid nitrogen and stored at –80°C. Three independent biological replicates were collected for each kind of sample.
Flower morphology and cytological observation of GMS lines
At the flowering stage, the four floral organs of a complete flower were observed under a stereomicroscope (Leica, Germany). Twenty flowers from different lines were collected for measurement. The difference was validated by t test (N=20, P<0.05). The flower buds at five pollen development stages based on the bud size (Huang et al., 2008) were collected and stored in 70% formalin–acetic acid–alcohol fixative solution at 4°C. Semi-thin section observation was performed using the flower buds at five pollen development stages at Wuhan Servicebio Technology Co., Ltd.
Library construction and illumina sequencing
Total RNA was extracted using TRIzol reagent (Invitrogen, USA). For each kind of sample, three biological replicates were employed. RNA samples were with an OD260/OD280 ratio of 2.0. Mix equal amounts of RNA from flower buds at five developmental stages. A total content of more than 2 µg were qualified for transcription library construction (Lin et al., 2019; Huang et al., 2020). Twenty-four sequencing libraries (three biological replicates each for “6251A”, “6251B”, “6284A”, “6284B”, “4001A”, “4001B”, “4006A”, and “4006B”) were constructed and then sequenced using Illumina Hiseq 2500/Miseq at Beijing Novogene Bioinformatics Technology Co. Ltd.
Data analysis
The clean reads were obtained by first filtering the reads with adapters, N-containing reads, and low-quality reads. Then, the sequencing error rate was checked. Lastly, the GC content distribution was checked. Then, the clean reads were mapped to the B. napus genome (http://brassicadb.agridata.cn/brad/) using HISAT2 software (Kim et al., 2015). The distribution of all the reads in genome was analyzed. The novel genes were predicted, and all the gene expression values (FPKM) were analyzed based on the read counts on genome. Pearson correlation analysis between samples was adopted to examine the reliability of experiment and rationality of sample selection. After the gene expression was quantified, the raw readcount was normalized. The p value and false discovery rate (FDR) were calculated using the DESeq2 software (Love et al., 2014). The readcount after normalization (FPKM) was used to screen the DEGs between different samples. P value < 0.05, FDR < 0.01, and |log2(FoldChange)| > 1 based on three biological replicates were considered as DEGs. The online software of Venny 2.1.0 was used to generate the Venn diagrams. To explore the related biological function or pathway of DEGs, gene ontology (GO) enrichment analysis of DEGs was implemented using the clusterProfiler R package, in which gene length bias was corrected. The FDR < 0.05 was considered as significantly enriched. KEGG is a database resource for understanding high-level functions and utilities of the biological system (http://www.genome.jp/kegg/). ClusterProfiler R package was used to test the statistical enrichment of DEGs in KEGG pathways. FDR < 0.05 was defined as significant differential expression.
QRT-PCR
Total RNA (2µg) was reverse-transcribed into cDNA by using reverse transcriptase (TaKaRa) with oligo (dT). The specific primers used in qPCR are listed in Table S8. The qRT-PCR reactions were conducted in the CFX96 real-time system (Bio-Rad, Hercules, CA, USA) by using ChamQ Universal SYBR qPCR Master Mix (Vazyme, China). The 20 µL reaction mixture contained 0.5 µL of cDNA, 7.9 µL of ddH2O, 10 µL of 2X SYBR mixture, 0.8 µL of forward primer, and 0.8 µL of reverse primer. Three biological replicates were employed with three technical replicates for each sample, and the relative expression levels were quantified using the 2−ΔΔCt method (Livak and Schmittgen, 2001).
Results
Morphological and cytological characterization of the fertile and sterile lines
The DGMS (4001AB, 4006AB) and RGMS (6251AB, 6284AB) lines were planted in the field under the same environment. The plant phenotypes of the A and B lines were observed along with their development processes. No significant difference was observed in the growth status during the vegetative growth among different GMS lines. For each GMS line, the flower development in A line was abnormal compared with that in B line (Figure 1). Five main traits of flower organs were examined at representative stages, including sepal length, petal length, petal width, stamen length, and pistil length. For all the four GMS materials (4001AB, 4006AB, 6251AB, and 6284AB), the A lines had shorter sepal, petal, stamen, and narrower petal than the B line. In 6251AB, the A line pistils were shorter, while in other GMS materials, the pistil lengths were not significantly different between the A and B lines (Figure 2).
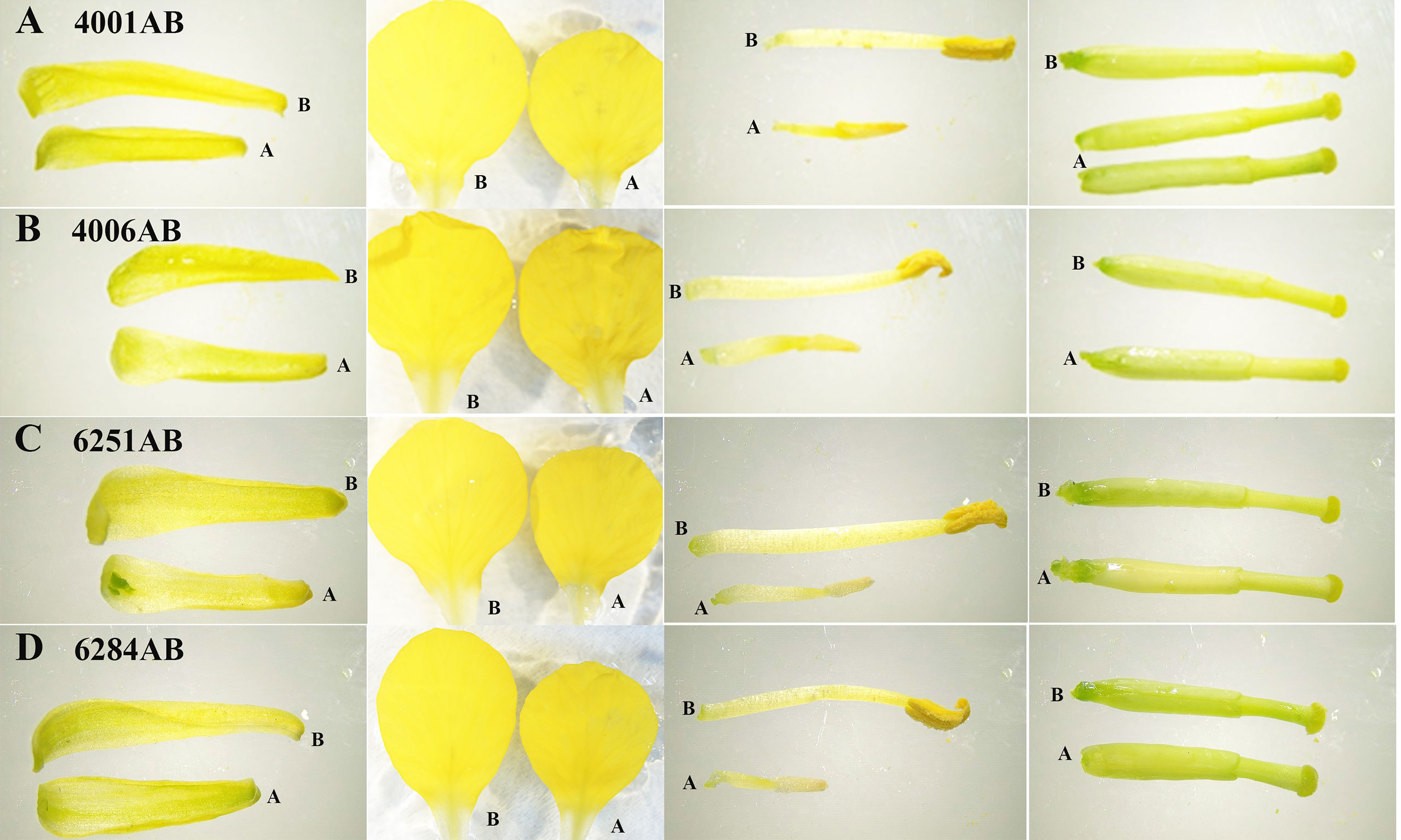
Figure 1 Phenotype observation of flower organs from 4001AB, 4006AB, 6251AB, and 6284AB. (A), flower organs of 4001AB; (B), flower organs of 4006AB; (C), flower organs of 6251AB; and (D), flower organs of 6284AB. The four types of flower organs were sepals, petals, stamens, and pistils. The “A” and “B” letters marked next to the flower organs indicate the organs from A or B line.
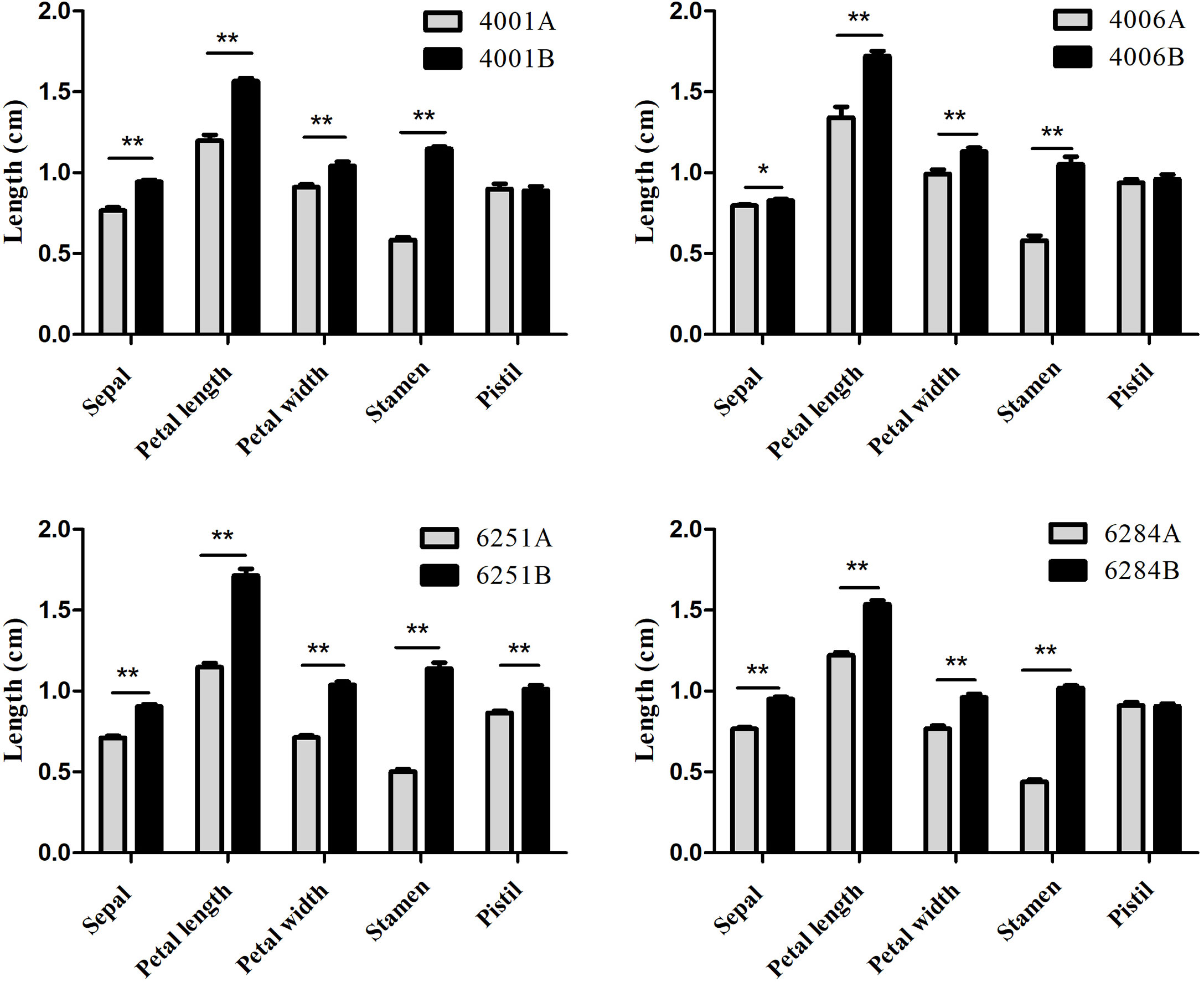
Figure 2 Average lengths of floral organs from 4001AB, 4006AB, 6251AB, and 6284AB. Error bars represent SD. The values in the figure are expressed as the means ± SD from 20 repeats. Differences between A and B line were considered significant (*) if p < 0.05 (Student’s t-test) and highly significant (**) if p < 0.01 (Student’s t-test).
To determine the stage at which the abnormality of pollen development began in the GMS lines, we chose 4001AB and 6251AB, which represent the DGMS and RGMS lines, respectively, and conducted semi-thin section analysis of anthers at five pollen development stages. For 4001AB, no difference and abnormality were detected between 4001A and 4001B at the pollen mother cell stage (Figures 3A, F). At the tetrad stage, 4001B could form normal tetrads, but 4001A did not (Figures 3B, G). At the uninucleate and binucleate microspore stages, the microspores in 4001B were round and full, showing one and two obvious nucleates, respectively (Figures 3H, I). At the trinucleate microspore stage, all the microspores in 4001B could form normal mature pollen grains (Figure 3J). However, in 4001A, the tetrad stage and later stages were not observed. The abnormal pollen mother cells were gradually degraded, and eventually only a little inclusion remained in the pollen sac of 4001A (Figures 3B, C, D, E). Therefore, abnormal pollen development in 4001A might start at the meiotic stage.
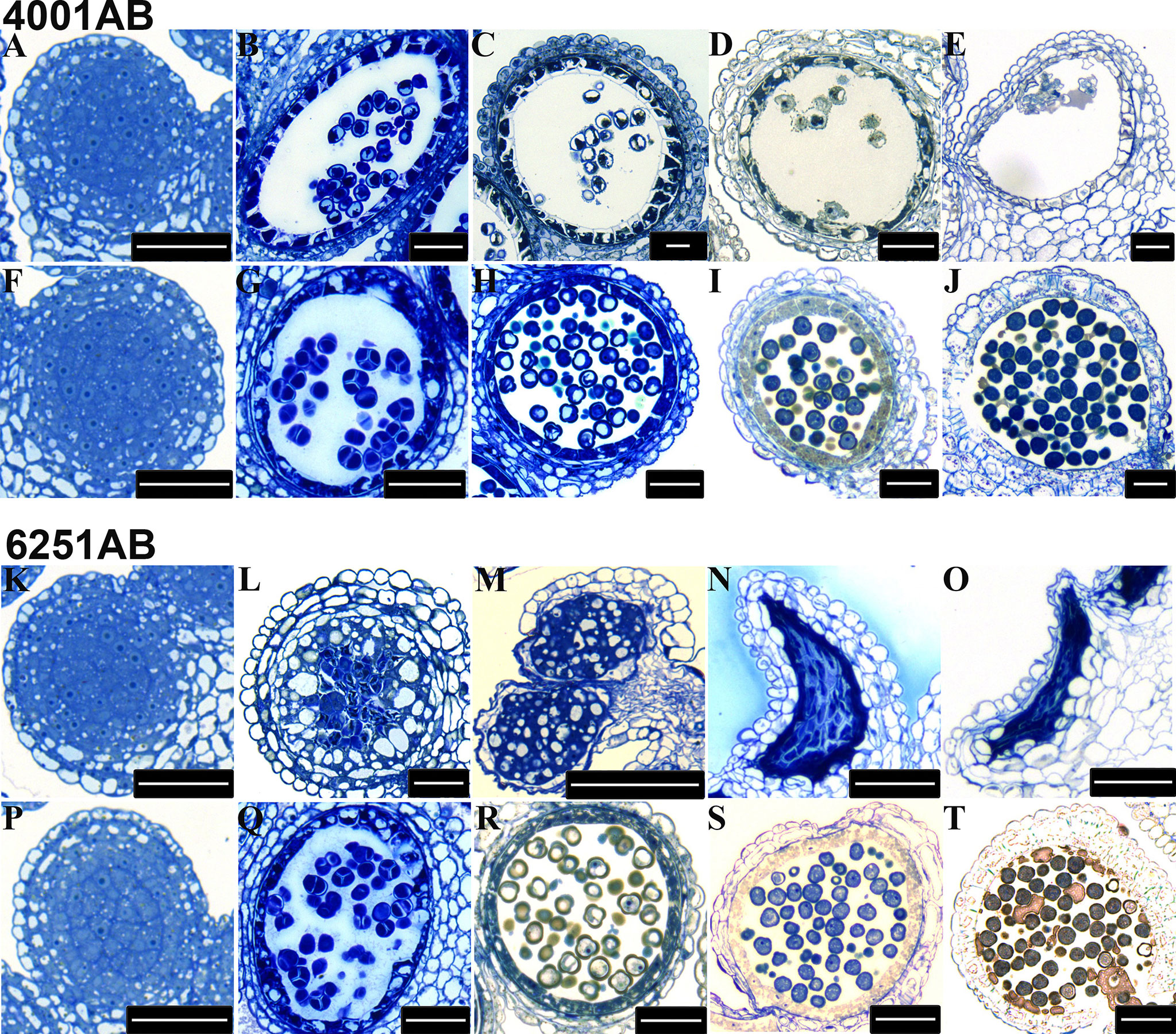
Figure 3 Transverse sections of anthers from the 4001AB and 6251AB lines. The semi-thin sections of anthers from 4001A (A–E) and 4001B (F–J) are shown above. The semi-thin sections of anthers from 6251A (K–O) and 6251B (P–T) are shown below. The microspores at the pollen mother cell stage (A, F, K, and P), tetrad stage (B, G, L, and Q), uninucleate microspore stage (C, H, M, and R), binucleate microspore stage (D, I, N, and S), and trinucleate microspore stage (E, J, O, and T) were observed. Scale bars were 50 µm.
For 6251AB, no abnormality was detected in the A and B line at the pollen mother cell stage (Figures 3K, P). At the tetrad stage, 6251B could form normal tetrads (Figure 3Q). While no tetrads were observed in 6251A, and the tapetal cells were aberrant, swollen and had excess vacuolization (Figure 3L). And the cell contents were messed up in pollen sac (Figure 3L). At the uninucleate microspore stage, the microspores in 6251B developed normally (Figure 3R), while the anther cells showed structural defects and the tapetal cells were degraded and messed up with other degradants in 6251A (Figure 3M). At the binucleate and trinucleate microspore stage, the anthers shrunk seriously and stained into dark blue with degradants (Figures 3N, O). In 6251B, the anthers and microspores were round and full. The microspores developed well and eventually formed normal mature pollen grains (Figures 3S, T).Therefore, abnormal pollen development in 6251A might occur before the tetrad stage.
Transcriptome sequencing and correlation analysis
To identify mRNAs related to pollen and fertile development in DGMS and RGMS lines, transcriptome sequencing was conducted. The flower buds from the sterile (4001A, 4006A) and fertile (4001B, 4006B) lines were collected. The flower buds of the sterile (6251A, 6284A) and fertile (6251B and 6284B) lines were also collected. Three biological replicates were employed for each sample. In total, 24 cDNA libraries were prepared for transcriptome sequencing. As shown in Table 1, 186 G clean bases were obtained, and the clean bases of each library ranged from 5.88 G to 9.46 G with Q30 > 92.86% and GC > 44.41%. The clean reads were then mapped to the B. napus reference genome (http://brassicadb.cn/) with a mapping rate of 89.73%–91.07%. The mapping rate of the unique reads ranged from 86.23% to 87.32%, while the mapping rate of the multi-mapped reads was less than 5% (Table 1). To visualize the underlying structure of the RNA-seq data and emphasize the high degree of correlation between biological replicates, principal component analysis (PCA) and Pearson correlation were performed using the fragments per kilobase of transcript per million mapped reads (FPKM) value. Excellent correlations were observed between A and B lines for each GMS material (Figure S1A). Moreover, PCA analysis show that the data collected from the fertile and sterile lines of DGMS and RGMS lines could be clearly separated. The three biological replications were concentrated well (Figure S1B). Therefore, the RNA-seq data were considered reliable for subsequent analysis.
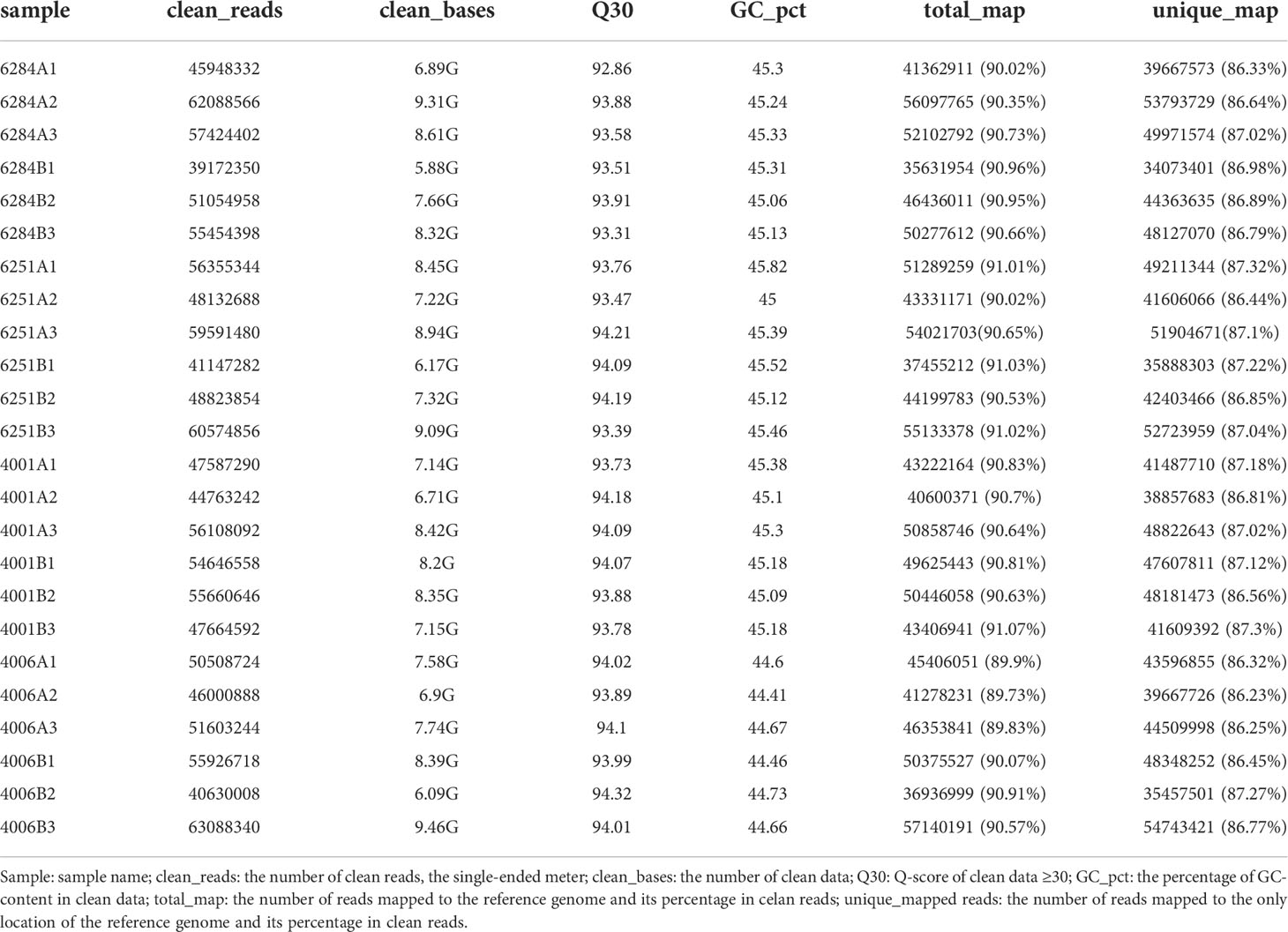
Table 1 The alignment statistics result with the reference genome for all samples.
Differentially expressed genes analysis
The FPKM value of transcripts was used for identifying DEGs (|log2 (fold change)| ≥1, q value < 0.05 and FDR< 0.01) between the fertile and sterile lines (4001B vs. 4001A, 4006B vs. 4006A, 6251B vs. 6251A, and 6284B vs. 6284A). Finally, 23,554 redundant DEGs were obtained. Among these DEGs, 4,820 DEGs were obtained in 4001AB, including 4,251 upregulated genes and 569 downregulated genes in 4001B, compared with 4001A. In total, 4,257 DEGs were obtained in 4006AB, including 3,857 upregulated genes and 400 downregulated genes in 4006B, compared with 4006A. In addition, 6,093 DEGs (5,230 upregulated and 863 downregulated) and 8,384 DEGs (7,347 upregulated and 1,037 downregulated) were obtained in 6251AB and 6284AB, respectively (Figure 4; Table S1). The number of upregulated genes was higher than that of downregulated genes. To explore the DEGs involved in the DGMS and RGMS lines, we conducted a Venn diagram analysis. Based on the comparison between 4001AB and 4006AB, 2,214 (37.6%) genes were commonly upregulated, and 52 (5.7%) genes were commonly downregulated (Figures 5A, B). In addition, 4,196 (50.1%) genes were commonly upregulated, and 283 (17.5%) genes were commonly downregulated in the two RGMS lines (6251AB and 6284AB) (Figures 5C, D). To intuitively show the differences and similarities of the DEGs in flower buds from different GMS lines, we performed hierarchical clustering to represent the expression of DEGs (Figure 6; Table S2). The results showed a significant difference in gene expression profile among the four GMS groups. Most of the DEGs were obviously upregulated in B lines. Some genes were barely expressed in A lines, but they were highly expressed in B lines (Figure S2A). Therefore, a large number of DEGs may play a positive regulatory role in pollen development.
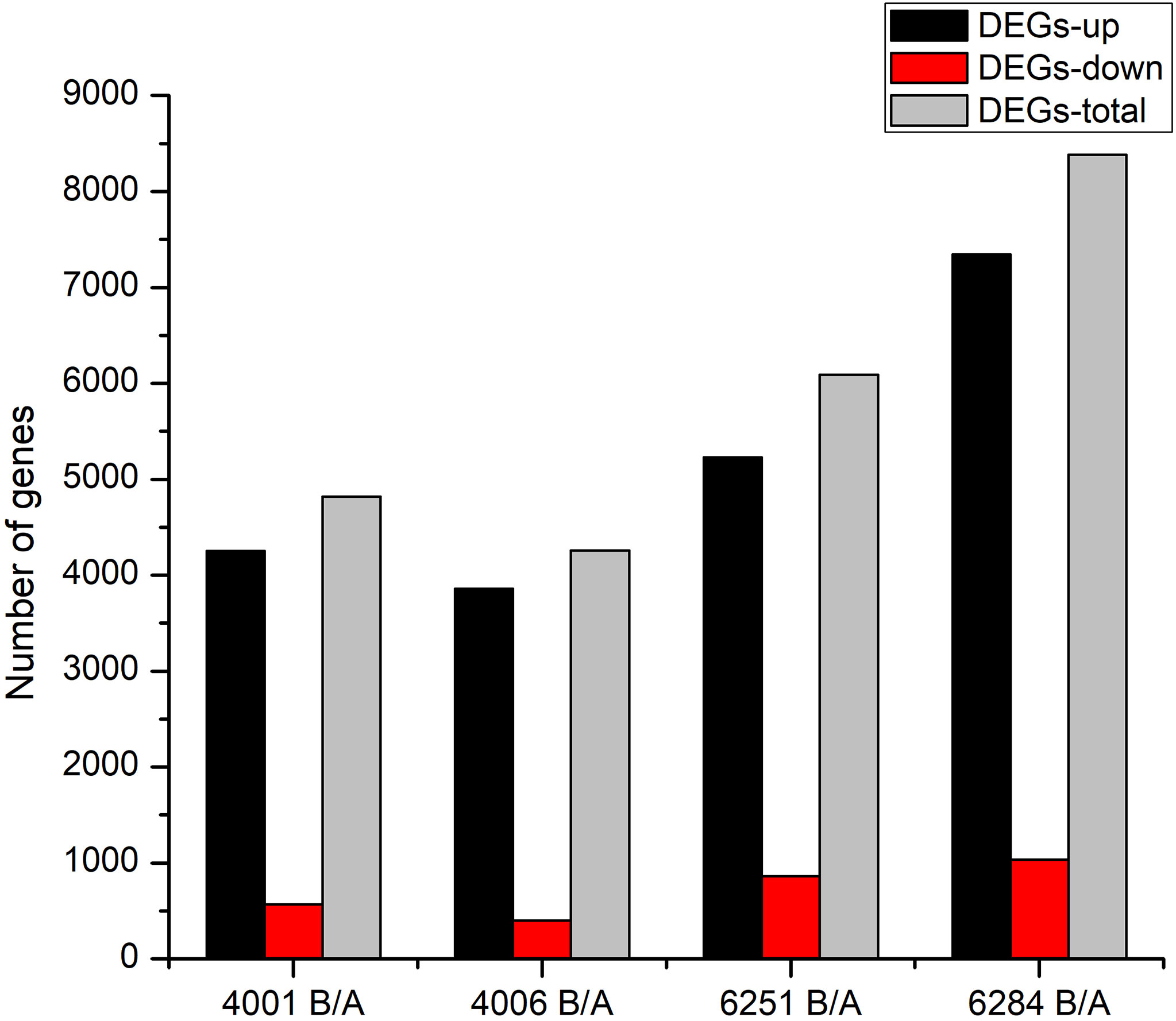
Figure 4 Column diagram shows the numbers of DEGs in four GMS materials. The dark and red columns represent the up- and downregulated DEGs in B lines. The gray columns represent the total DEGs in the corresponding GMS material.
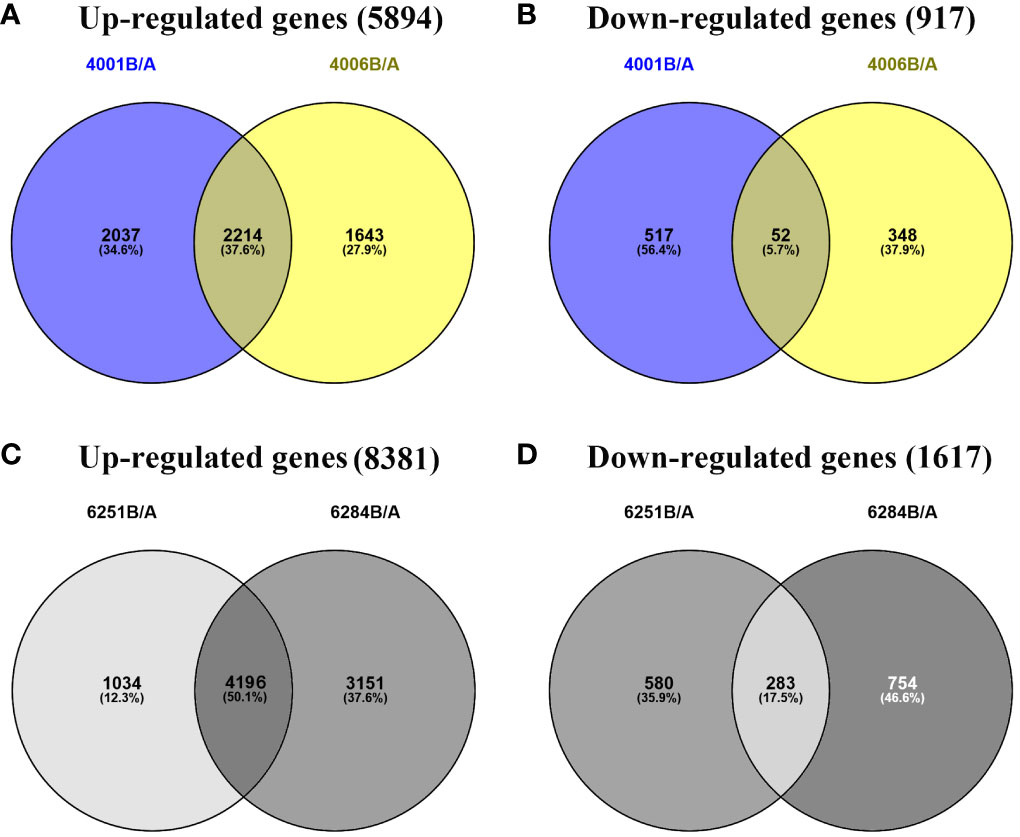
Figure 5 Venn diagrams representing the numbers of DEGs and the overlaps of sets obtained across four comparisons. The up- (A) and downregulated genes (B) in DGMS. The up- (C) and downregulated genes (D) in RGMS.
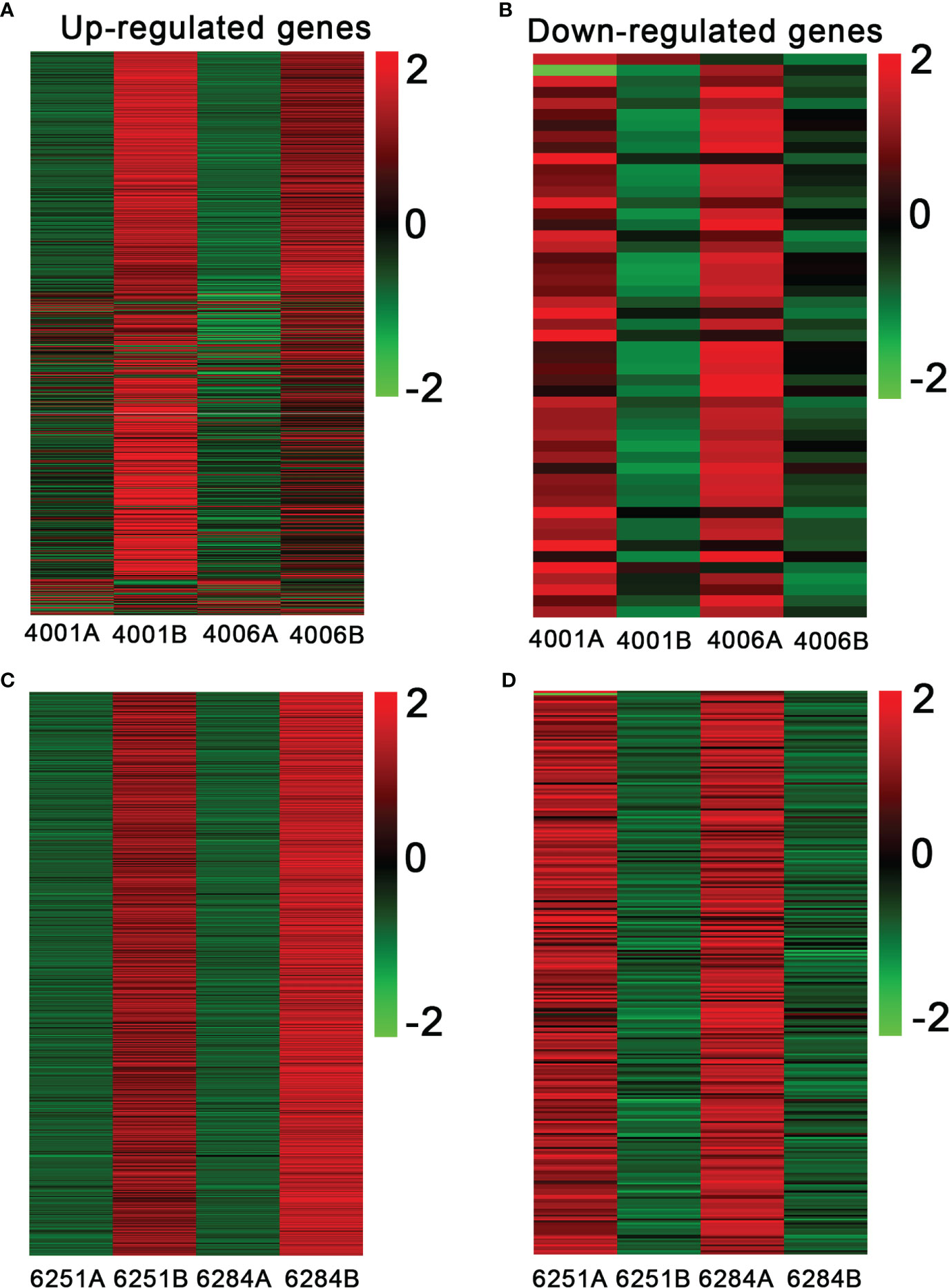
Figure 6 Heatmap showing the expression patterns of differentially expressed genes in the fertile and sterile lines. The up- (A) and downregulated genes (B) in DGMS. The up- (C) and downregulated genes (D) in RGMS. The legend represents the fold change level.
GO function and KEGG pathway analysis of differentially expressed gene
In the present study, an FDR of < 0.05 was used to pick significantly enriched GO terms. The annotated genes were divided into three major functional categories, namely, biological processes (BP), cellular components (CC), and molecular functions (MF, Table S3). The main GO terms were significantly enriched in biological processes. For the DEGs in DGMS (4001AB, 4006AB), the GO terms were significantly enriched in cell wall organization or biogenesis (GO: GO:0071554), external encapsulating structure organization (GO: GO:0045229), cell wall organization (GO: GO:0071555), single-organism carbohydrate metabolic process (GO:0044723), macromolecule catabolic process (GO:0009057), cellular macromolecule catabolic process (GO:0044265), and lipid biosynthetic process (GO:0008610, Figures 7A, B). Interestingly, for the down-regulated DEGs in 4001B, some GO terms related to pollen development, pollination, and fertilization were significantly enriched, including pollination (GO:0009856), pollen-pistil interaction (GO:0009875), and recognition of pollen (GO:0048544) (Figure 7A). The relevant genes included SDR1 (BnaA07g25970D), ARK2 (BnaA07g25960D), RKS2-1 (BnaC08g42100D), RKS2-2 (BnaC08g42090D), and SD1-29 (BnaC01g29480D, Table S3). For the DEGs in RGMS (6251AB, 6284AB), the GO terms were mainly enriched in cell wall organization or biogenesis (GO:0071554), external encapsulating structure organization (GO:0045229), cell wall organization (GO:0071555), cell wall modification (GO:0042545), and single-organism carbohydrate metabolic process (GO:0044723, Figures 7C, D).
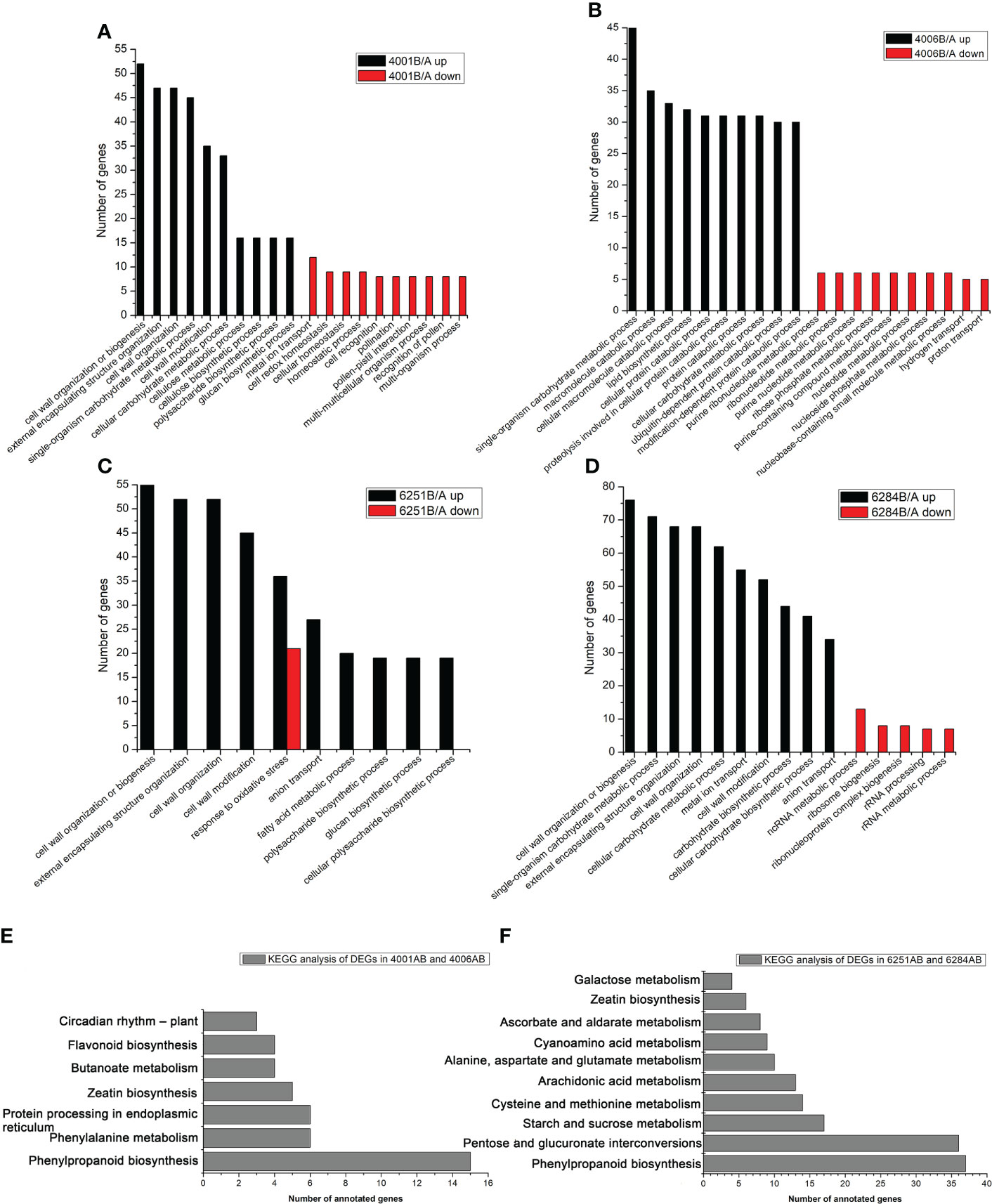
Figure 7 GO and KEGG classification of differentially expressed genes. The enriched GO categories of DEGs in 4001AB (A), 4006AB (B), 6251AB (C), and 6284AB (D). KEGG pathway analysis of DEGs in DGMS (E) and RGMS (F) based on the KEGG data.
KEGG pathway analysis was conducted to investigate whether the DEGs were involved in special pathways. For all the four GMS lines, the most significantly enriched pathway was phenylpropanoid biosynthesis (ath00940). For DGMS lines (4001AB and 4006AB), the most significantly enriched pathway was phenylpropanoid biosynthesis (ath00940), followed by phenylalanine metabolism (ath00360), protein processing in endoplasmic reticulum (ath04141), zeatin biosynthesis (ath00908), and butanoate metabolism (ath00650, Figure 7E). For RGMS lines (6251AB and 6284AB), the most significantly enriched pathways were highlighted in phenylpropanoid biosynthesis (ath00940), pentose and glucuronate interconversions (ath00040), starch and sucrose metabolism (ath00500), cysteine and methionine metabolism (ath00270), and arachidonic acid metabolism (ath00590) (Figure 7F). Based on the comparison between DGMS and RGMS, two KEGG terms were shared, namely, phenylpropanoid biosynthesis (ath00940) and zeatin biosynthesis (ath00908, Table S4). Therefore, a subset of genes potentially played roles in both DGMS and RGMS lines.
Identification of DEGS specifically related to DGMS or RGMS
In the present study, a subset of DEGs was obtained between the A and B line of DGMS lines. However, these genes did not show differential expression between the A and B line in RGMS. These genes were identified as DGMS-related genes, which might specifically function in DGMS. A total of 346 genes were DGMS-related genes, such as BnaC07g11890D (HRS1 HOMOLOG3, HHO3) and BnaA02g29090D (Figures S2B, C). According to the similar screening method as above, 1,553 genes were identified as RGMS-related genes, such as BnaA03g22110D and BnaA07g32020D. Both of the two genes were differentially expressed between the A and B line in RGMS, but they showed similar expression levels between the A and B line in DGMS (Figures S2D, E).
Identification of shared DEGs related to both DGMS and RGMS
To identify the candidate genes involved in both DGMS and RGMS, we compared the DEGs identified in DGMS (4001AB and 4006AB) and RGMS (6251AB and 6284AB). In total, 1,545 DEGs were shared between DGMS and RGMS (Figure 8A). GO enrichment analysis of the 1,545 DEGs showed that they were mainly enriched in single-organism carbohydrate metabolic process (GO:0044723), external encapsulating structure organization (GO:0045229), and cell wall organization (GO:0071555, Figure 8B). To display the expression differences of the shared DEGs in RGMS and DGMS, we selected eight genes and calculated their expression levels by using FPKM values, which were AGL18, AGL66, AGL104, LBD27, ACA7, BnaAnng17790D, BnaC08g05830D, and BnaA07g33840D (Figures 9 and S3).
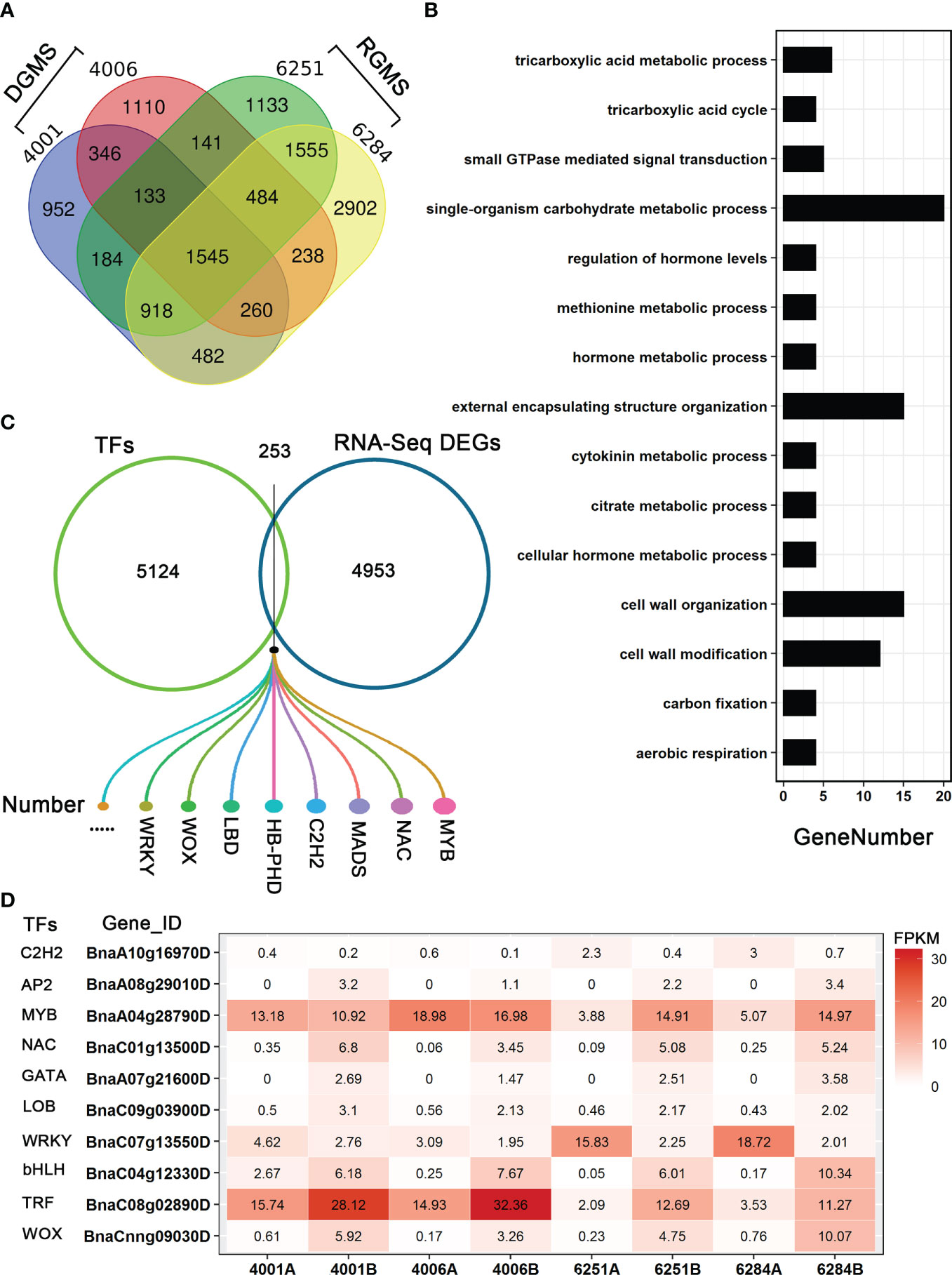
Figure 8 Analysis of DEGs in GMS. (A) The Venn diagram shows the overlap of DEGs in RGMS and DGMS. (B) GO classification of shared DEGs between RGMS and DGMS. (C) Differentially expressed TFs. (D) Ten differentially expressed TFs, which were identified from RNA-seq data. Numbers in each box indicate the FPKM values.
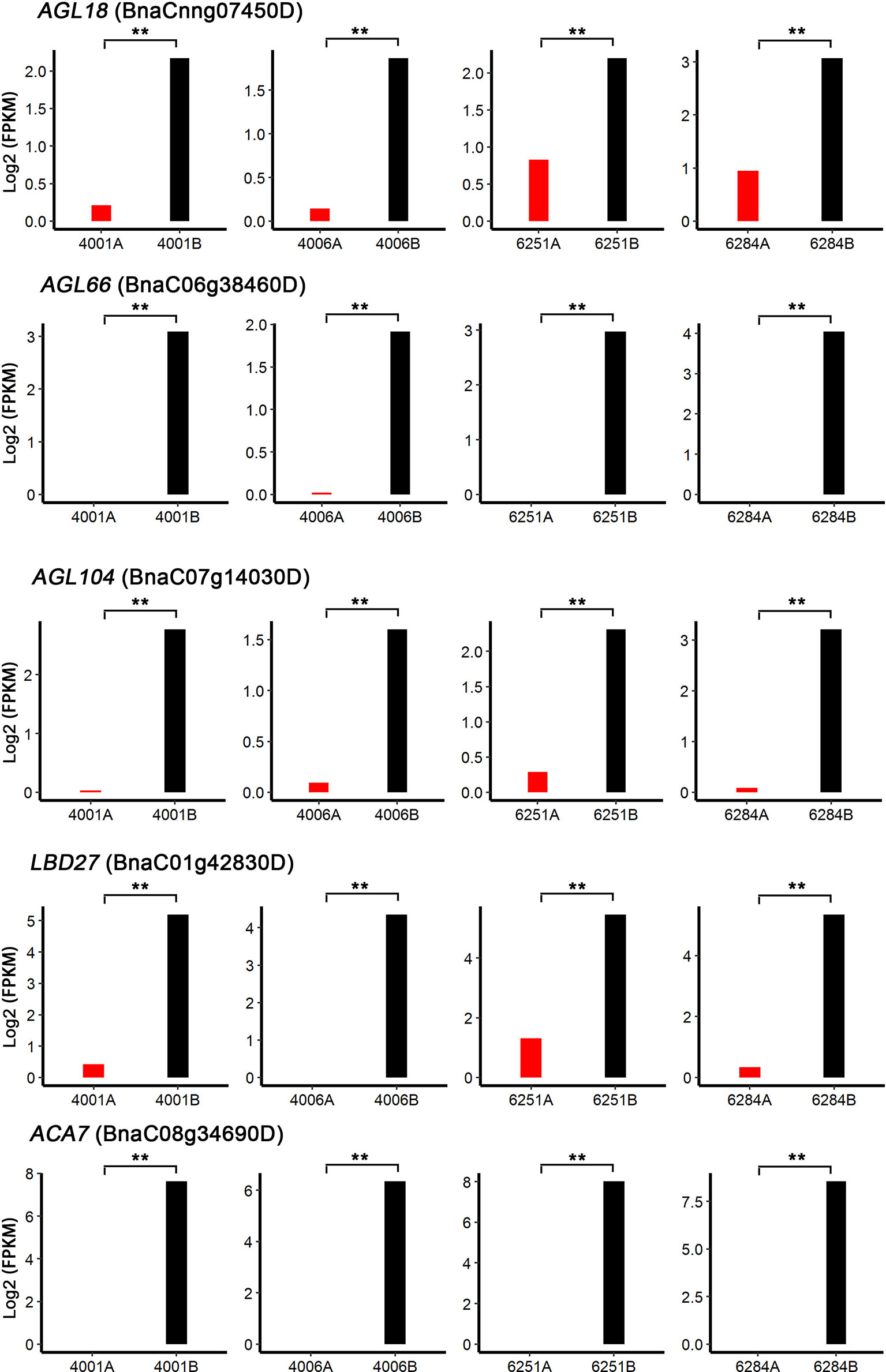
Figure 9 Shared DEGs possibly involved in anther and microspore development. AGL18, AGL66, AGL104, LBD27, and ACA7 were selected to calculate their expression levels by using FPKM values. Significant differences were calculated by performing paired t-test between FPKM values of A and B lines. **P < 0.01.
Identification of differentially expressed TFs
To explore the differentially expressed TFs in GMS lines, we downloaded all the B. napus transcription factors from BnTIR (http://yanglab.hzau.edu.cn/bntir), and they were compared to all the DEGs. As a result, 253 differentially expressed TFs were found, and they belong to several gene families, such as C2H2, WOX, LBD, WRKY, MYB, and NAC (Figures 8C, D; Table S5). As DEGs, some had higher expression levels in A lines than in B lines. For example, BnaC07g13550D (a WRKY family TF) was significantly differentially expressed between the A and B lines in the four GMS lines, and much more transcripts accumulated in the A lines (Figure 8D). In addition, 48 MYB TFs were differentially expressed between A and B lines, and the proportion (48/253) was larger than the other TFs. BnaA04g28790D was an MYB TF with significantly different expression level in 6251AB and 6284AB lines (Figure 8D). Some transcription factors may be involved in the regulation of anther and microspore development.
Analysis of potential key DEGs involved in GMS
To investigate the potential DEGs related to pollen development processes, 13,031 Arabidopsis genes related to anther, pollen, and microspore development were downloaded from TAIR website (https://www.arabidopsis.org/index.jsp). And 6,099 homologous DEGs in B. napus were identified, which might be involved in anther, pollen and microspore development (Table S6). KEGG enrichment analysis of these 6,099 DEGs was performed. A total of 1,073 DEGs were enriched in 27 significantly pathways (p-value <0.05, p-adjusted <0.05, q-value <0.05) (Table S7). The Top 20 pathways were used to draw the KEGG map (Figure 10). In Table 2, we excluded genes with lower expression in advance, and listed 74 highly credible candidate genes related to GMS. Their homologous genes in Arabidopsis have been reported in previous studies to be involved in anther and microspore developmental processes, such as anther cell differentiation, tapetum development, pollen mother cell and microspore division, meiosis process, sporopollenin biosynthesis, pollen exine formation, and pollen maturation process. Based on the functions of these Arabidopsis genes, we speculated the 74 DEGs might be also involved in the anther and microspore developmental processes.
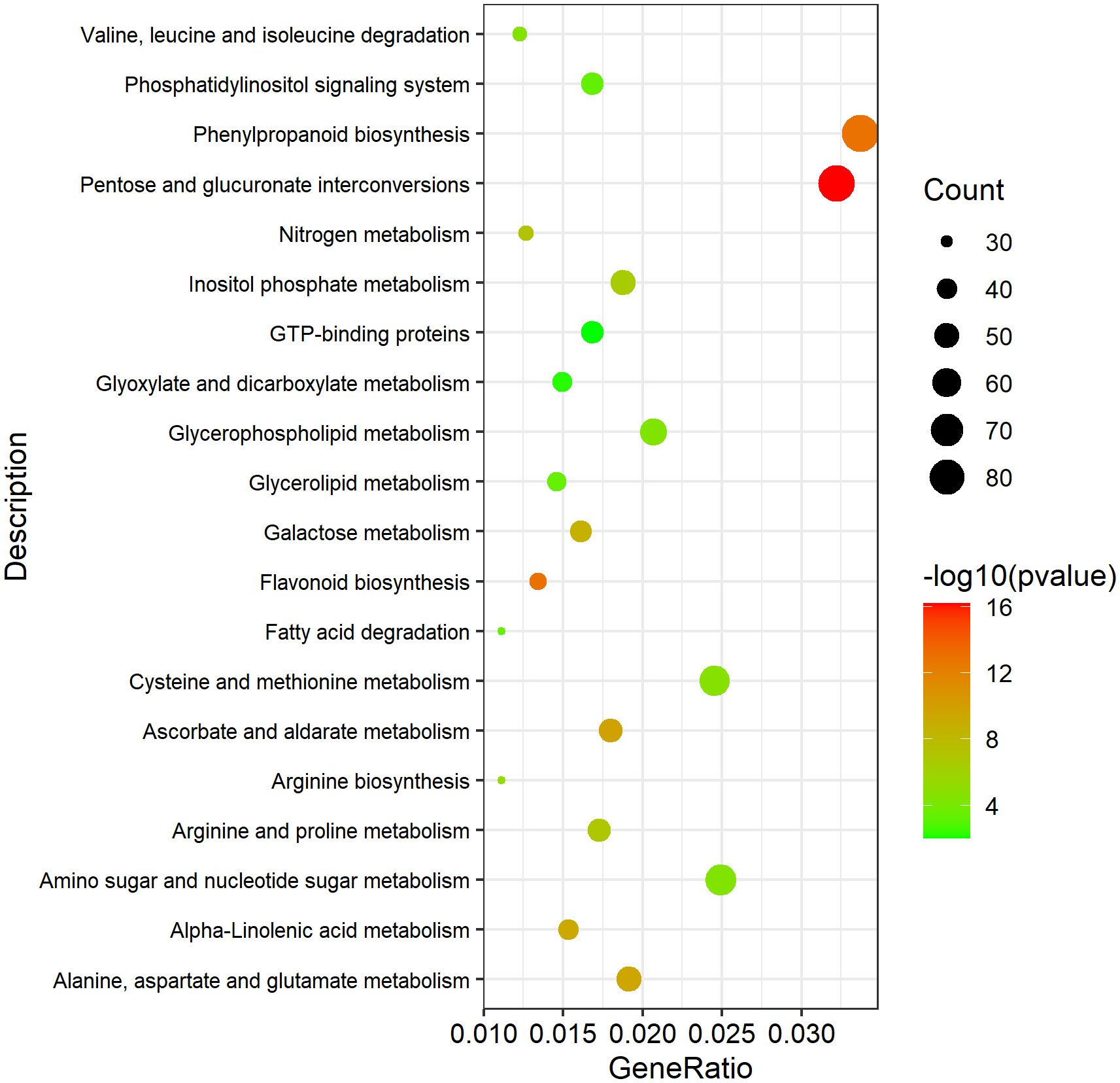
Figure 10 The Top 20 significantly enriched KEGG pathways of the 1073 DEGs possibly involved in pollen development. A total of 1073 DEGs were enriched in 27 significantly pathways (p-value <0.05, p-adjusted <0.05, q-value <0.05). The Top 20 pathways were used to draw the KEGG map.
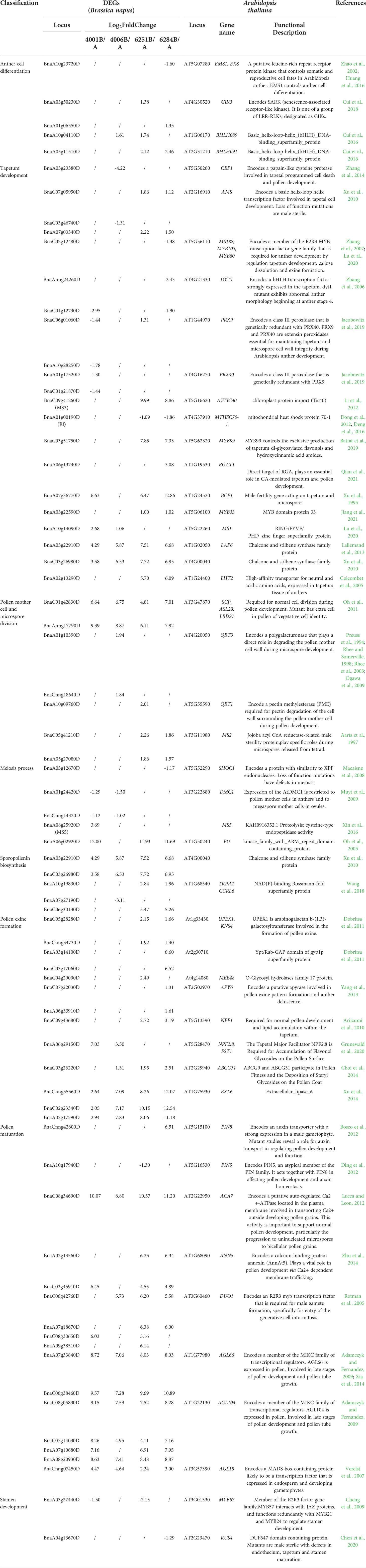
Table 2 Potential functional analysis of DEGs possibly involved in anther and microspore development.
In previous studies, MS5 (BnaA08g25920D) was a key gene that caused sterility in B. napus. It participated in meiosis progression by regulating chromosome configuration during early prophase I (Xin et al., 2016). DMC1 and FU played important roles during meiosis process in Arabidopsis (Oh et al., 2005; Muyt et al., 2009). In our transcriptome data, MS5, DMC1, and Fu were differentially expressed in DGMS (Table 2). And the abnormal pollen development in our sterile lines of DGMS arose from the meiosis process. Therefore, MS5, DMC1, and Fu may be key genes involved in the fertility regulation of DGMS. MS3 and Rf were key genes controlling fertility in RGMS of B. napus, and the abnormal vacuolization of the tapetum during the tetrad stage was associated with male sterility (Wan et al., 2010; Deng et al., 2016). We found the expression level of MS3 (BnaC09g41260D) decreased in both 6251A and 6284A. And Rf was also differentially expressed in 6251AB and 6284AB. Coincidentally, abnormal vacuolization of the tapetum was also found in 6251A. So MS3 and Rf were probably key genes in RGMS. In addition, many other genes involved in tapetum development were specifically differentially expressed in RGMS. These genes included AMS (BnaC07g05950D, BnaA07g03340D), MYB99 (BnaC03g51750D), MYB33 (BnaA03g22590D), and LHT2 (BnaA02g13290D). Therefore, MS3 (BnaC09g41260D), Rf, AMS, MYB99, MYB33, and LHT2 may be key genes involved in the fertility regulation of RGMS.
Validation of differentially expressed genes by qRT-PCR
qRT-PCR was conducted to verify the expression levels of three randomly selected DEGs, namely, BnaA03g02100D, BnaA03g52460D, and BnaAnng37210D in 4001AB, 4006AB, 6251AB, and 6284AB, respectively. These three genes were upregulated in the fertile lines, which largely agreed with the expression trend in transcriptome results (Figure 11). The qRT-PCR results validated the accuracy and reliability of the obtained transcriptome data.
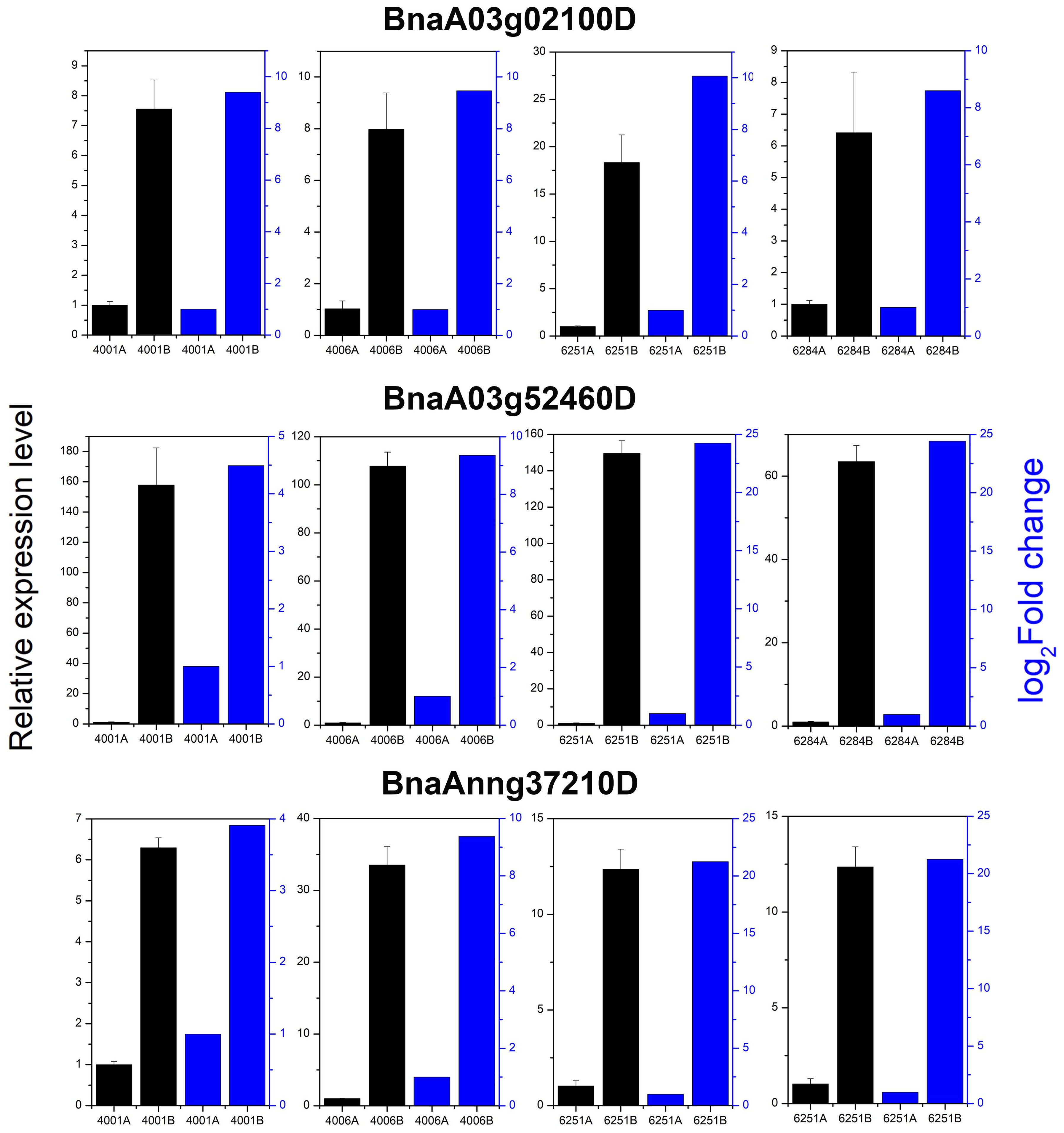
Figure 11 Relative expression analysis of three DEGs by transcriptome and qRT-PCR analysis. All qRT-PCR reactions were prepared in triplicate for each sample. The blue columns indicate the gene relative expression level from transcriptome sequencing. The black columns indicate the gene relative expression level obtained from qRT-PCR analysis.
Discussion
Rapeseed is one of the primary sources of vegetable oil for human nutrition in the world, which occupies an important position in China’s edible oil supply. In recent years, transcriptome sequencing has been widely used for the identification of candidate genes related to various agronomic traits of rapeseed, which greatly facilitate the identification of functional genes underlying important yield and quality traits and offer abundant resources for breeding excellent rapeseed varieties. By using transcriptome sequencing, developing pods of German cultivar Sollux and Chinese inbred line Gaoyou were studied to explore the molecular mechanism of oil-related biological processes. A total of 33 genes such as Sac domain-containing phosphoinositide phosphatase (Sac-PIP) were identified as candidate genes that affect seed oil content. Sac-PIP is a lipid-related gene, which participates in phospholipid signaling pathway (Xu et al., 2015). By transcriptome and metabolome correlation analysis, Tan et al. revealed the regulatory network for lipid synthesis in developing B. napus embryos (Tan et al., 2019). Transcriptome analysis was also widely used to identify the abiotic stress-related genes. By using transcriptomics, the mechanism of seed response to heat stress at 40 or 60°C was explored. The results show that 442 DEGs were identified in seeds after heat stress (Gao et al., 2021). By using transcriptome and proteome analyses, the molecular mechanisms underlying changes in oil storage under drought stress in B. napus L. were revealed. Lots of major genes involved in ABA signal transduction, such as BnaA06g40220D, BnaA03g13020D, BnaA01g23120D, and BnaA05g08020D, were highly expressed under drought stress (Li et al., 2021). Long et al. used transcriptome analysis to explore salt-stress-responding genes by using digital gene expression (DGE) at 0, 3, 12 and 24 h after H2O (control) and NaCl treatments on B. napus roots at the germination stage. A total of 163 genes were differentially expressed at all the time points. These genes were new candidate salt-stress-responding genes, which may function in novel putative nodes in the molecular pathways of salt stress resistance (Long et al., 2015). Li et al. performed a global dynamic transcriptome programming of rapeseed anther at different development stages. The transcriptome sequencing results revealed that 35,470 transcripts were expressed in at least one of the anther development stages from the pollen mother cell stage to the mature pollen stage (Li et al., 2016). Therefore, much more genes that are involved in pollen development need to be discovered.
In our study, the potential genes related to pollen development and the formation of genetic male sterility were identified by constructing and sequencing 24 transcriptome libraries for the flower buds from the fertile and sterile lines of two RGMS and two DGMS lines. A total of 23,554 redundant DEGs with over two-fold change between sterile and fertile lines were obtained, including 4,820 DEGs (4,251 upregulated and 569 downregulated) in 4001AB, 4,257 DEGs (3,857 upregulated and 400 downregulated) in 4006AB, 6,093 DEGs (5,230 upregulated and 863 downregulated) in 6251AB, and 8,384 DEGs (7,347 upregulated and 1,037 downregulated) in 6284AB. The number of DEGs identified in our study was much more than those in previous studies. Our findings provide a global view of genes that are potentially involved in GMS occurrence. In the four GMS lines, the number of upregulated genes was much greater than the number of downregulated genes. A similar phenomenon was also observed in previous reports (Wu et al., 2007). Maybe this phenomenon in our study was caused by the following reasons. The fertile and sterile plants of the homozygous GMS two-type line have similar genetic background (Li et al., 1988). Consequently, the fertile and sterile plants should have a similar gene expression pattern before the key stage (tetrad stage) for fertility control. Thereafter, the male gametes of the sterile lines began to stop its development at the meiotic stage or pre-tetrad stage, and its tapetum and pollen mother cell also began to degrade to form empty pollen sacs. As a result, a large number of pollen development-related genes had low or undetectable expression in the sterile lines, while most of them were normally expressed in fertile lines. Therefore, the number of up-regulated genes in fertile lines was significantly greater than the down-regulated genes. To verify the expression of genes in transcriptome sequencing, three genes were randomly selected, and their expression levels were analyzed by qRT-PCR. The results of qRT-PCR largely agreed with the transcriptome sequencing results.
In our sequencing results, 2,214 upregulated DEGs and 52 downregulated DEGs were shared between 4001AB and 4006AB. And 4,196 upregulated DEGs and 283 downregulated DEGs were shared between 6251AB and 6284AB. In total, 1,545 DEGs were shared between the two DGMS and RGMS lines. We found the DEGs shared between two DGMS lines or two RGMS lines were not too much. In theory, the same dominant sterility genes cause sterility of 4001A and 4006A. And the same recessive sterility genes cause sterility of 6251A and 6284A. However, the genetic backgrounds of 4001AB and 4006AB, or the genetic backgrounds of 6251AB and 6284AB, are very different. In addition, their flowering period and agronomic characters are also quite different. Perhaps these factors lead to the small number of DEGs shared between two fertile and sterile lines.
In previous studies, many Arabidopsis genes have been reported to be involved in anther and microspore development processes. To investigate the potential DEGs related to pollen development processes, 13,031 Arabidopsis genes related to anther, pollen, and microspore development were downloaded from TAIR website. And 6,099 homologous DEGs in B. napus were identified, which might be involved in anther, pollen and microspore development. We further screened 47 Arabidopsis genes that have been reported in previous studies to be involved in anther and microspore developmental processes, such as AGL18 (Verelst et al., 2007), AGL66 (Adamczyk and Fernandez, 2009; Xia et al., 2014), AGL104 (Adamczyk and Fernandez, 2009), SCP (Oh et al., 2011), ACA7 (Lucca and Leon, 2012), MS2 (Aarts et al., 1997), and QRT3 (Rhee et al., 2003). And 74 DEGs were identified to be their homologous genes, containing MS2, QRT1, and QRT3, etc. It is worth noting that the MS5 (BnaA08g25920D) (Xin et al., 2016), DMC1, FU play specific roles in meiosis process (Oh et al., 2005; Muyt et al., 2009). And MS3 (BnaC09g41260D), Rf, AMS, MYB99, MYB33, and LHT2 play important roles in tapetum development (Colcombet et al., 2005; Xu et al., 2010; Dong et al., 2012; Li et al., 2012; Deng et al., 2016; Battat et al., 2019; Jiang et al., 2021). Coincidentally, we found the abnormal pollen development in our sterile lines of DGMS arose from the meiosis process, and the abnormal pollen development in RGMS occurred before tetrad stage. Therefore, we speculated that the abnormal meiosis or tapetum development might be an important cause of sterility. And MS5 (BnaA08g25920D), DMC1, and Fu may be key genes involved in the fertility regulation of DGMS. MS3 (BnaC09g41260D), Rf (BnaA01g00190D), AMS, MYB99, MYB33, and LHT2 may be key genes involved in the fertility regulation of RGMS.
MYB in rice (OsGAMYB) and Arabidopsis (AtMYB33 and AtMYB65) were identified as plant GMS genes. Loss-of-function mutations of OsGAMYB failed to produce normal pollen grains and resulted in a sterile panicle of rice (Tsuji et al., 2006). B. napus MIR159 overexpression in A. thaliana resulted in reduced transcripts of AtMYB33 and AtMYB65 and decreased seed setting rate (Jiang et al., 2021). AtMYB101 participates in pollen tube reception possibly by controlling the expression of downstream genes (Liang et al., 2013). In the present study, the transcripts of MYB33, MYB65, and MYB101 were analyzed between fertile and sterile lines. More transcripts of MYB33 and MYB101 accumulated in the fertile lines (Figure S3D). Other TFs such as NAC and WRKY family genes may play critical roles in pollen development and plant fertility (Wang et al., 2019; Liu et al., 2020).
Conclusions
This study revealed that the abnormal pollen development in DGMS lines might start at the meiotic stage, and abnormal pollen development in RGMS lines probably occurred before the tetrad stage. In total, 24 transcriptome libraries were constructed and sequenced for the flower buds from the fertile and sterile lines of RGMS and DGMS. Based on the results, a large number of DGEs was obtained. Some DEGs were candidate genes for rapeseed breeding, and they are expected to provide new insight into the molecular mechanisms underlying GMS.
Data availability statement
The datasets presented in this study can be found in online repositories. The name of the repository and accession number can be found below: SRA, NCBI; PRJNA879090.
Author contributions
JJ and LY conceived the project and research plan. JJ drafted the manuscript. JJ and PX analyzed the sequencing data, performed the experiments, and finished the manuscript. JYZ, XZ, and JFZ planted and observed the DGMS plants. YL, MJ, and WW planted and observed the RGMS plants. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Key Research and Development Program of China (Grant No.2018YFD0100602), National Modern Agricultural Industry Technology System (Grant No. CARS-12), and Shanghai Agriculture Applied Technology Development Program, China (Grant No.T20210219).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1004781/full#supplementary-material
Supplementary Figure 1 | Pearson correlation and principal component analysis. (A) Representative correlation plots between A and B lines. Plots display between log10(FPKM), normalized read counts. (B) PCA of the transcriptome expression profiler among samples. Genotype is represented by different color. The relationships are calculated by FPKM values.
Supplementary Figure 2 | Analysis of the expression levels of DEGs. (A) The downregulated genes in A lines of GMS, numbers in each box are FPKM values. Genes specifically involved in DGMS (B and C) and RGMS (D and E). The expression levels are calculated by FPKM values. Significant differences were calculated by performing paired t-test between the FPKM values of A and B lines. **P < 0.01, “ns” means no significant difference.
Supplementary Figure 3 | Shared DEGs possibly involved in GMS. (A–C) BnaAnng17790D, BnaC08g05830D, and BnaA07g33840D were selected, and their expression levels were calculated using FPKM values. Significant differences were calculated by performing a paired t-test between FPKM values of A and B lines. **P < 0.01. (D) Analysis of the expression levels of MYB TFs. Numbers in each box are FPKM values.
Abbreviations
GMS, Genic male sterility; RGMS, recessive genic male sterility; DGMS, dominant genic male sterility; CMS, cytoplasmic male sterility; GO, Gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; DEGs, Differentially expressed genes; qRT-PCR, Quantitative real-time polymerase chain reaction.
References
Aarts, M. G., Hodge, R., Kalantidis, K., Florack, D., Wilson, Z. A., Mulligan, B. J., et al. (1997). The arabidopsis MALE STERILITY 2 protein shares similarity with reductases in elongation/condensation complexes. Plant J. 12, 615–623. doi: 10.1046/j.1365-313X.1997.00615.x
Adamczyk, B. J., Fernandez, D. E. (2009). MIKC* MADS domain heterodimers are required for pollen maturation and tube growth in arabidopsis. Plant Physiol. 149 (4), 1713–1723. doi: 10.1104/pp.109.135806
Allender, C. J., King, G. J. (2010). ). origins of the amphiploid species Brassica napus l. investigated by chloroplast and nuclear molecular markers. BMC Plant Biol. 10, 54. doi: 10.1186/1471-2229-10-54
Ariizumi, T., Hatakeyama, K., Hinata, K., Inatsugi, R., Nishida, I., Sato, S., et al. (2010). Disruption of the novel plant protein NEF1 affects lipid accumulation in the plastids of the tapetum and exine formation of pollen, resulting in male sterility in arabidopsis thaliana. Plant J. 39, 170–181. doi: 10.1111/j.1365-313X.2004.02118.x
Battat, M., Eitan, A., Rogachev., I., Hanhineva, K., Fernie, A., Tohge, T., et al. (2019). A MYB triad controls primary and phenylpropanoid metabolites for pollen coat patterning. Plant Physiol. 180 (1), 87–108. doi: 10.1104/pp.19.00009
Bosco, C. D., Dovzhenko, A., Liu, X., Woerner, N., Rensch, T., Eismann, M., et al. (2012). The endoplasmic reticulum localized PIN8 is a pollen-specific auxin carrier involved in intracellular auxin homeostasis. Plant J. 71, 860–870. doi: 10.1111/j.1365-313X.2012.05037.x
Chalhoub, B., Denoeud, F., Liu, S., Parkin, I. A., Tang, H., Wang, X., et al. (2014). Early allopolyploid evolution in the post-neolithic brassica napus oilseed genome. Science 345, 950–953. doi: 10.1126/science.1253435
Cheng, H., Song, S., Xiao, L., Soo, H. M., Cheng, Z., Xie, D., et al. (2009). Gibberellin acts through jasmonate to control the expression of MYB21, MYB24, and MYB57 to promote stamen filament growth in arabidopsis. PloS Genet. 5 (3), e1000440. doi: 10.1371/journal.pgen.1000440
Chen, F. X., Hu, B. C., Li, C., Li, Q. S., Chen, W. S., Zhang, M. L. (1998). Genetic studies on GMS in brassica napus l.: I. inheritance of recessive GMS line 9012A (in Chinese). Acta Agron. Sin. 24, 431–438.
Chen, Y. J., Yang, X. X., Li, W. C., Zhao, S. Q. (2020). Knockdown of the DUF647 family member RUS4 impairs stamen development and pollen maturation in arabidopsis. Plant Sci. 301, 110645. doi: 10.1016/j.plantsci.2020.110645
Choi, H., Ohyama, K., Kim, Y. Y., Jin, J. Y., Lee, S. B., Yamaoka, Y., et al. (2014). The role of arabidopsis ABCG9 and ABCG31 ATP binding cassette transporters in pollen fitness and the deposition of steryl glycosides on the pollen coat. Plant Cell 26 (1), 310–324. doi: 10.1105/tpc.113.118935
Colcombet, J., Boisson-Dernier, A., Ros-Palau, R., Vera, C. E., Schroeder, J. I. (2005). Arabidopsis SOMATIC EMBRYOGENESIS RECEPTOR KINASES1 and 2 are essential for tapetum development and microspore maturation. Plant Cell 17 (12), 3350–3361. doi: 10.1105/tpc.105.036731
Cui, Y., Hu, C., Zhu, Y., Cheng, K., Li, X., Wei, Z., et al. (2018). CIK receptor kinases determine cell fate specification during early anther development in arabidopsis. Plant Cell 30 (10), 2383–2401. doi: 10.1105/tpc.17.00586
Cui, J., You, C. J., Zhu, E. G., Huang, Q., Ma, H., Chang, F. (2016). Feedback regulation of DYT1 by interactions with downstream bHLH factors promotes DYT1 nuclear localization and anther development. Plant Cell 28 (5), 1078–1093. doi: 10.1105/tpc.15.00986
Deng, Z. H., Li, X., Wang, Z. Z., Jiang, Y. F., Wan, L. L., Dong, F. M., et al. (2016). Map−based cloning reveals the complex organization of the BnRf locus and leads to the identification of BnRfb , a male sterility gene, in brassica napus. Theor. Appl. Genet. 129, 53–64. doi: 10.1007/s00122-015-2608-8
Ding, Z., Wang, B., Moreno, I., Dupláková, N., Simon, S., Carraro, N., et al. (2012). ER-localized auxin transporter PIN8 regulates auxin homeostasis and male gametophyte development in arabidopsis. Nat. Commun. 3, 941. doi: 10.1038/ncomms1941
Dobritsa, A. A., Geanconteri, A., Shrestha, J., Carlson, A., Kooyers, N., Coerper, D., et al. (2011). A large-scale genetic screen in arabidopsis to identify genes involved in pollen exine production. Plant Physiol. 157 (2), 947–970. doi: 10.1104/pp.111.179523
Dong, F. M., Hong, D. F., Xie, Y. Z., Wen, Y. P., Dong, L., Liu, P. W., et al. (2012). Molecular validation of a multiple-allele recessive genic male sterility locus (BnRf) in brassica napus l. Mol. Breed. 30, 1193–1205. doi: 10.1007/s11032-012-9708-9
Fu, T. D. (2000). Breeding and utilization of rapeseed hybrid. 2nd ed (Wuhan: Hubei science and technology press).
Gao, G. Z., Hu, J. H., Zhang, X. J., Zhang, F. G., Li, M., Wu, X. M. (2021). Transcriptome analysis reveals genes expression pattern of seed response to heat stress in brassica napus l. Oil Crop Sci. 6 (2), 87–96. doi: 10.1016/j.ocsci.2021.04.005
Gibalova, A., Renak, D., Matczuk, K., Dupl'akova, N., Chab, D., Twell, D., et al. (2009). AtbZIP34 is required for arabidopsis pollen wall patterning and the control of several metabolic pathways in developing pollen. Plant Mol. Biol. 70, 581–601. doi: 10.1007/s11103-009-9493-y
Gómez, J. F., Talle, B., Wilson, Z. A. (2015). Anther and pollen development: A conserved developmental pathway. J. Inter. Plant Biol. 57, 876–891. doi: 10.1111/jipb.12425
Grunewald, S., Marillonnet, S., Hause, G., Haferkamp, I., Neuhaus, H. E., Veß, A., et al. (2020). The tapetal major facilitator NPF2.8 is required for accumulation of flavonol glycosides on the pollen surface in arabidopsis thaliana. Plant Cell 32, 5, 1727–1748. doi: 10.1105/tpc.19.00801
Gu, J. N., Zhu, J., Yu, Y., Teng, X. D., Lou, Y., Xu, X. F., et al. (2014). DYT1 directly regulates the expression of TDF1 for tapetum development and pollen wall formation in arabidopsis. Plant J. 80, 1005–1013. doi: 10.1111/tpj.12694
Honys, D., Twell, D. (2004). ). transcriptome analysis of haploid male gametophyte development in arabidopsis. Genome Biol. 5, R85. doi: 10.1186/gb-2004-5-11-r85
Huang, L., Cao, J., Ye, W., Liu, T., Jiang, L., Ye, Y. (2008). Transcriptional differences between the male-sterile mutant bcms and wild-type brassica campestris ssp. chinensis reveal genes related to pollen development. Plant Biol. 10, 342–355. doi: 10.1111/j.1438-8677.2008.00039.x
Huang, L., Cao, J. S., Zhang, A. H., Ye, Y. Q., Zhang, Y. C., Liu, T. T. (2009). The polygalacturonase gene BcMF2 from brassica campestris is associated with intine development. J. Exp. Bot. 60, 301–313. doi: 10.1093/jxb/ern295
Huang, S. N., Peng, S. L., Liu, Z. Y., Li, C. Y., Tian, C., Yao, R. P., et al. (2020). Investigation of the genes associated with a male sterility mutant (msm) in Chinese cabbage (Brassica campestris ssp. pekinensis) using RNA-seq. Mol. Genet. Genomics 295, 233–249. doi: 10.1007/s00438-019-01618-z
Huang, L., Ye, Y., Zhang, Y., Zhang, A., Liu, T., Cao, J. (2009). BcMF9, a novel polygalacturonase gene, is required for both brassica campestris intine and exine formation. Ann. Bot. 104, 1339–1351. doi: 10.1093/aob/mcp244
Huang, J., Zhang, T., Linstroth, L., Tillman, Z., Otegui, M. S., Owen, H. A., et al. (2016). Control of anther cell differentiation by the small protein ligand TPD1 and its receptor EMS1 in arabidopsis. PloS Genet. 12 (8), e1006147. doi: 10.1371/journal.pgen.1006147
Jacobowitz, J. R., Doyle, W. C., Weng, J. K. (2019). PRX9 and PRX40 are extensin peroxidases essential for maintaining tapetum and microspore cell wall integrity during arabidopsis anther development. Plant Cell 31 (4), 848–861. doi: 10.1105/tpc.18.00907
Jiang, J. X., Xu, P. F., Li, Y. J., Li, Y. L., Zhou, X. R., Jiang, M. Y., et al. (2021). Identification of miRNAs and their target genes in genic male sterility lines in brassica napus by small RNA sequencing. BMC Plant Biol. 21, 520. doi: 10.1186/s12870-021-03306-w
Jiang, J. J., Yu, Y. J., Dong, H., Yao, L. N., Zhang, Z. X., Cao, J. S. (2014). BcMF21 is important for pollen development and germination in brassica campestris ssp. chinensis. Mol. Biol. Rep. 41, 537–544. doi: 10.1007/s11033-013-2889-4
Kaul, M. L. H. (1988). Male Sterility in higher plants (New York: Springer-Verlag Berlin Heidelberg).
Kim, D., Langmead, B., Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Lallemand, B., Erhardt, M., Heitz, T., Legrand, M. (2013). Sporopollenin biosynthetic enzymes interact and constitute a metabolon localized to the endoplasmic reticulum of tapetum cells. Plant Physiol. 162 (12), 616–625. doi: 10.1104/pp.112.213124
Liang, J. L., Ma, Y., Wu, J., Cheng, F., Liu, B., Wang, X. W. (2017). Map-based cloning of the dominant genic male sterile Ms-cd1 gene in cabbage (Brassica oleracea). Theor. Appl. Genet. 130, 71–79. doi: 10.1007/s00122-016-2792-1
Liang, Y., Tan, Z. M., Zhu, L., Niu, Q. K., Zhou, J. J., Li, M., et al. (2013). MYB97, MYB101 and MYB120 function as Male factors that control pollen tube-synergid interaction in arabidopsis thaliana fertilization. PloS Genet. 9 (11), e1003933. doi: 10.1371/journal.pgen.1003933
Li, J., Hong, D. F., He, J. P., Ma, L., Wan, L. L., Liu, P. W., et al. (2012). Map-based cloning of a recessive genic male sterility locus in brassica napus l. and development of its functional marker. Theor. Appl. Genet. 125, 223–234. doi: 10.1007/s00122-012-1827-5
Lin, S., Huang, L., Yu, X. L., Xiong, X. P., Yue, X. Y., Liu, T. T., et al. (2017). Characterization of BcMF23a and BcMF23b, two putative pectin methylesterase genes related to pollen development in brassica campestris ssp. chinensis. Mol. Biol. Rep. 44, 139–148. doi: 10.1007/s11033-016-4090-z
Lin, S., Miao., Y. J., Su, S. W., Xu, J., Jin, L., Sun, D., et al. (2019). Comprehensive analysis of ogura cytoplasmic male sterility-related genes in turnip (Brassica rapa ssp. rapifera) using RNA sequencing analysis and bioinformatics. PloS One 14 (6), e0218029. doi: 10.1371/journal.pone.0218029
Li, S. L., Qian, Y. X., Wu, Z. H. (1988). Genetic male sterility in rape (B.napus l.) conditioned by interaction of genes at two loci. Can. J. Plant Sci. 68, 1115–1118. doi: 10.4141/cjps88-132
Liu, L. C., Cao, J. S., Yu, X. L., Xiang, X., Fei, Y. J. (2006). Expression of an antisense BcMF3 affects microsporogenesis and pollen tube growth in arabidopsis. Agri. Sci. China 5, 339–345. doi: 10.1016/S1671-2927(06)60059-X
Liu, J., Lu, W., He, Q. B. (2008). Genetic analysis and molecular mapping of gene associated with dominant genic Male sterility in rapeseed (Brassica napus l.). Gene Genomics 30 (6), 523–532. doi: 10.1016/S0039-128X(03)00098-9
Liu, M., Tan, X. L., Yang, Y., Liu, P., Zhang, X. X., Zhang, Y. C., et al. (2020). Analysis ofthe genetic architecture of maize kernel size traits by combined linkage andassociation mapping. Plant Biotechnol. J. 18, 207–221. doi: 10.1111/pbi.13188
Liu, L. C., Xiang, X., Cao, J. S. (2006). BcMF4 gene, encoding a leucine-rich repeat protein, plays a role in male fertility in Chinese cabbage-pak-choi]. Yi Chuan 28, 1428–1434. doi: 10.1360/yc-006-1428
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2– ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Li, Y. Y., Zhang, L. X., Hu, S., Zhang, J. F., Wang, L., Ping, X. K., et al. (2021). Transcriptome and proteome analyses of the molecular mechanisms underlying changes in oil storage under drought stress in brassica napus l. GCB-Bioenergy 13 (7), 1071–1086. doi: 10.1111/gcbb.12833
Li, Z., Zhang, P., Lv, J., Cheng, Y., Cui, J., Zhao, H., et al. (2016). Global dynamic transcriptome programming of rapeseed (Brassica napus l.) anther at different development stages. PloS One 11 (5), e0154039. doi: 10.1371/journal.pone.0154039
Long, W. H., Zou, X. L., Zhang, X. K. (2015). Transcriptome analysis of canola (Brassica napus) under salt stress at the germination stage. PloS One 10 (2), e0116217. doi: 10.1371/journal.pone.0116217
Lou, P., Kang, J. G., Zhang, G. Y., Bonnema, G., Fang, Z. Y., Wang, X. W. (2007). Transcript profiling of a dominant male sterile mutant (Ms-cd1) in cabbage during flower bud development. Plant Sci. 171 (1), 111–119. doi: 10.1016/j.plantsci.2006.07.015
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 1–21. doi: 10.1186/s13059-014-0550-8
Lucca, N., León, G. (2012). Arabidopsis ACA7, encoding a putative auto-regulated Ca2 + -ATPase, is required for normal pollen development. Plant Cell Rep. 31, 651–659. doi: 10.1007/s00299-011-1182-z
Lu, J. Y., Xiong, S. X., Yin, W., Teng, X. D., Lou, Y., Zhu, J., et al. (2020). MS1, a direct target of MS188, regulates the expression of key sporophytic pollen coat protein genes in arabidopsis. J. Exp. Bot. 71 (16), 4877–4889. doi: 10.1093/jxb/eraa219
Macaisne, N., Novatchkova, M., Peirera, L., Vezon, D., Jolivet, S., Froger, N., et al. (2008). SHOC1, an XPF endonuclease-related protein, is essential for the formation of class I meiotic crossovers. Curr. Biol. 18, 1432–1437. doi: 10.1016/j.cub.2008.08.041
McCormick, S. (2004). Control of male gametophyte development. Plant Cell 16, S142–S153. doi: 10.1105/tpc.016659
Muyt, A. D., Pereira, L., Vezon, D., Chelysheva, L., Gendrot, G., Chambon, A., et al. (2009). A high throughput genetic screen identifies new early meiotic recombination functions in arabidopsis thaliana. PloS Genet. 5, e1000654. doi: 10.1371/journal.pgen.1000654
Ogawa, M., Kay, P., Wilson, S., Swain, S. M. (2009). ARABIDOPSIS DEHISCENCE ZONE POLYGALACTURONASE1 (ADPG1), ADPG2, and QUARTET2 are polygalacturonases required for cell separation during reproductive development in arabidopsis. Plant Cell 21 (1), 216–233. doi: 10.1105/tpc.108.063768
Oh, S. A., Johnson, A., Smertenko, A., Rahman, D., Park, S. K., Hussey, P. J., et al. (2005). A divergent cellular role for the FUSED kinase family in the plant-specific cytokinetic phragmoplast. Curr. Biol. 15 (23), 2107–2111. doi: 10.1016/j.cub.2005.10.044
Oh, S. A., Twell, D., Park, S. K. (2011). SIDECAR POLLEN suggests a plant-specific regulatory network underlying asymmetric microspore division in arabidopsis. Plant Signal Behav. 6 (3), 416–419. doi: 10.4161/psb.6.3.14385
Preuss, D., Rhee, S. Y., Davis, R. W. (1994). Tetrad analysis possible in arabidopsis with mutation of the QUARTET (QRT) genes. Science 264, 1458–1460. doi: 10.1126/science.8197459
Qian, Q., Yang, Y., Zhang, W., Hu, Y., Li, Y., Yu, H., et al. (2021). A novel arabidopsis gene RGAT1 is required for GA-mediated tapetum and pollen development. New Phytol. 231 (1), 137–151. doi: 10.1111/nph.17314
Rhee, S. Y., Osborne, E., Poindexter, P. D., Somerville, C. R. (2003). Microspore separation in the quartet 3 mutants of arabidopsis is impaired by a defect in a developmentally regulated polygalacturonase required for pollen mother cell wall degradation. Plant Physiol. 133, 1170–1180. doi: 10.1104/pp.103.028266
Rhee, S. Y., Somerville, C. R. (1998). Tetrad pollen formation in quartet mutants of arabidopsis thaliana is associated with persistence of pectic polysaccharides of the pollen mother cell wall. Plant J. 15 (1), 79–88. doi: 10.1046/j.1365-313X.1998.00183.x
Rotman, N., Durbarry, A., Wardle, A., Yang, W. C., Chaboud, A., Faure, J. E., et al. (2005). Novel class of MYB factors controls sperm-cell formation in plants. Curr. Biol. 15, 244–248. doi: 10.1016/j.cub.2005.01.013
Shen, X. P., Xu, L. A., Liu, Y. H., Liu, Y. H., Dong, H., Zhou, D., et al. (2019). Comparative transcriptome analysis and ChIP-sequencing reveals stage-specific gene expression and regulation profiles associated with pollen wall formation in brassica rapa. BMC Genomics 20, 264. doi: 10.1186/s12864-019-5637-x
Song, L. Q., Fu, T. D., Tu, J. X., Ma, C. Z., Yang, G. S. (2006). Molecular validation of multiple allele inheritance for dominant genic male sterility gene in brassica napus l. Theor. Appl. Genet. 113, 55–62. doi: 10.1007/s00122-006-0271-9
Sorensen, A. M., Kröber, S., Unte, U. S., Huijser, P., Dekker, K., Saedler, H. (2003). The arabidopsis ABORTED MICROSPORES (AMS) gene encodes a MYC class transcription factor. Plant J. 33, 413–423. doi: 10.1046/j.1365-313X.2003.01644.x
Tan, H. L., Zhang, J. H., Qi, X., Shi, X. L., Zhou, J. G., Wang, X. C., et al. (2019). Correlation analysis of the transcriptome and metabolome reveals the regulatory network for lipid synthesis in developing brassica napus embryos. Plant Mol. Biol. 99 (1-2), 31–44. doi: 10.1007/s11103-018-0800-3
Tsuji, H., Aya, K., Ueguchi-Tanaka, M., Shimada, Y., Nakazono, M., Watanabe, R., et al. (2006). GAMYB controls different sets of genes and is differentially regulated by microRNA in aleurone cells and anthers. Plant J. 47, 427–444. doi: 10.1111/j.1365-313X.2006.02795.x
Verelst, W., Twell, D., Folter, S. D., Immink, R., Saedler, H., Münster, T. (2007). MADS-complexes regulate transcriptome dynamics during pollen maturation. Genome Biol. 8, R249. doi: 10.1186/gb-2007-8-11-r249
Wan, L. L., Xia, X. Y., Hong, D. F., Li, J., Yang, G. S. (2010). Abnormal vacuolization of the tapetum during the tetrad stage is associated with male sterility in the recessive genic male sterile brassica napus L. Line 9012A. J. Plant Biol. 53, 121–133.
Wang, H. Z., Guan, C. Y., Zhang, C. L. (2007). Studies on rapeseed production and cultivation science and technology in China. Agron.: Cultivation, 2–7.
Wang, K., Guo, Z. L., Zhou, W. T., Zhang, C., Zhang, Z. Y., Lou, Y., et al. (2018). The regulation of sporopollenin biosynthesis genes for rapid pollen wall formation. Plant Physiol. 178 (1), 283–294. doi: 10.1104/pp.18.00219
Wang, Y., Li, Y., He, S. P., Gao, Y., Wang, N. N., Lu, R., et al. (2019). A cotton (Gossypium hirsutum) WRKY transcription factor (GhWRKY22) participates in regulating anther/pollen development. Plant Physiol. Bioch. 141, 231–239. doi: 10.1016/j.plaphy.2019.06.005
Wang, J., Lydiate, D. J., Parkin, I. A. P., Falentin, C., Delourme, R., Carion, P. W. C., et al. (2011). ). integration of linkage maps for the amphidiploid brassica napus and comparative mapping with arabidopsis and brassica rapa. BMC Genomics 12, 101. doi: 10.1186/1471-2164-12-101
Wu, J., Shen, J., Mao, X., Liu, K., Wei, L., Liu, P., et al. (2007). Isolation and analysis of differentially expressed genes in dominant genic male sterility (dgms) brassica napus l. using subtractive PCR and cDNA microarray. Plant Sci. 172 (2), 204–211. doi: 10.1016/j.plantsci.2006.08.010
Xia, C., Wang, Y. J., Liang, Y., Niu, Q. K., Tan, X. Y., Chu, L. C., et al. (2014). The ARID-HMG DNA-binding protein AtHMGB15 is required for pollen tube growth in arabidopsis thaliana. Plant J. 79 (5), 741–756. doi: 10.1111/tpj.12582
Xin, Q., Shen, Y., Li, X., Lu, W., Wang, X., Han, X., et al. (2016). MS5 mediates early meiotic progression and its natural variants may have applications for hybrid production in brassica napus. Plant Cell 28, 1263–1278. doi: 10.1105/tpc.15.01018
Xu, J., Ding, Z. W., Vizcay-Barrena, G., Shi, J. X., Liang, W. Q., Yuan, Z., et al. (2014). ABORTED MICROSPORES acts as a master regulator of pollen wall formation in arabidopsis. Plant Cell 26 (4), 1544–1556. doi: 10.1105/tpc.114.122986
Xu, H., Knox, R. B., Taylor, P. E., Singh, M. B. (1995). Bcp1, a gene required for male fertility in arabidopsis. PNAS 92 (6), 2106. doi: 10.1073/pnas.92.6.2106
Xu, H. M., Kong, X. D., Chen, F., Huang, J. X., Lou, X. Y., Zhao, J. Y. (2015). Transcriptome analysis of brassica napus pod using RNA-seq and identification of lipid-related candidate genes. BMC Genomics 16, 858. doi: 10.1186/s12864-015-2062-7
Xu, J., Yang, C., Yuan, Z., Zhang, D., Gondwe, M. Y., Ding, Z., et al. (2010). The ABORTED MICROSPORES regulatory network is required for postmeiotic male reproductive development in arabidopsis thaliana. Plant Cell 22, 91–107. doi: 10.1105/tpc.109.071803
Yang, C., Vizcay-Barrena, G., Conner, K., Wilson, Z. A. (2007). MALE STERILITY 1 is required for tapetal development and pollen wall biosynthesis. Plant Cell 19, 3530–3548. doi: 10.1105/tpc.107.054981
Yang, J., Wu, J., Romanovicz, D., Clark, G., Roux, S. J. (2013). Co-Regulation of exine wall patterning, pollen fertility and anther dehiscence by arabidopsis apyrases 6 and 7. Plant Physiol. Bioch. 69, 62–73. doi: 10.1016/j.plaphy.2013.04.022
Zhang, Q., Huang, L., Liu, T., Yu, X., Cao, J. S. (2008). Functional analysis of a pollen-expressed polygalacturonase gene BcMF6 in Chinese cabbage (Brassica campestris l. ssp. chinensis makino). Plant Cell Rep. 27, 1207–1215. doi: 10.1007/s00299-008-0541-x
Zhang, D., Liu, D., Lv, X., Wang, Y., Xun, Z., Liu, Z., et al. (2014). The cysteine protease CEP1, a key executor involved in tapetal programmed cell death, regulates pollen development in arabidopsis. Plant Cell 26, 2939–2961. doi: 10.1105/tpc.114.127282
Zhang, W., Sun, Y., Timofejeva, L., Chen, C., Grossniklaus, U., Ma, H. (2006). Regulation of arabidopsis tapetum development and function by DYSFUNCTIONAL TAPETUM1 (DYT1) encoding a putative bHLH transcription factor. Development 133, 3085–3095. doi: 10.1242/dev.02463
Zhang, Z. B., Zhu, J., Gao, J. F., Wang, C., Li, H., Li, H., et al. (2007). Transcription factor AtMYB103 is required for anther development by regulating tapetum development, callose dissolution and exine formation in arabidopsis. Plant J. 52, 528–538. doi: 10.1111/j.1365-313X.2007.03254.x
Zhao, D. Z., Wang, G. F., Speal, B., Ma, H. (2002). The excess microsporocytes1 gene encodes a putative leucine-rich repeat receptor protein kinase that controls somatic and reproductive cell fates in the arabidopsis anther. Genes Dev. 16, 2021–2031. doi: 10.1101/gad.997902
Zhu, J., Chen, H., Li, H., Gao, J. F., Jiang, H., Wang, C., et al. (2008). Defective in tapetal development and function 1 is essential for anther development and tapetal function for microspore maturation in arabidopsis. Plant J. 55, 266–277. doi: 10.1111/j.1365-313X.2008.03500.x
Keywords: Brassica napus L., rapeseed, transcriptome, genic male sterility, pollen development
Citation: Jiang J, Xu P, Zhang J, Li Y, Zhou X, Jiang M, Zhu J, Wang W and Yang L (2022) Global transcriptome analysis reveals potential genes associated with genic male sterility of rapeseed (Brassica napus L.). Front. Plant Sci. 13:1004781. doi: 10.3389/fpls.2022.1004781
Received: 27 July 2022; Accepted: 09 September 2022;
Published: 21 October 2022.
Edited by:
Kun Lu, Southwest University, ChinaReviewed by:
Chaobo Tong, Oil Crops Research Institute, (CAAS), ChinaFengqing Han, Insititute of Vegetables and Flowers, (CAAS), China
Copyright © 2022 Jiang, Xu, Zhang, Li, Zhou, Jiang, Zhu, Wang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liyong Yang, yangliyong@saas.sh.cn
†These authors have contributed equally to this work and share first authorship