 Jia-Yi Wu1
Jia-Yi Wu1 Jia-Yu Xue
Jia-Yu Xue- 1College of Horticulture, Academy for Advanced Interdisciplinary Studies, Nanjing Agricultural University, Nanjing, China
- 2State Key Laboratory of Palaeobiology and Stratigraphy, Nanjing Institute of Geology and Palaeontology (CAS), Nanjing, China
- 3Department of Plant Biotechnology and Bioinformatics, VIB-UGent Center for Plant Systems Biology, Ghent University, Ghent, Belgium
- 4Department of Biochemistry, Genetics and Microbiology, University of Pretoria, Pretoria, South Africa
Magnoliids are the third-largest group of angiosperms and occupy a critical position in angiosperm evolution. In the past years, due to the lack of sequenced genomes, the disease resistance gene (R gene) profile of magnoliids remains poorly understood. By the genome-wide identification of 1,832 NLR genes from seven magnoliid genomes, we built a framework for the evolution of magnoliid R genes. TNL genes were completely absent from five magnoliids, presumably due to immune pathway deficiencies. A total of 74 ancestral R genes (70 CNLs, 3 TNLs, and 1 RNL) were recovered in a common ancestor of magnoliids, from which all current NLR gene repertoires were derived. Tandem duplication served as the major drive for NLR genes expansion in seven magnoliid genomes, as most surveyed angiosperms. Due to recent rapid expansions, most magnoliids exhibited “a first expansion followed by a slight contraction and a further stronger expansion” evolutionary pattern, while both Litsea cubeba and Persea americana showed a two-times-repeated pattern of “expansion followed by contraction.” The transcriptome analysis of seven different tissues of Saururus chinensis revealed a low expression of most NLR genes, with some R genes displaying a relatively higher expression in roots and fruits. Overall, our study sheds light on the evolution of NLR genes in magnoliids, compensates for insufficiency in major angiosperm lineages, and provides an important reference for a better understanding of angiosperm NLR genes.
Introduction
Plants have faced numerous destructive pathogens throughout their evolutionary history, such as bacteria, viruses, and fungi. Over time, plants have adapted and employed a two-tiered versatile immune system to ward off the perturbations of pathogens. The first tier, described as the pathogen-associated molecular pattern (PAMP)-triggered immunity (PTI), recognizes PAMPs with cell membrane-localized receptors. The second tier, known as effector-triggered immunity (ETI), perceives pathogen-derived effectors through diverse strategies via intracellular disease resistance genes (R genes), ultimately resulting in resistance responses typically accompanied by hypersensitive reactions (HRs) in infected parts of the plants (Cui et al., 2015; Jones et al., 2016; Wang and Chai, 2020).
Nucleotide-binding site-leucine-rich repeat (NLR or NBS-LRR) genes comprise the largest group of plant R genes (Kourelis and van der Hoorn, 2018). The encoded NLR proteins are usually found in a signaling-competent, autoinhibited state with the LRR domain folding back onto the central NBS domain (Hu et al., 2013). NLR proteins are activated on the recognition of invading pathogens, and the NBS domain undergoes conformational alterations, as it possesses exposed N-terminal domains that trigger downstream HRs that elicit the apoptosis of infected cells to prevent pathogen transmission and proliferation (Andersen et al., 2018).
Angiosperm NLR genes can be divided into three subclasses, namely, TIR-NBS-LRR (TNL), CC-NBS-LRR (CNL), and RPW8-NBS-LRR (RNL), based on the identity of the N-terminal domain, which can be one of the three types, namely, Toll/Interleukin-1 receptor-like (TIR), coiled-coil (CC), and resistance to powdery mildew 8 (RPW8; Parker et al., 1997; Pan et al., 2000; Shao et al., 2014). Most TNL and CNL proteins function as pathogen sensors, either directly recognizing pathogenic effector proteins or indirectly monitoring the status shift of host proteins targeted by effectors (Kourelis and van der Hoorn, 2018). RNL proteins assist the downstream immune signal transduction of TNL and CNL proteins and are thus termed as the “helper” NLR (Jubic et al., 2019; Wang and Chai, 2020). Additionally, with regard to the molecular mechanism of NLR proteins that regulate cell death and resistance downstream, it has been well documented that CNL and RNL proteins act as Ca2+-permeable channels that trigger immunity and cell necrosis (Bi et al., 2021; Jacob et al., 2021).
The NLR genes originated from the common ancestor of all green plants and have been found in green algae and bryophytes (Xue et al., 2012; Yue et al., 2012; Shao et al., 2019). The divergence of NLR genes occurred early on, and the CNL and TNL subclasses have also been found in green algae and bryophytes, while other plant taxa have specific subclasses of their own [e.g., a/b-hydrolase-NBS-LRR (HNL) in liverworts and protein-kinase-NBS-LRR (PNL) in mosses] (Xue et al., 2012). Studies on lycophyte and fern NLRs are rare, and no new subclasses have been found in these taxa. Gymnosperms possess three similar NLR subclasses as angiosperms, namely, CNL, TNL, and RNL (Shao et al., 2019). The evolutionary history of NLR genes in angiosperms proceeded in two stages. The first was a salient stage starting at the origin of angiosperms until the Cretaceous-Paleogene (K-Pg) boundary when NLR genes were kept in low gene numbers. The second was a drastic expanding stage after the K-Pg boundary that led to the large NLR gene numbers we observed at present (Shao et al., 2016).
Due to frequent gene duplication and loss events, different angiosperm taxa possess different NLR gene numbers [e.g., rice has 498 NLRs (497 CNLs and 1 RNL but no TNLs); Arabidopsis thaliana has 165 NLRs (52 CNLs, 106 TNLs, and 7 RNLs), and tomato has 255 NLRs (222 CNLs, 31 TNLs, and 2 RNLs)] (Shao et al., 2016; Liu et al., 2021). The absence of TNLs can be observed in several angiosperms, such as most species of Ranunculales and Lamiales, and likely all monocots, suggesting that multiple and independent losses of TNLs have occurred throughout angiosperm evolution (Tarr and Alexander, 2009; Shao et al., 2016; Liu et al., 2021).
Different species are favored by different pathogens; therefore, NLR genes in different species evolved under different selection pressures and have distinct evolutionary patterns. For instance, Brassicaceae exhibit a “first expansion and then contraction” evolutionary pattern (Zhang et al., 2016), Poaceae manifest a “contraction” evolutionary pattern, and both Fabaceae and Rosaceae show a consistently expanding pattern (Shao et al., 2014; Jia et al., 2015). Moreover, it is not uncommon that the evolutionary patterns within the same family are distinct [e.g., potato, pepper, and tomato in Solanaceae (Qian et al., 2017); Gastrodia elata, Apostasia shenzhenica, Phalaenopsis equestris, and Dendrobium catenatum in Orchidaceae (Xue et al., 2020b); and Xanthoceras sorbifolium, Acer yangbiense, and Dinnocarpus longan in Sapindaceae (Zhou et al., 2020)].
Magnoliids (Magnoliidae), the third-largest group of angiosperms, are a clade of early diverging angiosperm lineages, encompassing over 10,000 living species that can be divided into four orders, namely, Piperales, Magnoliales, Laurales, and Canellales. Magnoliid plants are distributed throughout temperate and tropical zones of the world and are mainly found in the form of large trees, shrubs, vines, lianas, and occasionally herbs (Judd et al., 2002; Soltis et al., 2005). Traditionally, magnoliids have been classified as dicotyledonous plants, but the Angiosperm Phylogeny Group (APG) classification separates them from eudicots, and there exists a monophyletic branch with an unresolved phylogenetic position (Byng et al., 2016). Hence, magnoliids possess an important phylogenetic position that can be utilized for better comprehending the evolution of extant flowering plants.
Previous studies comprehensively clarified the evolutionary framework of angiosperm NLR genes exclusive to magnoliids (Shao et al., 2014, 2016; Zhang et al., 2016; Qian et al., 2017; Xue et al., 2020b; Zhou et al., 2020). In this study, we identified NLR genes in seven magnoliid genomes (Figure 1), performed a series of comprehensive analyses, and established an underlying framework of NLR gene evolution. This study uncovered the evolutionary features and patterns of the NLR gene family in magnoliids and investigated the mechanisms that molded these evolutionary changes. Collectively, our findings will serve as a fundamental resource for the mining of functional R genes in future investigations.
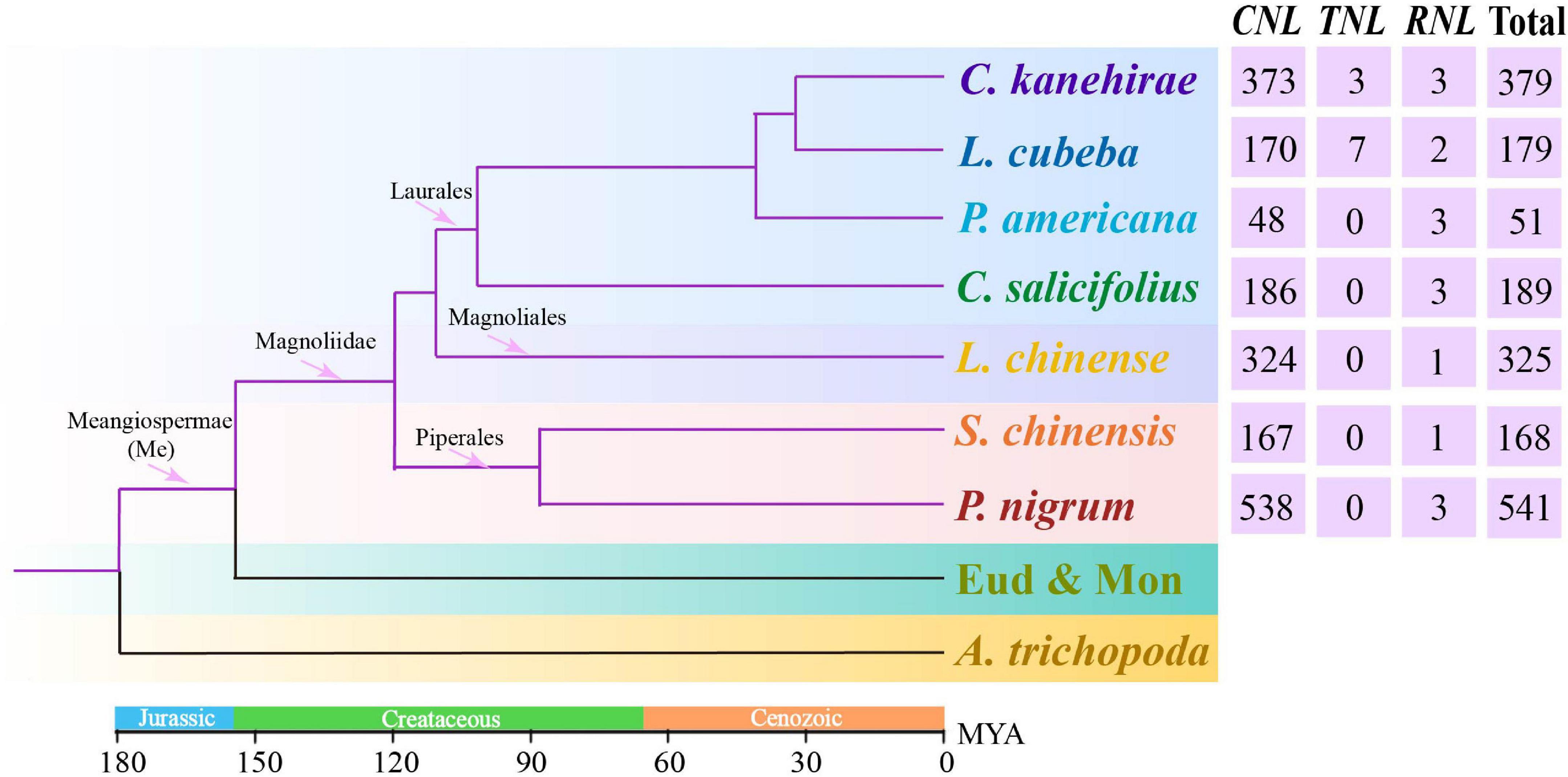
Figure 1. The phylogenetic tree of angiosperms, such as seven investigated magnoliids (Liriodendron chinense in Magnoliales, Persea americana, Chimonanthus salicifolius, Litsea cubeba, and Cinnamomum kanehirae in Laurales, Piper nigrum and Saururus chinensis in Piperales), was constructed according to the Angiosperm Phylogeny Group (APG) IV system (Byng et al., 2016). The divergence times at different nodes of angiosperms were combined from previous studies (Zhou et al., 2006, 2012; Nie et al., 2007; Magallón, 2010; Fiz-Palacios et al., 2011; Jiao et al., 2011).
Materials and Methods
Data Source
The whole genomes of seven Magnoliidae species, Liriodendron chinense in Magnoliales, P. americana, Chimonanthus salicifolius, Litsea cubeba, and Cinnamomum kanehirae in Laurales, and Piper nigrum and Saururus chinensis in Piperales were used in this study. Genomic sequences and annotation files of P. nigrum, C. salicifolius, L. chinense, and P. americana were obtained from the cotton database1, Xuehui Huang Lab2, Hardwood Genomics Database3, and Comparative Genomics Database4, respectively. Genomic sequences and annotation files of C. kanehirae and L. cubeba were downloaded from the National Center for Biotechnology Information (NCBI) database5 (accession Nos. PRJNA477266 and PRJNA562049, respectively). Genomic sequences and annotation files of S. chinensis were from our own unpublished data, and all S. chinensis NLR genes have been deposited at the NCBI database (BioProject ID: PRJNA764779). These seven genomes all have a high quality according to two parameters, namely, N50 and BUSCO completeness (Supplementary Table 1).
Identification and Classification of the NLR Genes
The NLR genes were identified in the seven magnoliid genomes as described previously (Shao et al., 2015). In brief, a two-step strategy was adopted to identify the NLR genes. The first step was to conduct simultaneously hidden Markov model searches (HMMsearch) and BLAST search using the amino acid sequence of the NB-ARC domain (Pfam accession number: PF00931) as a query to identify potential NLR genes. The threshold expectation value was set to 1.0 for the BLAST search. Then, the remaining candidate genes were merged, and the redundant hits were removed. For the sake of confirming the presence of the NBS domain, the remaining sequence hits were subjected to online Pfam analysis with an E-value of 10––4.6 All of the identified NLR genes were subjected to the conserved domain database (CDD) of NCBI7 using the default settings to determine whether they encoded CC, TIR, RPW8, or LRR domains.
Distribution of NLR Genes in Different Chromosomes
To determine the distribution of NLR genes on chromosomes or scaffolds of magnoliid genomes, GFF3 annotation file was anatomized to extract the genomic locations of all identified NLR genes. To detect the organization of NBS genes on chromosomes or scaffolds, a sliding window size of 250 kb was used to identify the number of genes that appear in a cluster on a chromosome or a scaffold as described in the study by Ameline-Torregrosa et al. (2008). Based on this criterion, NLR genes were assigned to clustered loci and singleton loci, which were mapped along the chromosomes.
Sequence Alignment and Conserved Motif Analysis
Amino acid sequences of the NBS domain were extracted from all identified NLR genes and used for multiple alignments using ClustalW integrated into MEGA 7.0 using default parameter settings (Kumar et al., 2016). Very short (<190 amino acids, less than two-thirds of a regular NBS domain) or very divergent sequences were removed to prevent interference with the alignments and subsequent phylogenetic inference. Then, the resulting alignments were manually corrected using MEGA 7.0 for further improvement (Kumar et al., 2016). The conserved protein motifs within the NBS domain of the three classes of NLR genes, along with the N-terminal domain plus the NBS domain of four subclasses of CNL genes, were analyzed by Multiple Expectation Maximization for Motif Elicitation (MEME) and WebLogo using default settings (Crooks et al., 2004; Bailey et al., 2006). Additionally, structural motif annotation was performed using the Pfam analysis8 and SMART tools9.
Phylogenetic Analysis and Reconciliation of Gene Loss/Duplication Events
To explore the relationships of NLR genes in the magnoliids, a phylogenetic tree was constructed based on the aligned amino acid sequences of the conserved NBS domains. The phylogenetic analyses were conducted using IQ-TREE and the maximum-likelihood method (Nguyen et al., 2015). The best-fit model was estimated by ModelFinder (Kalyaanamoorthy et al., 2017). Branch support values were assessed with UFBoot2 tests (Minh et al., 2013). The scale bar indicated genetic distance. To trace ancestral NLR genes, representative genes from different angiosperm lineages were used as references. In our earlier study (Shao et al., 2016), we identified 55 and 56 ancestral NLR genes in eudicots and monocots, respectively, representing 55 and 56 ancestral NLR genes in the genomes of their ancestors. Representative genes were picked from each of the ancestral genes, based on the following conditions: genes should be located in internal nodes with shorter branch lengths in each ancestral gene, and genes from species, such as Arabidopsis, rice, tomato, and Populus, were preferred because the genomes of these model plants are better assembled and genes are better studied. These picked genes are good representatives of ancestral NLR genes in eudicots and monocots. In addition, all NLRs of Amborella trichopoda, which is the earliest-diverging angiosperm, were used as the representatives of basal angiosperms. These genes represent the ancestral NLRs in early diverging angiosperms, and they were used as a reference and analyzed together with magnoliid genes. Additionally, gene loss/duplication events were recovered by reconciling the phylogenetic tree of the NLR gene with the real magnoliid species tree using Notung software (Stolzer et al., 2012).
Gene Duplication Type Determination
We employed the MCScanX package, which was developed and adopted by the Plant Genome Duplication Database (PGDD)10, to conduct the syntenic analysis of NLR genes among seven magnoliid genomes (Wang et al., 2012; Lee et al., 2013). In brief, pair-wise all-against-all BLAST was performed for the protein sequences within a genome. The obtained results and GFF annotation files were then subjected to MCScanX for microsynteny detection and determination of the gene duplication type (Wang et al., 2012). Microsynteny relationships were displayed using TBtools (Chen et al., 2020).
Gene Expression Analysis
To analyze the expression of the NLR genes of S. chinensis, the RNA-seq data were generated by ourselves and checked with FastQC software to remove low-quality reads or adapter. Clean reads in each sample were mapped to the reference genome of S. chinensis using HISAT2 with default settings (Pertea et al., 2016). The mapping results were subjected to Cufflinks to assemble transcripts in each sample and then merged into one cohesive set using Cuffmerge. The expression of each gene was evaluated using Cuffdiff (Trapnell et al., 2012). All analyses by Cufflinks were performed with default settings. The genes with the reads per kilobase per million (RPKM) value larger than 100 were recognized as a high expression gene in the analysis.
Results
Identification and Classification of NLR Genes in the Seven Magnoliid Genomes
A total of 1,832 NLR genes were identified from seven magnoliid genomes. Specifically, 325 NLR genes from L. chinense were identified in Magnoliales; 51 from P. americana, 179 from L. cubeba, 189 from C. salicifolius, and 379 from C. kanehirae in Laurales; and 541 from P. nigrum and 168 from S. chinensis in Piperales (Figure 1 and Supplementary Table 2). The number of CNL genes was much greater than 100, while the number of TNL and RNL genes was less than 10. Thus, the CNL genes accounted for over 98.5% (1,804) of all NLR genes and heavily outnumbered other classes. In fact, TNL genes were completely absent from five genomes, suggesting that the magnoliids are the characteristic of the loss of TNLs. Moreover, the RNL subfamily was found in all seven magnoliid genomes but in extremely low numbers in each genome.
Notably, the number of NLR genes varied within the same order among the seven magnoliid genomes investigated in this study. In Laurales, C. kanehirae possessed the greatest number of NLR genes (379), which was more than 7-fold higher than the number observed in P. americana. In Piperales, there was a 3-fold difference in the gene numbers between S. chinensis and P. nigrum. Furthermore, our study found a peculiar type of CNL gene with an Rx_N domain at the N-terminus, which consisted of a total of ∼400 members within the seven magnoliid genomes and could be considered a specialized CC domain, wherein a CC adopts a 4-helical-bundle-fold (Hao et al., 2013). The Rx_N domain was named after the potato R protein, Rx, which functions against potato virus X (PVX; Sacco et al., 2007; Tameling and Baulcombe, 2007). It is ∼83 amino acids in length, which is slightly shorter than a normal CC domain. On the pathogenic stimulation, the Rx_N domain will undergo an intramolecular interaction with the NBS domain and an intermolecular combination with the Trp-Pro-Pro (WPP) domain of the Rx cofactor, i.e., Ran GTPase-activating protein 2 (RanGAP2), primarily through hydrophobic interactions and form a heterodimer that transfers signals downstream (Hao et al., 2013).
Of the seven magnoliid genomes surveyed, the intact NLR genes with all three domains (i.e., CC, TIR, and RPW8-NBS-LRR) accounted for only 12.5% (229) of the total, while other genes either lacked an N-terminal or LRR domain at the C-terminus or entirely lacked the domains at both termini. S. chinensis had the highest proportion of intact genes (24.4%), while P. nigrum had the lowest (2.8%). Other than genomic changes (e.g., recombination, fusion, and pseudogenization) that result in real truncated genes, other subjective factors, such as sequencing errors, assembly errors, and false annotations, also artificially elicit “truncated” genes.
Occasionally, NLR proteins fuse with other domains (e.g., WRKY). The unusually integrated domains (IDs) were identified and counted, which are presented in a pie chart (Supplementary Figure 1). A variety of unusual IDs were found, and the associated pentatricopeptide repeat (PPR) protein occupied a fairly large proportion (13.2%). Other IDs, comprising a negligible component, demonstrated that many kinds of domains were integrated.
Distribution and Organization of NLR Genes in Magnoliid Genomes
The NLR genes were scattered unevenly among magnoliid chromosomes (Supplementary Figure 2). For instance, among the 24 chromosomes in P. nigrum, Chromosome 3 contained the most genes (135; 25.0%), while Chromosomes 6, 10, and 25 contained the least (only one gene in each chromosome) (Supplementary Figure 2A). Magnoliid NLR genes in clustered loci were much more abundant than in singleton loci, except for P. americana, where their ratio ranged from 1.4 to 9.6 among the seven genomes (Table 1). The majority of NLR genes in P. americana were organized into singletons. Moreover, the number of clusters with 10 or more NLR genes varied tremendously among these species. L. cubeba and P. americana had no NLR genes, and P. nigrum had the most with 18 loci. Furthermore, on average, a cluster in P. nigrum (8.5) and C. kanehirae (5.9) contained more NLR genes than clusters from the other five genomes. The two largest clusters were found in P. nigrum and C. kanehirae, containing 51 and 35 NLR genes, respectively (Table 1).
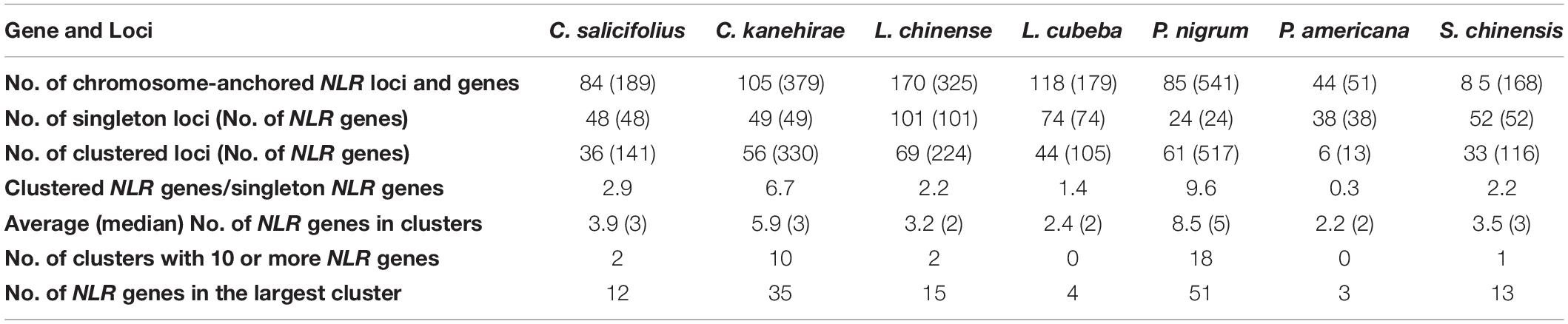
Table 1. Organization of nucleotide-binding site (NBS)-encoding genes in the seven magnoliid genomes.
Reconstructing the NLR Gene Phylogenies
To reconstruct the evolutionary relationship of magnoliid NLR genes, a phylogenetic tree was constructed based on the amino acid sequences of the conserved NBS domain and alignments, along with representative angiosperm NLR genes extracted from the study by Shao et al. (2016). The phylogenetic tree was composed of three monophyletic clades, namely, RNL, TNL, and CNL, with support values of 100%; many internal nodes had high (over 70%) support values. Compared to TNLs and CNLs, RNL genes had a relatively lower evolutionary rate, which was mirrored by the shorter branch lengths (Figure 2 and Supplementary Figure 3). In contrast, CNL genes exhibited an extremely active evolutionary pattern with far more gene duplications and losses, as well as faster evolutionary rates, which was reflected by the longer branch lengths (Figure 2). The CNL class could be further divided into four major subclasses. Every CNL subclass contained genes from all seven magnoliid species and representatives from eudicots and/or monocots, suggesting that the topology of the four subclasses should be stable at the whole angiosperm scale. However, the genes did not evenly fall into the four subclasses. Specifically, the CNL-1 subclass contained the fewest with only 38 genes, while CNL-2, -3, and -4 contained 346, 385, and 527 genes, respectively, which were at least 9-fold higher when compared to CNL-1. Genes with the Rx_N domain are mainly clustered into CNL-3 (94.9%) and with a small number falling into CNL-1 (1.9%) and -2 (3.2%) (Supplementary Table 3). The distribution of Rx_N domains containing genes in the phylogenetic tree suggests that this specialized CC domain has multiple origins.
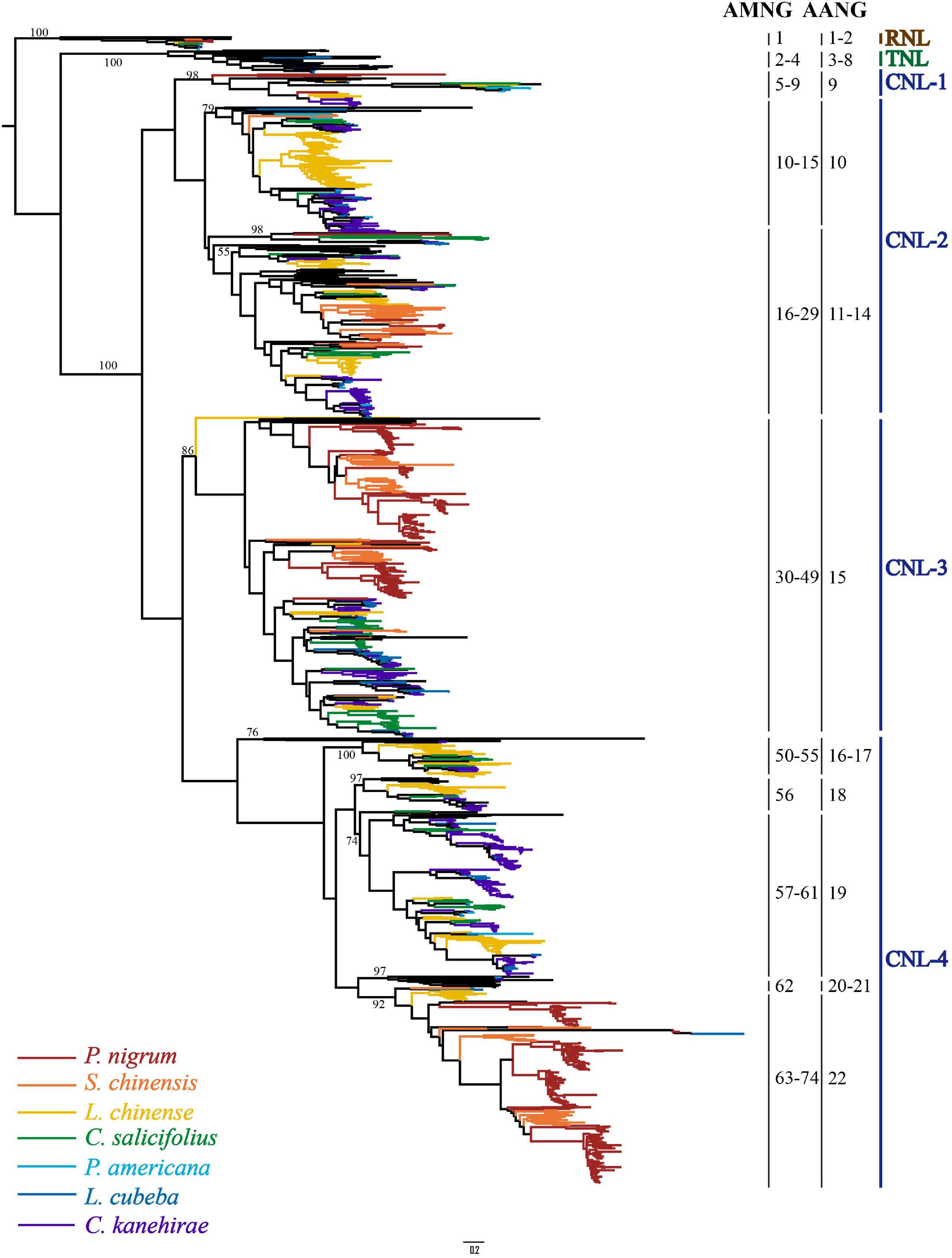
Figure 2. The phylogenetic relationships of NLR genes from seven magnoliid genomes. Predicted ancestral genes in the common ancestor of the seven magnoliids are indicated at the right of the phylogeny. NLR genes from different species are indicated with different colors in accordance with that of the species tree shown in Figure 1. Branch support values for three NLR classes (i.e., CNL, TNL, and RNL) and each gene are shown. The detailed phylogenetic tree is shown in Supplementary Figure 3.
Based on the reconstructed NLR gene phylogeny, 22 angiosperm ancestral NLR genes (AANGs), such as 14 CNLs, 6 TNLs, and 2 RNLs, were reconciled (Figure 2 and Supplementary Figure 4). Notably, not all ancestral NLR genes were preserved in all angiosperm clades, where only 4 out of the 22 AANGs (i.e., R2, T6, C5, and C6) were inherited by four early diverging angiosperm branches (i.e., magnoliids, eudicots, monocots, and A. trichopoda). No branch successfully maintained all 22 AANGs. Magnoliids lost seven AANGs (1 RNL, 2 TNLs, and 12 CNLs), eudicots and monocots together lost five genes, and A. trichopoda lost up to 10 AANGs (Figure 3A).
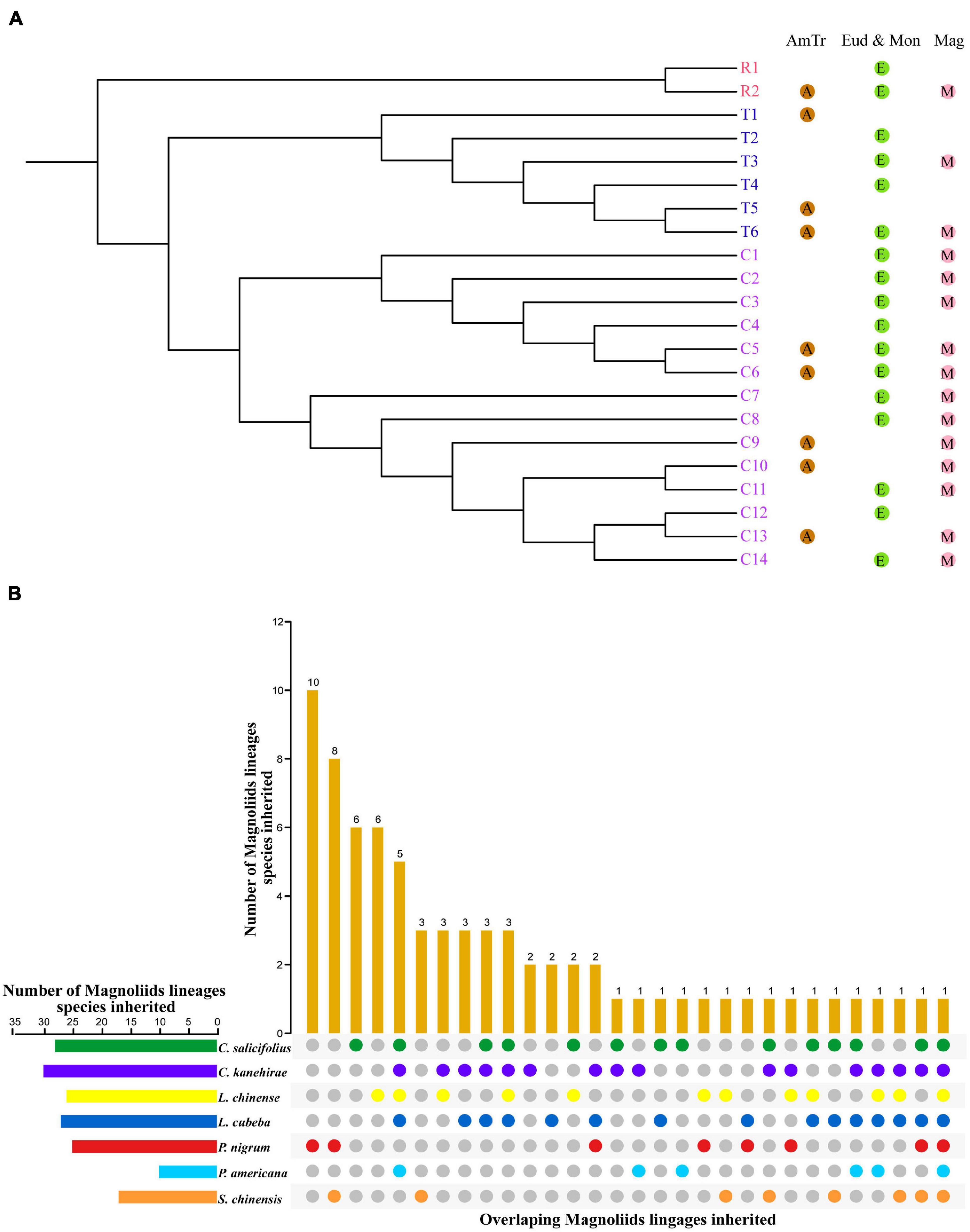
Figure 3. Inheritance of ancestral NLR genes. (A) Inheritance of ancestral angiosperm NLR genes in Amborella trichopoda, eudicots + monocots, and magnoliids. (B) Inheritance of ancestral magnoliid NLR genes in seven magnoliids.
Reconciling the magnoliid NLR genes recovered 74 ancestral magnoliid NLR genes (AMNGs), such as 1 RNL, 3 TNLs, and 70 CNLs, in the common ancestor of all magnoliids, indicating that the 15 AANGs inherited by magnoliids intensively expanded to 74 genes before further taxa divergences in the magnoliid ancestor, among which, two RNLs dropped to one, two TNLs slightly expanded to three, and CNLs largely expanded from 12 to 70 genes. Further analysis revealed that 28, 30, 26, 27, 25, 10, and 17 of the 74 AMNGs were inherited by C. salicifolius, C. kanehirae, L. chinense, L. cubeba, P. nigrum, P. americana, and S. chinensis, respectively (Figure 3B and Supplementary Table 4). Notably, none of the genes were reserved in all seven magnoliid genomes, just as the magnoliid ancestor did not inherit all 22 AANGs due to independent gene losses that occurred throughout magnoliid evolution. The differential inheritance patterns suggest that AMNGs experienced distinct gene duplication/loss events. Both gene loss and duplication events resulted in the observed gene number variations among the seven different species.
Conserved Motifs of the NBS Domain in Magnoliids
To explore the structural components and confirm the homology of all NLR genes in the magnoliid genomes, we searched for conserved motifs in the NBS domains via the MEME analysis (Figure 4A; DeYoung and Innes, 2006). From the N-terminus to the C-terminus, a total of five conserved motifs were identified, such as P-loop, Kinase-2, RNBS-B, GLPL, and RNBS-D. The P-loop, GLPL, and RNBS-B motifs exhibited high similarity among the three subclasses of NLR genes, suggesting that the homology of NBS domains with critical functions regulates immune responses. The other two motifs, especially RNBS-D, had distinct sequences among the three subclasses of NLR genes. These motifs can be utilized to distinguish the classes of magnoliid NLR genes without conducting phylogenetic analyses. The CNLs extensively duplicated throughout the evolution of magnoliids and had considerable sequence diversity, while RNLs and TNLs possessed remaining copies with hardly any detectable duplication events, which may explain their highly conserved motifs. Comparatively, the Kinase-2 motif had a conserved “DDVW” sequence in the RNL and CNL genes but frequently appeared as “DDVD” in the TNL genes, which is in agreement with previous angiosperm studies. However, in contrast to these previous studies, the “SR” sequence in the RNBS-B motif of RNLs was conserved, which may be a trait of magnoliids, while CNL and TNL proteins frequently appeared as “TTR” and “TTRD,” respectively (Shao et al., 2016).
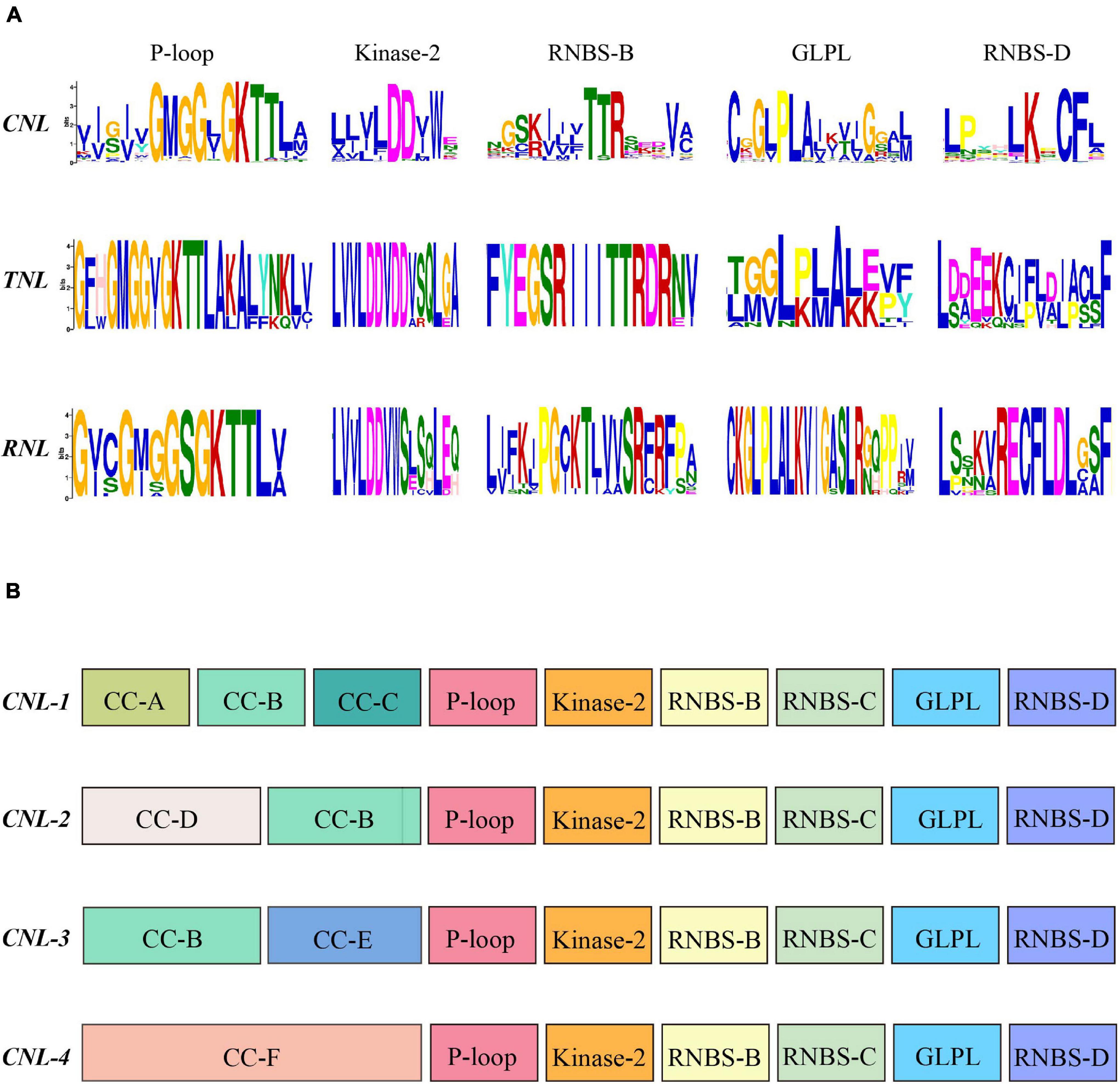
Figure 4. (A) Conserved motifs in the nucleotide-binding site (NBS) domain of the seven magnoliids. (B) Conserved motifs in the N-terminal domain and NBS domain of the four CNL subclasses. The amino acids of the 11 conserved motifs are extracted. Larger letters indicate higher frequency.
Further analysis revealed that four CNL subclasses had unique, distinctive motifs at the N-terminal domain (e.g., CC-A, CC-C, and CC-D) (Figure 4B). Additionally, the more conserved amino acid sequence of “YDAED” at the CC-B motif shared three CNL subclasses (Supplementary Figure 5A). With regard to the NBS domain, although four CNL subclasses shared five conserved motifs, some amino acids at specific sites showed some discrepancies among the four subclasses and could be used as preliminary labels for classification, such as the “ELP” sequence in RNBS-D of the CNL-1 subclass, “GSR” in RNBS-B of CNL-3, and “DD” in Kinase-2 of CNL-4. More importantly, in CNL-3, the RNBS-D motif had a conserved “CF” sequence but no “PED” sequence, while other subclasses appeared as “CFL” and had a “PED” sequence (Supplementary Figure 5B).
Differential Losses and Frequent Duplication Events of NLR Genes During Magnoliid Evolution
Based on the phylogenetic tree, we deduced that numerous independent gene duplication and loss events occurred at different stages of magnoliid evolution (Figure 5). The 74 ancestral NLR genes in the magnoliid ancestor should have undergone considerable complicated evolutionary processes to result in the current NLR genes observed in the seven magnoliid genomes of this study. The detailed evolutionary processes of the TNL and CNL genes were reconstructed, and species-specific gene duplication and loss events were detected, which reflected the diverse NLR gene number and the evolutionary patterns of the NLR genes in magnoliids (Figure 5).
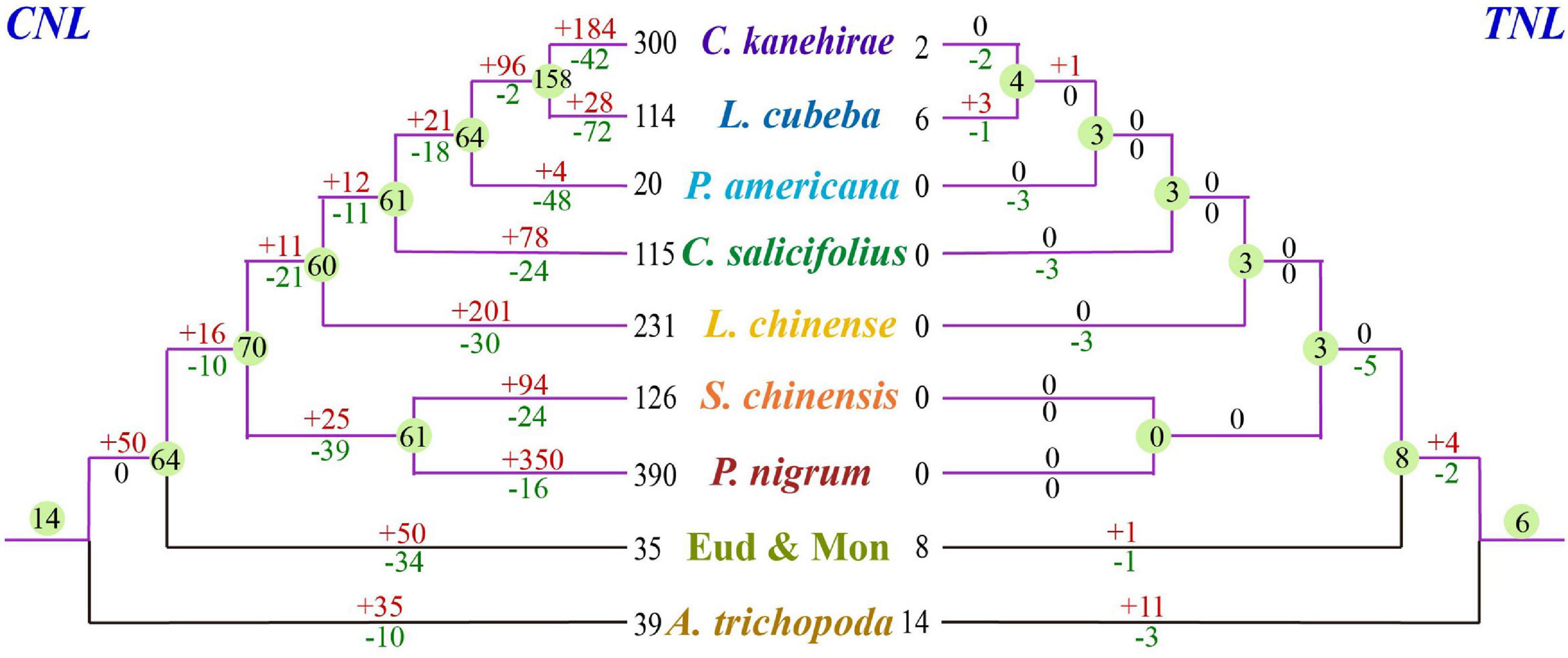
Figure 5. Loss and gain events of NLR genes across magnoliid evolution. Gene losses and gains are indicated by numbers with “–” or “+” on each branch. Detailed information for gain and loss events of NLR genes is shown in Supplementary Figure 4.
Five species exhibited similar evolutionary patterns: a first expansion, followed by a slight contraction, and another expansion; among the two expansions, the most recent expansion appeared to be stronger (Figures 6A–D,F). Specifically, P. nigrum duplicated 350 genes and lost 16 genes, and the gene number accordingly sharply increased in the genome, which was likely due to a recent whole-genome duplication event (Figure 6B). Additionally, both L. cubeba and P. americana showed a two-times-repeated pattern of “expansion followed by contraction” (Figures 6E,G). In summary, the seven magnoliid genomes exhibited two dynamic and discrepant patterns of NLR gene evolution, and the discrepancy was dependent on whether a given taxon underwent a recent expansion/contraction.
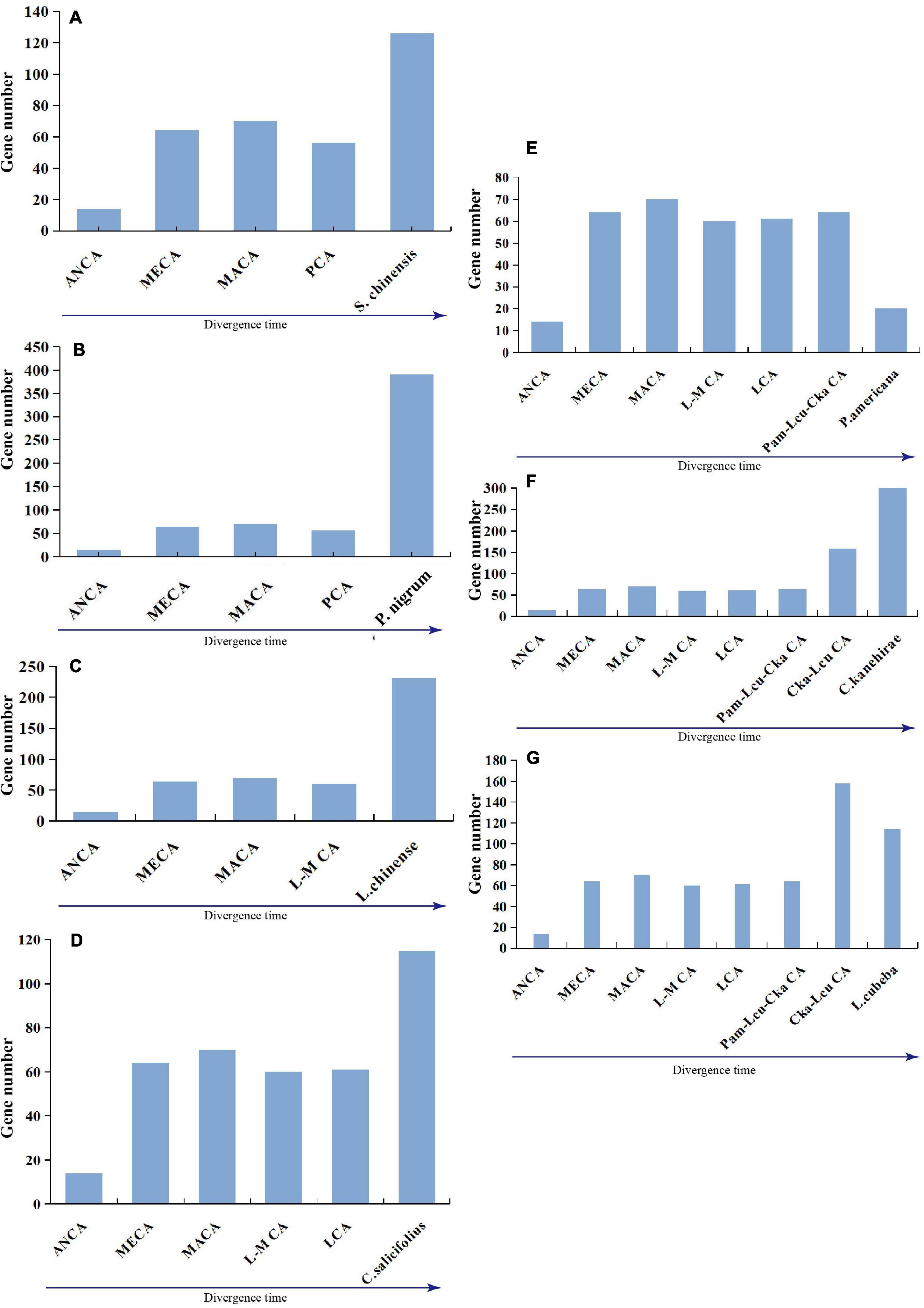
Figure 6. Evolutionary patterns of NLR genes in seven magnoliids: (A) S. chinensis, (B) P. nigrum, (C) L. chinense, (D) C. salicifolius, (E) P. americana, (F) C. kanehirae, and (G) L. cubeba. Pam-Lcu-Cka indicates the common ancestor of P. americana, L. cubeba, and C. kanehirae; Cka-Lcu indicates the common ancestor of C. kanehirae and L. cubeba.
Three types of NLR gene duplications, namely, local tandem, ectopic, and segmental duplications, have been defined (Leister, 2004), indicating that the tandem duplication events played major roles in NLR gene expansion in the seven magnoliid genomes. Ectopic and segmental duplication events were the main contributors to NLR gene expansion in P. americana and P. nigrum genomes (Table 2).
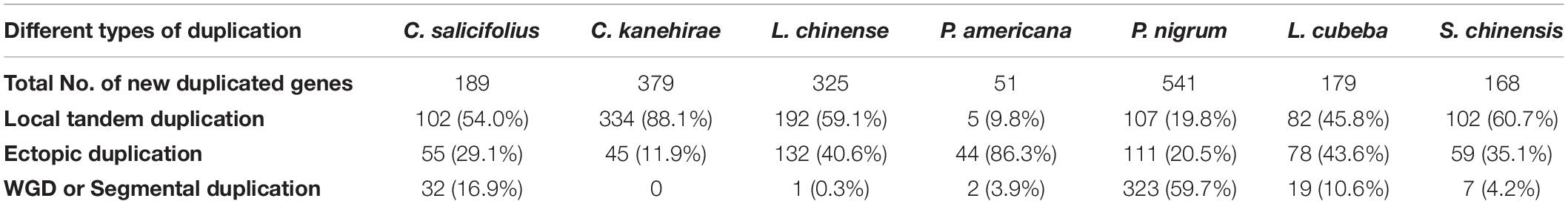
Table 2. Contributions of three duplication types in producing NLR genes during the evolution of magnoliids.
Expression Profile of NLR Genes in S. chinensis
To obtain the expression profile of NLR genes in S. chinensis, the transcriptomes of seven S. chinensis tissues were analyzed. Results indicated that most NLR genes were only expressed at very low levels in all of the tissues [i.e., flowers, green leaves, white leaves, mixed color leaves (half green half white), roots, fruits, and stems]. Nonetheless, the expression of some particular genes reached 100 or up to 200 RPKM, showing unusually high expression levels (Figure 7 and Supplementary Table 5).
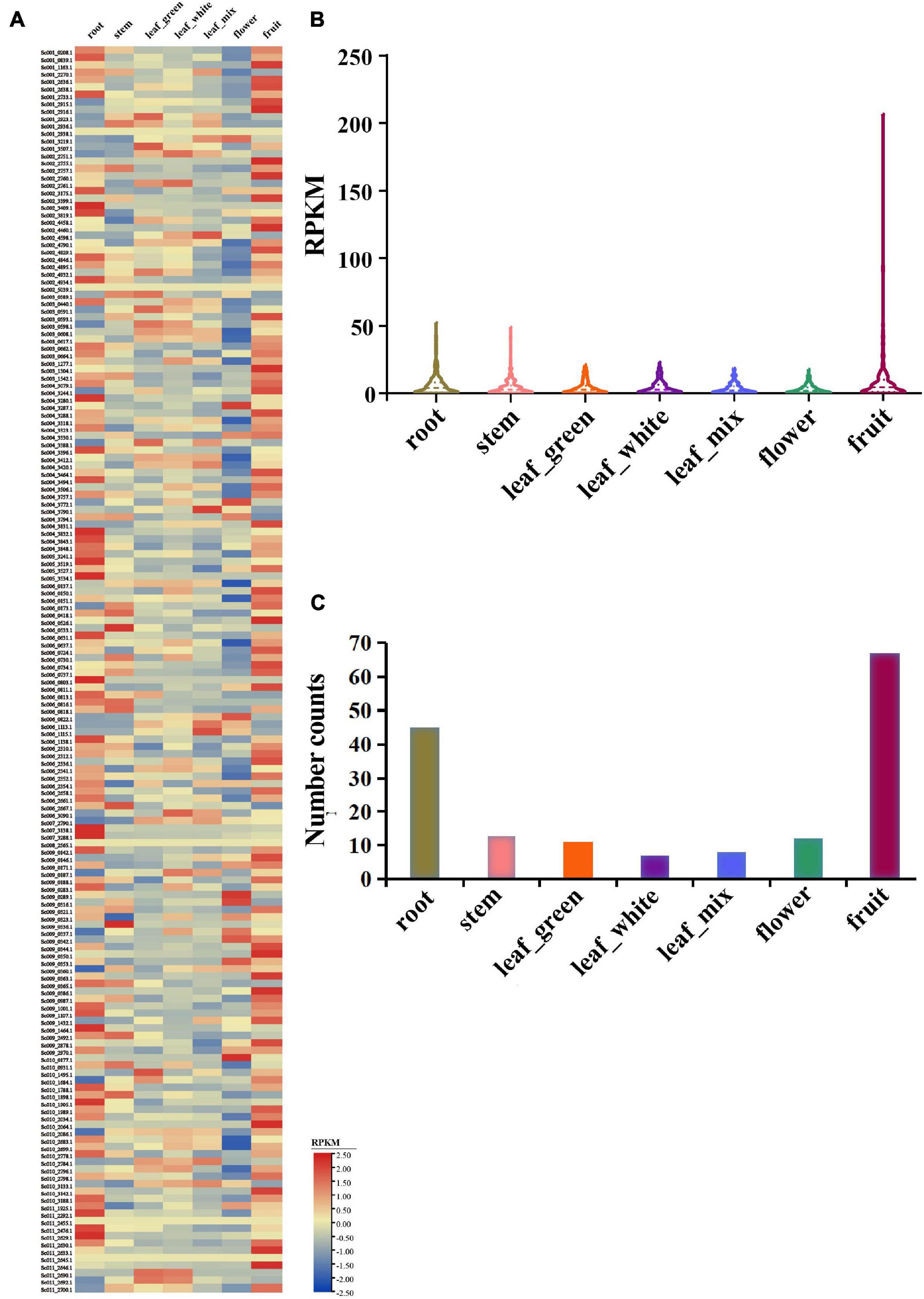
Figure 7. Expression pattern of S. chinensis NLR genes. (A) Heatmap of the expression of 168 NLR genes in seven different plant tissues. (B) Average expression of 168 NLR genes in the seven tissues. (C) Distribution of the top expression tissue for 168 NLR genes.
The average expression values of 168 genes were 6.4, 4.7, 4.3, 4.4, 3.7, 3.0, and 9.3 RPKM in the roots, stems, green leaves, white leaves, mixed color leaves, flowers, and fruits, respectively (Figure 7B). The roots and fruits had higher average R gene expression levels than the other tissues, which may be because the roots have greater direct physical interactions with soil microorganisms, providing easier access for infection, while the fruits may attract microorganisms and pathogens. The highest expression value of each gene was detected in the seven tissues. Results revealed that 168 genes were expressed in at least one tissue, 45 genes had the highest expression value in the roots, 13 in the stems, 11 in green leaves, 7 in white leaves, 8 in mixed color leaves, 12 in flowers, and 67 in the fruits (Figure 7C). Overall, the expression analysis indicated that NLR genes in S. chinensis were expressed at low levels with only some genes, showing high expression levels in specific tissues; these results are in accordance with the expressional trait of R genes, which usually remain silent but are highly expressed on exposure to pathogen stimuli.
Discussion
Dramatic Variations in NLR Gene Numbers Among Different Species
The discrepancy of NLR gene expression among angiosperms is extremely prominent. A recent study investigated 305 angiosperm genomes and discovered that the number of NLR genes per genome ranged from 5 to more than 2,000, except for one genome (Utricularia gibba), representing the only known land plant genome completely lacking in NLR genes (Liu et al., 2021). The NLR gene number varies greatly within the same family. Previous studies on Fabaceae, Solanaceae, Poaceae, and Brassicaceae identified 2- to 6-fold differences in NLR gene number among species in the same family, and up to a 20-fold difference was found in Orchidaceae (Luo et al., 2012; Shao et al., 2014; Zhang et al., 2016; Qian et al., 2017; Tirnaz et al., 2020; Xue et al., 2020b). As expected, among the seven magnoliid genomes examined in this study, the NLR gene numbers varied considerably. Our analyses covered three orders, and some taxa diverged more than 100 million years ago (One Thousand Plant Transcriptomes Initiative, 2019; Xue et al., 2020a; Yang et al., 2020). In Laurales, P. americana contained 51 NLR genes, while all the other three species had at least three times the number of genes. For example, C. kanehirae possessed 379 genes. The few NLR genes in P. americana may be related to its poor genome assembly and annotation (Rendón-Anaya et al., 2019); comparatively, all other magnoliids were better assembled.
According to the reconciled NLR gene gains and losses, CNL genes contributed much more to the current gene numbers and discrepancy among species than other classes (Figure 5). CNL genes (70/74) vastly outnumbered other classes in the common magnoliid ancestor and played an essential role in the discrepancy of NLR gene numbers we observed at present. Magnoliid TNL and RNL genes started with low copy numbers, and no follow-up vast expansions were detected, which led to minimal or overlooked effects on the whole NLR gene number variation. In actuality, the common magnoliid ancestor only inherited 14 CNL AANGs but quickly expanded up to 70 AMNGs before further species divergences. Among all four subclasses, CNL-1, -2, and -4 expanded 4- to 5-fold more, while CNL-3 genes increased up to 20-fold (from 1 to 20) and greatly contributed to the overall expansion. CNL-3 also appeared to be a magnoliid-specific expanded subclass.
In terms of the underlying mechanism, tandem duplications largely accounted for most NLR gene expansions, which is consistent with other investigated angiosperm genes (e.g., legumes, Brassicaceae, orchids, Sapindaceae, and Solanaceae) (Shao et al., 2014; Zhang et al., 2016; Qian et al., 2017; Xue et al., 2020b; Zhou et al., 2020).
Independent TNL Losses in Magnoliids
The hypothesis of TNL losses in the common monocot ancestor continues to receive increasing support from growing genomic data (Shao et al., 2016; Xue et al., 2020b). Recent studies reported the absence of TNL genes in monocots and certain eudicots, such as one basal eudicot (Aquilegia coerulea) and two Lamiales (Sesamum indicum and Mimulus guttatus) species (Collier et al., 2011; Shao et al., 2016). Therefore, these independent TNL losses may be the consequence of convergent evolution, which could be explained by inner factors, such as TNL genes starting with a few genes present in the common angiosperm ancestor that did not expand during the long-term evolution (Shao et al., 2016). Influences from outside particular habitats or lifestyles reduce the pathogenic threat, and some plants can afford the loss of several R genes (Liu et al., 2021).
The TNL gene loss has been increasingly observed in major angiosperm lineages, such as eudicots, monocots, and now in magnoliids, as well as more frequently among closely related taxa (e.g., within Lamiales) (Liu et al., 2021). Syntenic and phylogenetic evidence has shown that a whole-genome duplication event in the common angiosperm ancestor resulted in two RNL subclasses in angiosperms, namely, activated disease resistance 1 (ADR1) and N requirement gene 1 (NRG1; Shao et al., 2016; Van Ghelder et al., 2019). Growing evidence has also revealed that nearly all TNL proteins functionally rely on RNL proteins predominantly from the NRG1 lineage to confer resistance (Qi et al., 2018; Castel et al., 2019; Wu et al., 2019; Saile et al., 2020). Interestingly, RNL-NRG1 is synchronously absent with TNLs, supporting the hypothesis that TNLs rely on this indispensable downstream gene, i.e., RNL-NRG1, to transfer resistance signals (Collier et al., 2011).
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found here: National Center for Biotechnology Information (NCBI) BioProject database under accession number PRJNA764779.
Author Contributions
J-YW, J-YX, and YV designed the study and analyzed the data. J-YW wrote the manuscript. J-YX and YV participated in the revision of the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by grants from the State Key Laboratory of Palaeobiology and Stratigraphy (Nanjing Institute of Geology and Palaeontology, CAS) (No. 213124).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Zhu-Qing Shao and Yang Liu at the Nanjing University for their assistance in extracting protein-coding sequences and statistics of domain combinations by PERL scripts.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.777157/full#supplementary-material
Footnotes
- ^ http://cotton.hzau.edu.cn/EN/download.php
- ^ http://xhhuanglab.cn
- ^ https://hardwoodgenomics.org
- ^ https://genomevolution.org
- ^ https://www.ncbi.nlm.nih.gov/
- ^ http://pfam.sanger.ac.uk/
- ^ https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi
- ^ http://pfam.janelia.org/
- ^ http://smart.embl-heidelberg.de/
- ^ http://chibba.agtec.uga.edu/duplication/
References
Ameline-Torregrosa, C., Wang, B. B., O’Bleness, M. S., Deshpande, S., Zhu, H., Roe, B., et al. (2008). Identification and characterization of nucleotide-binding site-leucine-rich repeat genes in the model plant Medicago truncatula. Plant Physiol. 146, 5–21. doi: 10.1104/pp.107.104588
Andersen, E. J., Ali, S., Byamukama, E., Yen, Y., and Nepal, M. P. (2018). Disease Resistance mechanisms in plants. Genes 9:339. doi: 10.3390/genes9070339
Bailey, T. L., Williams, N., Misleh, C., and Li, W. W. (2006). MEME: discoverind and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 34, W369–W373.
Bi, G., Su, M., Li, N., Liang, Y., Dang, S., Xu, J., et al. (2021). The ZAR1 resistosome is a calcium-permeable channel triggering plant immune signaling. Cell 184, 3528–3541.e12. doi: 10.1016/j.cell.2021.05.003
Byng, J. W., Chase, M., Christenhusz, M., Fay, M. F., Judd, W. S., Mabberley, D., et al. (2016). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20. doi: 10.1111/boj.12385
Castel, B., Ngou, P. M., Cevik, V., Redkar, A., Kim, D. S., Yang, Y., et al. (2019). Diverse NLR immune receptors activate defence via the RPW8-NLR NRG1. New Phytol. 222, 966–980. doi: 10.1111/nph.15659
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Collier, S. M., Hamel, L. P., and Moffett, P. (2011). Cell death mediated by the N-terminal domains of a unique and highly conserved class of NB-LRR protein. Mol. Plant Microbe Interact. 24, 918–931. doi: 10.1094/MPMI-03-11-0050
Crooks, G. E., Hon, G., Chandonia, J.-M., and Brenner, S. E. (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. doi: 10.1101/gr.849004
Cui, H., Tsuda, K., and Parker, J. E. (2015). Effector-triggered immunity: from pathogen perception to robust defense. Annu. Rev. Plant Biol. 66, 487–511. doi: 10.1146/annurev-arplant-050213-040012
DeYoung, B. J., and Innes, R. W. (2006). Plant NBS-LRR proteins in pathogen sensing and host defense. Nat. Immunol. 7, 1243–1249. doi: 10.1038/ni1410
Fiz-Palacios, O., Schneider, H., Heinrichs, J., and Savolainen, V. (2011). Diversification of land plants: insights from a family-level phylogenetic analysis. BMC Evol. Biol. 11:341. doi: 10.1186/1471-2148-11-341
Hao, W., Collier, S. M., Moffett, P., and Chai, J. (2013). Structural basis for the interaction between the potato virus X resistance protein (Rx) and its cofactor Ran GTPase-activating protein 2 (RanGAP2). J. Biol. Chem. 288, 35868–35876. doi: 10.1074/jbc.M113.517417
Hu, Z., Yan, C., Liu, P., Huang, Z., Ma, R., Zhang, C., et al. (2013). Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science 341, 172–175. doi: 10.1126/science.1236381
Jacob, P., Kim, N. H., Wu, F., El-Kasmi, F., Chi, Y., Walton, W. G., et al. (2021). Plant “helper” immune receptors are Ca2+-permeable nonselective cation channels. Science 373, 420–425. doi: 10.1126/science.abg7917
Jia, Y., Yuan, Y., Zhang, Y., Yang, S., and Zhang, X. (2015). Extreme expansion of NBS-encoding genes in Rosaceae. BMC Genet. 16:48. doi: 10.1186/s12863-015-0208-x
Jiao, Y., Wickett, N. J., Ayyampalayam, S., Chanderbali, A. S., Landherr, L., Ralph, P. E., et al. (2011). Ancestral polyploidy in seed plants and angiosperms. Nature 473, 97–100. doi: 10.1038/nature09916
Jones, J. D., Vance, R. E., and Dangl, J. L. (2016). Intracellular innate immune surveillance devices in plants and animals. Science 354:aaf6395. doi: 10.1126/science.aaf6395
Jubic, L. M., Saile, S., Furzer, O. J., El Kasmi, F., and Dangl, J. L. (2019). Help wanted: helper NLRs and plant immune responses. Curr. Opin. Plant Biol. 50, 82–94. doi: 10.1016/j.pbi.2019.03.013
Judd, W. S., Campbell, C. S., Kellogg, E. A., Stevens, P. F., and Donoghue, M. J. (2002). Plant Systematics: A Phylogenetic Approach. Sunderland, MA: Sinauer Associates, Inc.
Kalyaanamoorthy, S., Minh, B. Q., Wong, T., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Kourelis, J., and van der Hoorn, R. A. L. (2018). Defended to the nines: 25 years of resistance gene 40 cloning identifies nine mechanisms for R protein function. Plant Cell 30, 285–299. doi: 10.1105/tpc.17.00579
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lee, T. H., Tang, H., Wang, X., and Paterson, A. H. (2013). PGDD: a database of gene and genome duplication in plants. Nucleic Acids Res. 41, D1152–D1158. doi: 10.1093/nar/gks1104
Leister, D. (2004). Tandem and segmental gene duplication and recombination in the evolution of plant disease resistance gene. Trends Genet. 20, 116–122. doi: 10.1016/j.tig.2004.01.007
Liu, Y., Zeng, Z., Li, Q., Jiang, X. M., Jiang, Z., Tang, J. H., et al. (2021). An angiosperm NLR atlas reveals that NLR gene reduction is associated with ecological specialization and signal transduction component deletion. Mol. Plant. doi: 10.1016/j.molp.2021.08.001 [Epub ahead of print].
Luo, S., Zhang, Y., Hu, Q., Chen, J., Li, K., Lu, C., et al. (2012). Dynamic nucleotide-binding site and leucine-rich repeat-encoding genes in the grass family. Plant Physiol. 159, 197–210. doi: 10.1104/pp.111.192062
Magallón, S. (2010). Using fossils to break long branches in molecular dating: a comparison of relaxed clocks applied to the origin of angiosperms. Syst. Biol. 59, 384–399. doi: 10.1093/sysbio/syq027
Minh, B. Q., Nguyen, M. A. T., and Von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. doi: 10.1093/molbev/mst024
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nie, Z. L., Wen, J., and Sun, H. (2007). Phylogeny and biogeography of Sassafras (Lauraceae) disjunct between eastern Asia and eastern North America. Plant Syst. Evol. 267, 191–203. doi: 10.1007/s00606-007-0550-1
One Thousand Plant Transcriptomes Initiative (2019). One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574, 679–685. doi: 10.1038/s41586-019-1693-2
Pan, Q., Wendel, J., and Fluhr, R. (2000). Divergent evolution of plant NBS-LRR resistance gene homologues in dicot and cereal genomes. J. Mol. Evol. 50, 203–213. doi: 10.1007/s002399910023
Parker, J. E., Coleman, M. J., Szabò, V., Frost, L. N., Schmidt, R., van der Biezen, E. A., et al. (1997). The Arabidopsis downy mildew resistance gene RPP5 shares similarity to the toll and interleukin-1 receptors with N and L6. Plant Cell 9, 879–894. doi: 10.1105/tpc.9.6.879
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., and Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667. doi: 10.1038/nprot.2016.095
Qi, T., Seong, K., Thomazella, D., Kim, J. R., Pham, J., Seo, E., et al. (2018). NRG1 functions downstream of EDS1 to regulate TIR-NLR-mediated plant immunity in Nicotiana benthamiana. Proc. Natl. Acad. Sci. U. S. A. 115, E10979–E10987. doi: 10.1073/pnas.1814856115
Qian, L. H., Zhou, G. C., Sun, X. Q., Lei, Z., Zhang, Y. M., Xue, J. Y., et al. (2017). Distinct patterns of gene gain and loss: diverse evolutionary modes of nbs-encoding genes in three solanaceae crop species. G3 (Bethesda) 7, 1577–1585. doi: 10.1534/g3.117.040485
Rendón-Anaya, M., Ibarra-Laclette, E., Méndez-Bravo, A., Lan, T., Zheng, C., Carretero-Paulet, L., et al. (2019). The avocado genome informs deep angiosperm phylogeny, highlights introgressive hybridization, and reveals pathogen-influenced gene space adaptation. Proc. Natl. Acad. Sci. U. S. A. 116, 17081–17089. doi: 10.1073/pnas.1822129116
Sacco, M. A., Mansoor, S., and Moffett, P. (2007). A RanGAP protein physically interacts with the NB-LRR protein Rx, and is required for Rx-mediated viral resistance. Plant J. 52, 82–93. doi: 10.1111/j.1365-313X.2007.03213.x
Saile, S. C., Jacob, P., Castel, B., Jubic, L. M., Salas-Gonzáles, I., Bäcker, M., et al. (2020). Two unequally redundant “helper” immune receptor families mediate Arabidopsis thaliana intracellular “sensor” immune receptor functions. PLoS Biol. 18:e3000783. doi: 10.1371/journal.pbio.3000783
Shao, Z. Q., Xue, J. Y., Wang, Q., Wang, B., and Chen, J. Q. (2019). Revisiting the Origin of Plant NBS-LRR Genes. Trends Plant Sci. 24, 9–12. doi: 10.1016/j.tplants.2018.10.015
Shao, Z. Q., Xue, J. Y., Wu, P., Zhang, Y. M., Wu, Y., Hang, Y. Y., et al. (2016). Large-scale analyses of angiosperm nucleotide-binding site-leucine-rich repeat genes reveal three anciently diverged classes with distinct evolutionary patterns. Plant Physiol. 170, 2095–2109. doi: 10.1104/pp.15.01487
Shao, Z. Q., Zhang, Y. M., Hang, Y. Y., Xue, J. Y., Zhou, G. C., Wu, P., et al. (2014). Long-term evolution of nucleotide-binding site-leucine-rich repeat genes: understanding gained from and beyond the legume family. Plant Physiol. 166, 217–234. doi: 10.1104/pp.114.243626
Shao, Z. Q., Zhang, Y. M., Wang, B., and Chen, J. Q. (2015). Computational identification of MicroRNA-targeted nucleotide-binding site-leucine-rich repeat genes in plants. Bio-protocol 5:e1637.
Soltis, D., Soltis, P., Endress, P., Chase, M., Manchester, S., Judd, W., et al. (2005). Phylogeny and Evolution of Angiosperms. Sunderland, MA: Sinauer Associates.
Stolzer, M., Lai, H., Xu, M., Sathaye, D., Vernot, B., and Durand, D. (2012). Inferring duplications, losses, transfers and incomplete lineage sorting with nonbinary species trees. Bioinformatics 28, i409–i415. doi: 10.1093/bioinformatics/bts386
Tameling, W. I., and Baulcombe, D. C. (2007). Physical association of the NB-LRR resistance protein Rx with a Ran GTPase-activating protein is required for extreme resistance to Potato virus X. Plant Cell 19, 1682–1694. doi: 10.1105/tpc.107.050880
Tarr, D. E., and Alexander, H. M. (2009). TIR-NBS-LRR genes are rare in monocots: evidence from diverse monocot orders. BMC Res. Notes 2:197. doi: 10.1186/1756-0500-2-197
Tirnaz, S., Bayer, P. E., Inturrisi, F., Zhang, F., Yang, H., Dolatabadian, A., et al. (2020). Resistance gene analogs in the brassicaceae: identification, characterization, distribution, and evolution. Plant Physiol. 184, 909–922. doi: 10.1104/pp.20.00835
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Van Ghelder, C., Parent, G. J., Rigault, P., Prunier, J., Giguère, I., Caron, S., et al. (2019). The large repertoire of conifer NLR resistance genes includes drought responsive and highly diversified RNLs. Sci. Rep. 9:11614. doi: 10.1038/s41598-019-47950-7
Wang, J., and Chai, J. (2020). Molecular actions of NLR immune receptors in plants and animals. Sci. China Life Sci. 63, 1303–1316. doi: 10.1007/s11427-019-1687-6
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Wu, Z., Li, M., Dong, O. X., Xia, S., Liang, W., Bao, Y., et al. (2019). Differential regulation of TNL-mediated immune signaling by redundant helper CNLs. New Phytol. 222, 938–953. doi: 10.1111/nph.15665
Xue, J. Y., Wang, Y., Wu, P., Wang, Q., Yang, L. T., Pan, X. H., et al. (2012). A primary survey on bryophyte species reveals two novel classes of nucleotide-binding site (NBS) genes. PLoS One 7:e36700. doi: 10.1371/journal.pone.0036700
Xue, J. Y., Zhao, T., Liu, Y., Liu, Y., Zhang, Y. X., Zhang, G. Q., et al. (2020b). Genome- wide analysis of the nucleotide binding site leucine-rich repeat genes of four orchids revealed extremely low numbers of disease resistance genes. Front. Genet. 10:1286. doi: 10.3389/fgene.2019.01286
Xue, J. Y., Dong, S. S., Wang, M. Q., Song, T. Q., Zhou, G. C., Li, Z., et al. (2020a). Mitochondrial genes from eighteen angiosperms fill sampling gaps for phylogenomic inferences of the early diversification of flowering plants. J. Syst. Evol. doi: 10.1111/jse.12708
Yang, L., Su, D., Chang, X., Foster, C., Sun, L., Huang, C. H., et al. (2020). Phylogenomic insights into deep phylogeny of angiosperms based on broad nuclear gene sampling. Plant Commun. 1:100027. doi: 10.1016/j.xplc.2020.100027
Yue, J. X., Meyers, B. C., Chen, J. Q., Tian, D., and Yang, S. (2012). Tracing the origin and evolutionary history of plant nucleotide-binding site-leucine-rich repeat (NBS-LRR) genes. New Phytol. 193, 1049–1063. doi: 10.1111/j.1469-8137.2011.04006.x
Zhang, Y. M., Shao, Z. Q., Wang, Q., Hang, Y. Y., Xue, J. Y., Wang, B., et al. (2016). Uncovering the dynamic evolution of nucleotide-binding site-leucine-rich repeat (NBS-LRR) genes in Brassicaceae. J. Integr. Plant Biol. 58, 165–177. doi: 10.1111/jipb.12365
Zhou, G. C., Li, W., Zhang, Y. M., Liu, Y., Zhang, M., Meng, G. Q., et al. (2020). Distinct Evolutionary patterns of NBS-encoding genes in three soapberry family (sapindaceae) species. Front. Genet. 11:737. doi: 10.3389/fgene.2020.00737
Zhou, L. L., Su, Y. C. F., Thomas, D. C., and Saunders, R. M. K. (2012). Out-of-Africa dispersal of tropical floras during the Miocene climatic optimum: evidence from Uvaria (Annonaceae). J. Biogeogr. 39, 322–335. doi: 10.1111/j.1365-2699.2011.02598.x
Keywords: magnoliids, NLR genes, phylogeny, evolution pattern, expression
Citation: Wu J-Y, Xue J-Y and Van de Peer Y (2021) Evolution of NLR Resistance Genes in Magnoliids: Dramatic Expansions of CNLs and Multiple Losses of TNLs. Front. Plant Sci. 12:777157. doi: 10.3389/fpls.2021.777157
Received: 15 September 2021; Accepted: 24 November 2021;
Published: 21 December 2021.
Edited by:
Changsoo Kim, Chungnam National University, South KoreaReviewed by:
Liangsheng Zhang, Zhejiang University, ChinaTong Wei, Beijing Genomics Institute (BGI), China
Copyright © 2021 Wu, Xue and Van de Peer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia-Yu Xue, xuejy@njau.edu.cn; xuejy@nju.edu.cn; Yves Van de Peer, yves.vandepeer@psb.ugent.be; yvpee@psb.vib-ugent.be