 Noha Alassaf
Noha Alassaf Hala Attia
Hala Attia- 1Department of Pharmacology and Toxicology, College of Pharmacy, King Saud University, Riyadh, Saudi Arabia
- 2Department of Biochemistry, College of Pharmacy, Mansoura University, Mansoura, Egypt
Cisplatin (CP) is a broad-spectrum antineoplastic agent, used to treat many different types of malignancies due to its high efficacy and low cost. However, its use is largely limited by acute kidney injury (AKI), which, if left untreated, may progress to cause irreversible chronic renal dysfunction. Despite substantial research, the exact mechanisms of CP-induced AKI are still so far unclear and effective therapies are lacking and desperately needed. In recent years, necroptosis, a novel subtype of regulated necrosis, and autophagy, a form of homeostatic housekeeping mechanism have witnessed a burgeoning interest owing to their potential to regulate and alleviate CP-induced AKI. In this review, we elucidate in detail the molecular mechanisms and potential roles of both autophagy and necroptosis in CP-induced AKI. We also explore the potential of targeting these pathways to overcome CP-induced AKI according to recent advances.
1 Introduction
Cancer represents a serious health threat and is considered one of the main deadly diseases worldwide (Benyamini et al., 2003; Nguyen et al., 2022). In 2019, approximately 23.6 million new cases and 10 million deaths were associated with cancer (Kocarnik et al., 2022). Cisplatin (CP) is one of the most effective antineoplastic drugs for the management of several solid-organ malignancies including Hodgkin’s and non-Hodgkin’s lymphomas, sarcomas, as well as head, lung, neck, bladder, breast, testicular, ovarian, and cervical cancers (Dasari and Bernard Tchounwou, 2014; Manohar and Leung, 2018). It has been reported that around 50% of all patients are treated with CP in their chemotherapeutic regimens (Chen and Chang, 2019). Interestingly, despite its earlier discovery in 1844 by the Italian chemist, Michele Peyrone, CP did not attract much attention until 1965 (Dasari and Bernard Tchounwou, 2014). During that year, Rosenberg et al. (1965) noticed that the platinum compound released from the platinum electrodes into the growth medium of Escherichia coli markedly affects its cellular division and filamentation. This observation has driven a strong wave of preclinical and clinical research for testing its anticancer effect afterward. In 1969, the anticancer effect of CP was evaluated in the sarcoma mouse model (Rosenberg et al., 1969), and by 1971, CP had entered phase I clinical trials and was finally approved by the US Food and Drug Administration for the management of cancers in 1978 (Andrea and Reddy, 2018).
It is currently well established that CP mediates its tumoricidal effects through binding to DNA, causing formation of intra- and interstrand cross-links. These cross-links distort the DNA structure and subsequently prevent DNA synthesis and replication in rapidly proliferating cancer cells (Dugbartey et al., 2016) (Figure 1). Despite its efficiency, the clinical use of CP is complicated by multiple side effects such as ototoxicity, neurotoxicity, cardiotoxicity, hepatotoxicity, and cancer cell resistance (Karasawa and Steyger, 2015; Ghosh, 2019) (Figure 2). However, the main serious, dose-limiting, and potentially irreversible side effect of CP is nephrotoxicity (Miller et al., 2010; Hägerström et al., 2019).
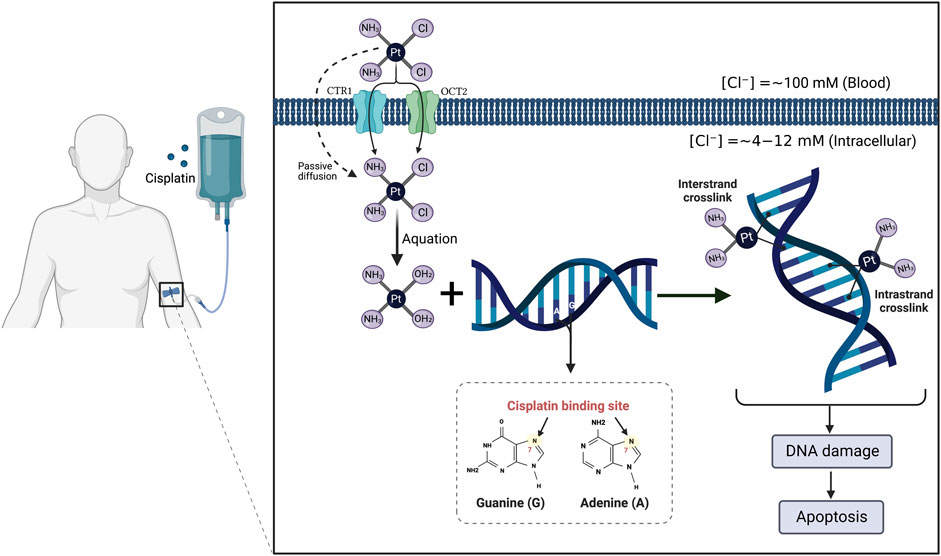
FIGURE 1. Mechanism of action of cisplatin (CP). The CP structure is composed of a platinum atom in the center, surrounded by two chloride atoms and two ammonia atoms in the cis configuration. Following its intravenous infusion, CP is passed through the cell membrane by passive diffusion or active transport. In the bloodstream, the high chloride concentration (∼100 mM) retains CP in a relatively stable structure and prevents its hydrolysis. Once it enters the cell where the chloride concentration is greatly reduced (∼4–12 mM), the two chlorides are replaced by water molecules in a process termed aquation to produce potent, positively charged electrophilic product. The resulting product covalently binds to the N7 atom of the purine bases, preferentially guanines, in the DNA causing formation of either intrastrand cross-linking when the two organic bases are located on the same DNA strand or interstrand cross-linking is produced if the organic bases are on opposite strands. These cross-links distort the DNA structure and subsequently lead to cell cycle arrest and apoptosis.

FIGURE 2. Side effects associated with cisplatin (CP) administration. Numerous deleterious side effects continue to restrict the therapeutic efficacy of CP; with nephrotoxicity especially acute kidney injury being the most serious issue.
CP-induced renal toxicity was originally reported by laboratory animal research in 1971 (Kociba and Sleight, 1971). In humans, CP nephrotoxicity reaches approximately one-third of patients undergoing treatment. Notably, at least 40% of these patients failed to complete the course of therapy owing to renal impairment, which, in turn, negatively affects patients’ survival rate (Shiraishi et al., 2000; Hamroun et al., 2019). CP nephrotoxicity may result in a wide range of manifestations including distal renal tubular acidosis, chronic kidney disease (CKD), defect in urinary concentrating ability, thrombotic microangiopathy, transient proteinuria, glucosuria, hyperuricemia, hypocalcemia, and other electrolyte imbalances, most importantly magnesium and potassium losses (Miller et al., 2010; Oh et al., 2014; Karasawa and Steyger, 2015). However, the most common presentation of CP-induced nephrotoxicity is acute kidney injury (AKI) (Miller et al., 2010), which is characterized by the rapid decline of renal excretory function leading to the accumulation of nitrogenous waste products, water, and electrolytes (Ostermann and Joannidis, 2016; Hu et al., 2021). AKI is a global health problem associated with high mortality which far exceeds those of heart failure, diabetes mellitus, breast, and prostate carcinoma collectively (Lewington et al., 2013).
The pathogenesis of CP-induced AKI is complex and involves multiple molecules and factors. Oxidative stress, mitochondrial dysfunction, inflammation, and apoptosis are all participating in the progression of CP-induced AKI (Xu et al., 2015; Liang et al., 2016). Despite significant progress in elucidation of molecular pathways underlying CP nephrotoxicity, there is no approved treatment currently available for managing such nephrotoxicity except amifostine, which is no longer recommended for this purpose (Sharp and Siskind, 2017). This necessitates further investigation of new molecular targets for preventing or treating this problem. Interestingly, necroptosis and autophagy pathways have emerged recently and have been shown to have a crucial role in CP-induced AKI (Rovetta et al., 2012; Holditch et al., 2019). While many reviews have addressed mechanisms of CP nephrotoxicity, none cover the specific role of both autophagy and necroptosis and their potential for therapeutic intervention. In this review article, we focus on necroptosis and autophagy pathways, giving a comprehensive understanding of their molecular mechanisms, roles in CP-induced AKI, and the potential of targeting necroptosis or autophagy pathways for attenuation of CP-induced AKI. The review also highlights the impact of such interventions on the effectiveness of CP chemotherapy.
2 Materials and methods
The PubMed/MEDLINE, Google Scholar, Wiley, and Science Direct databases were used to retrieve relevant publications published until October 2022. We conducted searches using the following search terms: “cisplatin”, “cancer”, “cisplatin nephrotoxicity”, “acute kidney injury”, “cisplatin-induced AKI”, “autophagy”, “AMPK-mTOR-mediated autophagy”, “necroptosis”, “RIPK1/RIPK3/MLKL-mediated necroptosis”, “necroinflammation”, “nephroprotective”, “autophagy activators”, and “necroptosis inhibitors”. The abstracts or full texts of English-written papers were reviewed to determine if they matched the relevant section. Additional studies were obtained from the reference sections of selected studies.
3 Renal cellular uptake and biotransformation of cisplatin (CP)
The kidney is the main route of CP excretion, since it has been demonstrated that the concentration of CP in the kidneys is five-fold greater than that in the blood, and therefore such accumulation of the drug within the kidney cells contributes to its nephrotoxicity (Ozkok and Edelstein, 2014; Abdel-Daim et al., 2019). CP is excreted via both glomerular filtration and tubular secretion, and it is subsequently accumulated in kidney cells, specifically in the proximal tubular cells. Regarding tubular secretion, CP is actively transported into tubular cells through two membrane transporters, organic cation transporter 2 (OCT2) and copper transporter 1 (CTR1) (Gómez-Sierra et al., 2018).
It is currently believed that CP is actively metabolized in the kidney cells to produce a potent nephrotoxic metabolite (Miller et al., 2010). This pathway begins with the binding of CP to reduced glutathione (GSH) by the action of the glutathione-S-transferase (GST) enzyme in circulation (Gómez-Sierra et al., 2018). The resulting GSH-conjugates are cleaved into a cysteinyl-glycine-conjugate by gamma glutamyltranspeptidase which is localized on the surface of the kidney proximal tubule (Miller et al., 2010; Gómez-Sierra et al., 2018). Aminodipeptidase, also present on the surface of these cells, further metabolized the resulting conjugates to yield cysteine conjugates (Gómez-Sierra et al., 2018). Then, cysteine conjugates are taken up into tubular cells, where they are converted by cysteine-S-conjugate beta-lyase to form highly reactive thiols (Miller et al., 2010). Finally, the formed thiols bind to several cellular proteins and ultimately cause damage and death of renal tubular cells (Zhang and Hanigan, 2003; Peres and da Cunha, 2013).
4 Necroptosis in CP-induced acute kidney injury (AKI)
4.1 Fundamentals of necroptosis
4.1.1 General overview of necroptosis
The term “programmed cell death” had been commonly used to describe caspase-dependent apoptosis until caspase-independent necrosis was identified (Linkermann et al., 2012). Necroptosis (also called regulated necrosis) is a recently recognized form of cell death that is activated during apoptosis-compromised conditions and shares characteristics of both accidental necrosis and apoptosis (Kaczmarek et al., 2013; Dhuriya and Sharma, 2018; Oliveira et al., 2018). Necroptosis is programmed (like apoptosis) and morphologically characterized by cellular swelling, rupture of the plasma membrane, and organelle dysfunction (like necrosis) (Weinlich et al., 2017; Dhuriya and Sharma, 2018). Additionally, biochemical features of necroptosis may include energy depletion, formation of reactive oxygen species (ROS), and accumulation of calcium (Ca2+) (Cho, 2018). Physiologically, necroptotic cell death is emerging as an important process for normal development and host defense against various pathogens (Fulda, 2013; Wu et al., 2014; Nailwal and Chan, 2019). However, dysregulation of necroptosis has been shown to be associated with several diseases and tissue damages including cancer (Dhuriya and Sharma, 2018), ischemia/reperfusion injury (Fulda, 2013), acute pancreatitis (Wang G. et al., 2016), colitis (Hou et al., 2019), drug-induced hepatotoxicity (Ramachandran et al., 2013), atherosclerosis (Coornaert et al., 2018), and neurodegeneration (Oliveira et al., 2018).
4.1.2 Molecular mechanism and regulation of necroptotic cell death
Necroptosis is considered the best and most studied form of regulated necrosis and its molecular mechanism is being thoroughly investigated in recent years (Linkermann, 2016; Li L. et al., 2021). A cascade of several kinases including receptor-interacting protein kinase (RIPK)-1, RIPK3, and mixed lineage kinase domain-like protein (MLKL) has been identified to play a critical role in the regulation of the necroptotic pathway (Wang X. et al., 2018; Shan et al., 2018). Necroptosis can be triggered by multiple endogenous and exogenous stimuli including anticancer agents, metabolic disturbance, ischemia/reperfusion injury, and activation of death receptors such as tumor necrosis factor-α (TNF-α) receptor (TNFR), toll-like receptors (TLRs) particularly TLR3/4, as well as interferon (IFN) receptors (Wang G. et al., 2016; Seifert and Miller, 2017; Dhuriya and Sharma, 2018). Despite diversity of stimuli that responsible for necroptosis initiation, TNF-α remains the best-characterized and probably the most important trigger of necroptosis (Vandenabeele et al., 2010; Shan et al., 2018).
The binding of TNF-α to its receptor, TNFR1, can lead to either cellular survival, apoptosis, or necroptosis (Seifert and Miller, 2017) (Figure 3). Stimulation of TNFR1 induces certain conformational changes in the receptor, enabling its cytosolic portion to recruit several proteins to form the prosurvival complex I consisting of TNFR-associated death domain (TRADD), TNFR-associated factor 2 and 5 (TRAF2/5), RIPK1, cellular inhibitor of apoptosis proteins 1/2 (cIAP1/2), and linear ubiquitin chain assembly complex (LUBAC) (Nikoletopoulou et al., 2013; Seifert and Miller, 2017; Dhuriya and Sharma, 2018). Within complex I, cIAPs and LUBAC trigger RIPK1 ubiquitination leading to stabilization of complex I, which in turn induces recruitment of TGF-β-activating kinase 1 (TAK1), TAK1-binding protein 2 (TAB2), and TAB3 to generate TAK1-TAB2-TAB3 complex (Liu et al., 2021; Seo et al., 2021). TAK1 phosphorylates and activates the IκB kinase (IKK) complex and MAPK kinases (MKKs), which in turn stimulates nuclear factor kappa B (NF-κB) and activator protein 1 (AP-1) transcriptional activity leading to inflammation and cell survival (Mihaly et al., 2014).
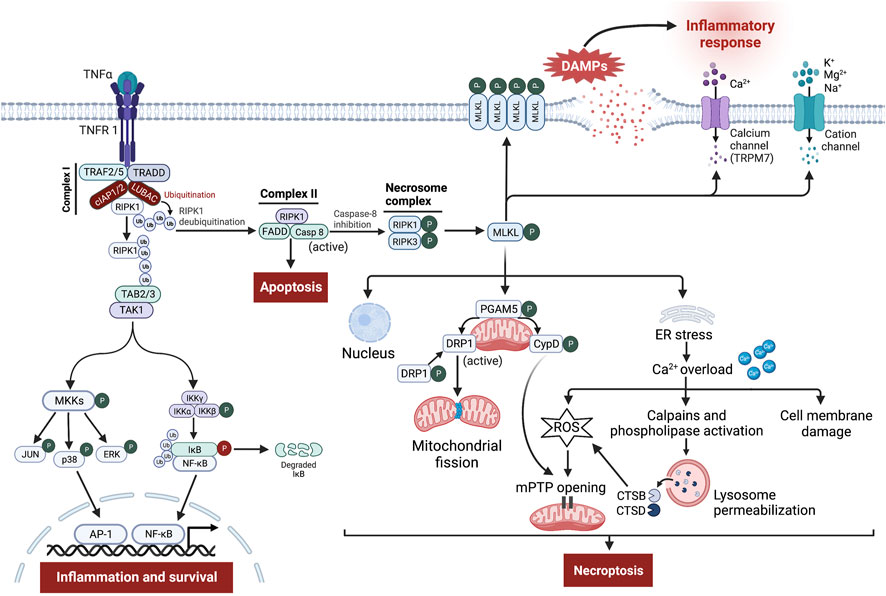
FIGURE 3. Signaling pathway leading to tumor necrosis factor-α (TNF-α)-receptor 1 (TNFR 1)-mediated necroptosis. Ligation of TNF-α to TNFR1 leads to the generation of the membrane-bound complex I, in which cellular inhibitor of apoptosis proteins (cIAPs) and linear ubiquitin chain assembly complex (LUBAC) polyubiquitinate receptor-interacting protein kinase (RIPK)-1 (RIPK1), directing it towards proteasomal degradation and stabilize complex I, which contributes to inflammation and cell survival through stimulation of nuclear factor kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) pro-survival pathways. However, the deubiquitination of RIPK1 induces the assembly of complex II involving Fas-associated death domain (FADD), RIPK1, and caspase-8. Normally, caspase-8 cleaves RIPK1 and RIPK3, thereby triggering the activation of apoptosis. Nevertheless, upon caspase-8 inhibition, RIPK1 binds to RIPK3 to form the necrosome complex, followed by phosphorylation and formation of mixed lineage kinase domain-like protein (MLKL) oligomers. These oligomers either form pores in the cell membrane or recruit cation channels and calcium (Ca2+) channels, namely transient receptor potential melastatin related 7 (TRPM7), thereby allowing the influx of extracellular ions, disrupting osmotic pressure, leading to cell rupture and release of damage-associated molecular patterns (DAMPs) which in turn provoke potent inflammatory responses. Besides, phosphoglycerate mutase family member 5 (PGAM5) on the mitochondrial membrane is recruited and phosphorylated upon necroptosis activation. Once activated, PGAM5 phosphorylates cyclophilin D (CypD) and reverses phosphorylation of dynamin-related protein 1 (Drp1), resulting in their activation where they contribute to mitochondrial fission and mitochondrial permeability transition pore (mPTP) opening, eventually leading to necroptosis. Moreover, a small part of MLKL oligomers translocates into the nucleus, facilitating necroptotic cell death, while other moves to the endoplasmic reticulum (ER), triggering stress response that causes intracellular Ca2+ overload. Increased Ca2+ concentration executes necroptosis through damage to the cell membrane, overproduction of reactive oxygen species (ROS), which promotes mPTP opening, and finally through stimulation of Ca2+-dependent enzymes like calpains and phospholipase leading to permeabilization of lysosome membrane and subsequent release of cathepsin B (CTSB) and D (CTSD) into the cytosol. These enzymes in turn lead to lysosomal dysfunction, and increased oxidative potential, all of which events result in necroptosis.
Alternatively, during specific conditions, where complex I is destabilized or the ubiquitination process is inhibited, RIPK1 dissociates from complex I and binds to Fas-associated death domain (FADD) and caspase-8, forming apoptotic complex II that results in induction of TNF-α-induced apoptosis (Seifert and Miller, 2017; Dhuriya and Sharma, 2018; Seo et al., 2021). Within complex II, caspase-8 inhibits necroptosis through proteolytic cleavage of RIPK1 and RIPK3 (Nikoletopoulou et al., 2013). However, exhaustion or reduced activity of caspase-8 shifts cellular fate from apoptosis towards necroptosis (Cho et al., 2009; Leeper, 2016; Seifert and Miller, 2017). Thus, necroptosis acts as a backup pathway for cell death during impairment of caspase-8-dependent apoptosis (Zhang et al., 2016; Dhuriya and Sharma, 2018).
From a molecular point of view, the necroptosis pathway is initiated by auto- and trans-phosphorylation of RIPK1 and RIPK3 at Ser-227, resulting in stimulation of their kinase activity and assembly of a heterodimer protein complex termed necrosome via their RIPK homotypic interaction motif domains (Fulda, 2013; Cho, 2018; Dhuriya and Sharma, 2018; Oliveira et al., 2018; Hou et al., 2019). Then, RIPK3 recruits and phosphorylates its substrate MLKL at Thr-357/Ser-358 (Oliveira et al., 2018; Hou et al., 2019). Following phosphorylation, MLKL oligomerizes and a small part of these oligomers moves towards the cell membrane where they mediate their effect in two different ways, either directly by facilitating the formation of pores across the cell membrane mediated through binding to membrane phospholipids like phosphatidylinositol phosphate and cardiolipin or indirectly by increasing influx of ions (Na+, K+, Mg2+, and Ca2+) through recruitment of cation channels and Ca2+ channels, the transient receptor potential melastatin related 7 (TRPM7), thereby increasing osmotic pressure and eventually leading to cell rupture (vanden Berghe et al., 2016; Seifert and Miller, 2017; Dhuriya and Sharma, 2018; Li L. et al., 2021). Meanwhile, another part of MLKL oligomers translocates into the membranes of various organelles e.g., nucleus, mitochondria, endoplasmic reticulum (ER), and lysosomes, causing their damage via necroptotic cell death (Li L. et al., 2021; Jantas and Lasoń, 2021).
4.2 Emerging role of necroptosis in CP-induced AKI
Cellular death and inflammation are largely found in the proximal tubular cells, which concur with the main target of CP accumulation (Xu et al., 2015; Ning et al., 2018). In the past, most of the previous research has investigated and characterized apoptosis as the primary cell death in the context of CP nephrotoxicity (Lau, 1999; Cummings and Schnellmann, 2002; Jiang et al., 2004). As a result, strategies targeting the apoptosis mediators have long been studied for the treatment of CP-induced renal injury (Wei et al., 2007; Molitoris et al., 2009; Pabla et al., 2011). However, inhibition of inflammation, tubular necrosis, and necroptosis was sufficient to protect against CP nephrotoxicity even in the presence of apoptosis (Kim et al., 2012; Ozkok et al., 2016; Yoon and Kim, 2018). Moreover, two reports surprisingly demonstrated that suppression of apoptosis alone fails to ameliorate or stop the development of CP-induced nephrotoxicity (Herzog et al., 2012; Sridevi et al., 2013). These data could reflect our limited knowledge of the complex cell death mechanisms involved in the pathogenesis of AKI. Recent advances suggest that in addition to apoptosis, other forms of programmed cell death co-exist and play a significant role in the pathogenesis of CP-induced renal injury. In recent years, tremendous interest has been focused on necroptosis as the most important mechanism of tubular death in CP-induced AKI. The implication of necroptosis in CP-induced AKI was first reported about a decade ago by Tristão’s group (Tristão et al., 2012). Following this observation, several in vitro and in vivo studies demonstrated that the expression of necroptosis-related proteins (RIPK1, RIPK3, and MLKL) was simultaneously induced following CP administration (Gao et al., 2016; Al-Salam et al., 2021). Interestingly, Xu et al. (2015) reported that the CP-induced tubular damage could be diminished by either knockout or inhibition of RIPK3 or MLKL activity. Later, Gao et al. (2016) and Gwon et al. (2021) further confirmed the implication of necroptosis in CP-induced AKI where pharmacological inhibition of RIPK1/RIPK3/MLKL axis by protocatechuic aldehyde and 6-shogaol, respectively significantly attenuates renal injury. Similarly, pretreatment with novel RIPK1 inhibitors, Cpd-71 and Cpd-2 mitigated CP nephropathy (Wang et al., 2019; Li C. et al., 2022). Likewise, Liu et al. (2022b) found that the novel heat shock protein (Hsp) 90 inhibitor, C-316-1 facilitates ubiquitination and degradation of RIPK1, thereby inhibiting RIPK1-mediated necroptosis and subsequently attenuating CP-induced AKI. Recently, several investigators further emphasized the significance of this contribution by showing that the protective and detrimental effects of various proteins in the CP-induced AKI model were mediated via modulation of necroptosis signaling. In this regard, Gao et al. (2018) showed that in vitro and in vivo enhancement of E-cadherin protein by PPBICA treatment protects against CP-induced AKI by attenuating necroptosis and necroptotic inflammation. In a comparable way, the protective role of Numb and galectin-3 proteins against CP injury has been primarily attributed to the inhibition of tubular necroptosis and/or its related inflammatory response (Liu et al., 2020; Al-Salam et al., 2021). In contrast, another preclinical study found that pyruvate dehydrogenase kinase 4 aggravated CP-induced AKI at least partially by activating necroptosis, while pharmacological or genetic disruption of its activity dampened CP-induced necroptosis, thereby attenuating AKI in mice (Oh et al., 2017). More recently, Yang et al. (2019); Yang et al. (2021) and Wang F. et al. (2022) showed that Smad 2/3 and stratifin proteins play a detrimental role in CP-induced AKI by inducing necroptosis and necroinflammation, and interestingly, targeting these proteins suppressed the necroptosis signaling and thereby alleviating AKI.
4.2.1 Necroptosis and oxidative stress
Intriguingly, how CP would activate tubular necroptosis is still largely unknown, but one possible explanation could be related to the prooxidant activity of CP. Upon administration, CP amplifies the formation of ROS including hydrogen peroxide, hydroxyl radical, and superoxide anion through stimulation of nicotinamide adenine dinucleotide phosphate oxidase enzymes (Chirino et al., 2008; Gómez-Sierra et al., 2018). In addition, CP also affects mitochondrial function by inhibiting activities of various antioxidant enzymes such as GST, GSH-peroxidase, and superoxide dismutase, resulting in an imbalance between oxidant production and endogenous antioxidant defense system, which constitutes oxidative stress (Karasawa and Steyger, 2015). CP may also directly disrupt the mitochondrial respiratory chain leading to the generation of ROS and impairment of mitochondrial function (Pabla and Dong, 2008; Manohar and Leung, 2018). Moreover, during its conversion to a more potent nephrotoxic metabolite, CP binds to GSH, a powerful antioxidant molecule leading to depletion of its cellular levels (Manohar and Leung, 2018). A growing number of studies have shown the reciprocal relationship between ROS and the necroptosis pathway. ROS directly stimulates RIPK1 autophosphorylation, thereby enabling RIPK3 recruitment and necrosome formation (Zhang Y. et al., 2017). In addition, Fan et al. (2022) recently found that increased levels of ROS induced necroptotic cell death in renal tubules, while antioxidants like N-acetylcysteine significantly attenuated this effect. Similarly, inhibition of ROS by butylated hydroxyanisole suppressed activation of TNF-induced necroptosis, suggesting the critical role of ROS in this form of cellular death (Lin et al., 2004). Conversely, following necroptotic stimuli, RIPK3 activates several metabolic regulatory enzymes, thereby increasing energy metabolism and ROS, which in turn further enhance necroptosis activation (Zhang et al., 2009). Similarly, Yang Z. et al. (2018) showed that the necrosome-containing RIPK3 and MLKL enhanced aerobic respiration, which in turn increased the production of mitochondrial ROS. These ROS act in a positive feedback circle to induce necroptosis. This connection was also confirmed by Zhu et al.’s (2018) group who found that upregulation of RIPK3 triggers ER stress which subsequently increased Ca2+ overload and xanthine oxidase activity leading to overproduction of ROS, all of which was reduced by RIPK3 deletion. Collectively, these data may suggest that ROS outbursts induced by CP treatment may represent a critical mediator in regulating necroptosis induction.
4.2.2 Necroptosis and inflammation
Notably, necroptosis rather than apoptosis provokes the inflammatory reaction in AKI (Meng et al., 2018). Dying of proximal tubular cells by necroptosis induced the release of endogenous components like damage-associated molecular patterns (DAMPs) including high-mobility group box 1, Hsps, uric acid, interleukin-33, etc., which in turn activate downstream inflammatory signaling like TLRs signaling, thereby triggering robust inflammatory responses (Gao et al., 2018; Seo et al., 2021). Previous studies using genetic or small-molecule inhibitors of necroptosis via RIPK1/RIPK3/MLKL axis have given accumulating evidence regarding the essential role of necroptosis in ameliorating multi-organ inflammation such as skin inflammation (Bonnet et al., 2011), vascular inflammation (Lin et al., 2013), and inflammation of the pancreas (Wu et al., 2013), liver (Gautheron et al., 2014), and intestine (Welz et al., 2011; Lee et al., 2020). A similar situation was observed in kidney research, where blockade of necroptosis signaling greatly attenuates inflammatory response associated with tubulointerstitial injury and nephrotoxic nephritis (Xiao et al., 2017; Hill et al., 2018). In addition, Mulay et al. (2016a) also demonstrated that RIPK3 and MLKL-deficient mice were protected from tubular injury and interstitial inflammation induced by crystal deposition. Furthermore, necroptotic inflammation is also considered an important driving factor for kidney graft failure. The proinflammatory DAMPs molecules released by necroptosis in renal allografts triggered the inflammatory injury, thereby accelerating allograft rejection. Conversely, RIPK3 deficiency under this setting prevented necroptosis and the subsequent release of DAMPs, thereby prolonging allograft survival following transplantation (Lau et al., 2013). Besides the direct cytotoxic and prooxidant effects of CP treatment, necroptosis could be induced indirectly through the combined action of cytokines, TNF-α, TNF-related weak inducer of apoptosis, and IFN-γ, which were upregulated upon CP administration. Interestingly, suppression of the necroptotic pathway significantly diminished cytokines upregulation and attenuated the inflammatory response in the CP-induced AKI model (Xu et al., 2015). These data indicate the positive feedback circle involving necroptosis and inflammation during CP-induced AKI, where induction of one factor activates another. Similarly in myocardial infarction, stroke, and acute tubular necrosis, the inflammatory response which is activated after initial insult further augmented necroptotic cell death (Mulay et al., 2016b). This augmentation occurs either directly through the stimulation of TNFR1 by TNF released from necrotic cells or indirectly through the recruitment and activation of leukocytes including macrophages, neutrophils, lymphocytes, and other proinflammatory cells that contribute to tissue injury (Linkermann et al., 2014; Anders, 2018).
4.2.3 Necroptosis in AKI–CKD transition
Importantly, recent data indicated that the reciprocal enhancement between necroptosis and inflammation in this auto-amplification loop could further promote kidney damage, leading to fibrosis and chronic organ failure. Under the conditions of unilateral ureteral obstruction and renal ischemia/reperfusion injury, tubular necroptosis is markedly upregulated, which in turn promotes NOD-like receptor protein 3 (NLRP3) inflammasome activation leading to renal fibrosis. Whereas genetic or pharmacologic inhibition of the necroptosis axis prevented activation of necroinflammation and subsequent development of renal fibrogenesis (Chen et al., 2018; Jiang et al., 2022; Xuan et al., 2022), implying a relationship between necroinflammation and renal fibrosis. Most importantly, in the CP-induced AKI model, Landau et al. (2019) found that continuous activation of regulated necrosis following CP treatment was shown to be the most important factor that drives the transition and progression of AKI to chronic irreparable kidney disease. These findings suggest involvement of necroptosis signaling in both CP-induced acute and chronic kidney diseases, further making necroptosis a valuable target for therapeutic intervention during CP chemotherapy.
4.2.4 Targeting necroptosis in CP-induced AKI
Given the central role of the necroptotic pathway and its associated inflammation in the pathogenesis of CP-induced AKI along with their potential role in AKI-to-CKD transition, multiple agents with anti-necroptotic activity were tested in the CP-induced AKI model. However, currently available agents are still limited (Table 1) and despite their considerable merits, multiple concerns were raised concerning the use of these inhibitors, specifically those targeting RIPK1. The first issue associated with RIPK1 inhibitors like wogonin and PPBICA is their low water solubility and limited bioavailability (Baek et al., 2018; Liu et al., 2022), together with cardiovascular problems of hydrogen sulfide (Beck and Pfeilschifter, 2022), all of which greatly limits the clinical application and translation of these agents. Likewise, the first identified RIPK1 inhibitor, Necrostatin-1 has somehow limited utility due to low solubility, short half-life of about 1 h along with narrow structure–activity relationship profile (Degterev et al., 2008; Oliveira et al., 2018; Wang et al., 2019). Another critical issue that was raised regarding Necrostatin-1 is its non-specificity. For example, it was shown that Necrostatin-1 blocked apoptosis and partially inhibits the two human kinases, PAK1 and PKAcα (Biton and Ashkenazi, 2011; Hill et al., 2018). Shortly thereafter, Takahashi N. et al. (2012) demonstrated that Necrostatin-1 also inhibits the activity of other enzymes as indoleamine 2,3-dioxygenase, which plays an important role in the innate and adaptive immune systems. Thus, this could necessitate careful interpretation of its biological effect in vivo and suggests importance of targeting and assessing other more specific downstream mediators, i.e., RIPK3 and MLKL for suppressing necroptosis. Other factors that should be taken into account are the dose and duration of treatment for these inhibitors. Necroptosis is usually induced after the administration of high doses of CP or following its long-time exposure at low concentrations. Therefore, the ability of these agents to produce notable protective effects and ameliorate renal injury is expected to observe several days after treatment (Deng et al., 2021).
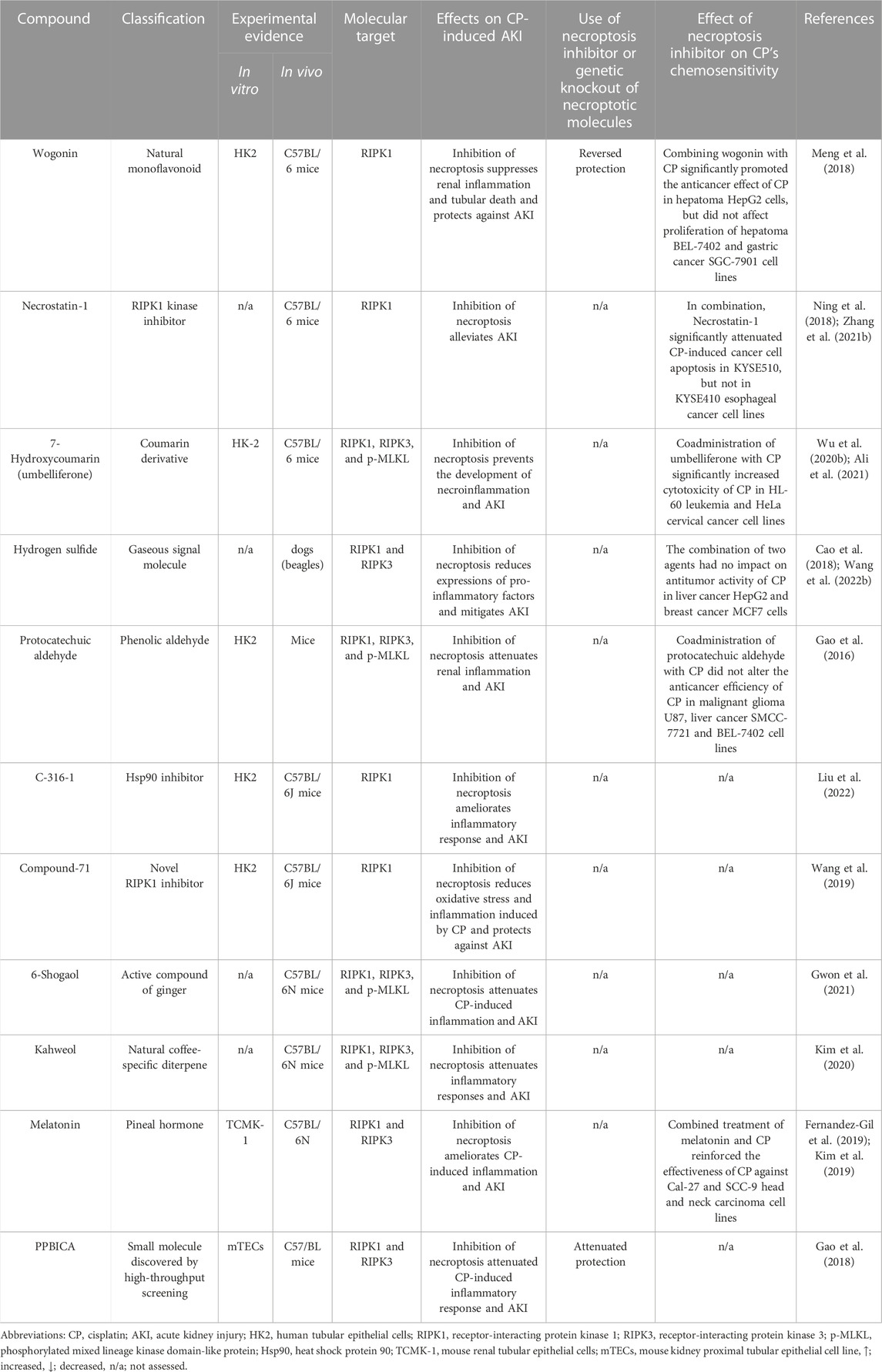
TABLE 1. Summary of molecules targeting necroptosis in CP-induced AKI.
4.2.5 Interplay between necroptosis and apoptosis in CP-induced AKI
Undoubtedly, both apoptotic and necroptotic cell death coexist in the pathophysiological course of AKI (Wang S. et al., 2016). In in vivo model of CP-induced nephrotoxicity, both apoptotic and necroptotic cell death pathways were shown to be concomitantly induced in kidney tubules following CP treatment; whereas in vitro various forms of regulated cell death are activated at different stages of renal injury depending on CP concentration (Kim et al., 2020; Deng et al., 2021; Gwon et al., 2021). Interestingly, these various forms, specifically necroptosis and apoptosis could interplay at various cellular and molecular levels and thus could mutually influence each other. A recent study by Zhang S. et al. (2019) revealed the important role of RIPK3 in mediating renal tubular cell apoptosis in endotoxin/sepsis-induced AKI. Reducing RIPK3 expression or inhibiting its activity significantly reversed the elevation of cleaved caspase-3 and proapoptotic protein Bax, thereby ameliorating AKI. Furthermore, this interplay is well exemplified by the findings of previous studies that showed that administration of pan-caspase inhibitor, zVAD-fmk potentially facilitated RIPK-mediated necroptosis in several renal models including AKI induced by CP (Linkermann et al., 2013; Tristão et al., 2016; Zhu et al., 2016; Zhu et al., 2018). Therefore, although the apoptosis pathway has been considered an important target for the attenuation of CP-induced AKI for many years, inhibition of apoptosis only could paradoxically sensitize tubular death through necroptosis signaling. Therefore, optimal protection against such injury may necessitate the antagonizing of both pathways.
In this regard, a previous report by Linkermann et al. (2013) compared RIPK3- to caspase 8/RIPK3–double knockout mice in the CP-induced AKI model. The authors found that caspase 8/RIPK3–double knockout mice showed a significant improvement in survival kinetics in this model, indicating that combined blockade of necroptotic and apoptotic pathways could provide additional protection. Later, multiple studies have further highlighted this concept by demonstrating that several agents attenuate CP-induced renal injury through dual suppression of both CP-induced apoptosis and necroptosis processes (Kim et al., 2019; Kim et al., 2020; Gwon et al., 2021). Recently, the impact of such synergism has been investigated and confirmed to be effective in mitigating CP-induced nephrotoxicity. Tristão’s group confirmed in 2 publications that the simultaneous use of apoptotic inhibitor, Z-VAD-fmk, and necroptotic inhibitor, necrostatin-1 synergistically attenuates CP nephrotoxicity (Tristão et al., 2012; 2016). Similar to the CP-induced AKI model, necroptosis inhibitors synergize apoptosis inhibitors to attenuate renal injury in the rat subtotal nephrectomy model (Zhu et al., 2016).
5 Autophagy in CP-induced AKI
5.1 Introduction of autophagy pathway
5.1.1 An overview of autophagy and its main types
In the late 1960s, Christian de Duve, the Nobel Laureate, first introduced the concept of autophagy. The expression derives from Greek words which originally mean self-eating (Deter and de Duve, 1967; Klionsky, 2008; Glick et al., 2010). Autophagy is a highly conserved and tightly controlled cellular process responsible for the degradation of the damaged cytoplasmic organelles, misfolded proteins, and other macromolecules within the lysosome (Miller et al., 2010; Levine and Klionsky, 2017). Once degraded, the resulting autophagic products including amino acids, fatty acids, and sugars are recycled for energy production and protein synthesis. In this manner, autophagy maintains cellular homeostasis during periods of stress and starvation (Kaushal, 2012; Sciarretta et al., 2018). In response to DNA damage or oxidative stress, autophagy is strongly induced, allowing for cell survival through utilization of autophagic products for energy production and protein synthesis (Levine and Yuan, 2005). In contrast, inhibition of autophagy under such conditions resulted in cellular damage and apoptosis (Amaravadi et al., 2007).
Various types of autophagy have been recognized in mammalian cells, involving i) macroautophagy, ii) microautophagy and iii) chaperone-mediated autophagy, all of which act through the lysosome-dependent pathway but differ in the machinery used to deliver autophagic cargo to the lumen of lysosomes (Ravikumar et al., 2009; Glick et al., 2010; Kaushik et al., 2011) (Table 2).
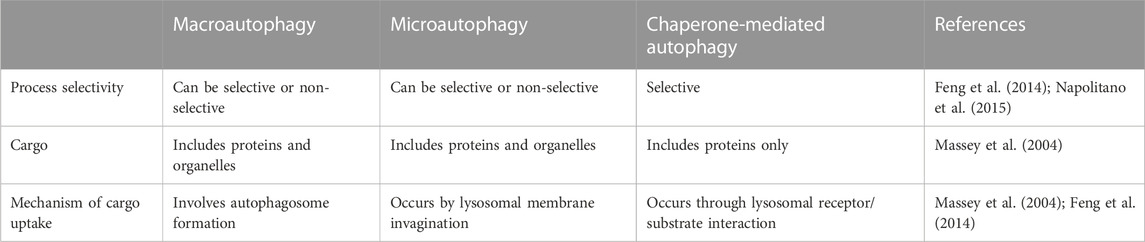
TABLE 2. The main characteristics of different types of autophagy.
The most common type of autophagy is macroautophagy (henceforth referred to as autophagy). It is a process in which cells sequester the autophagic cargoes within double-membrane vesicular structures, named autophagosomes, which finally fuse with lysosomes, where sequestered materials are degraded and recycled for reuse (Yang et al., 2008; Ravikumar et al., 2009; Levine and Klionsky, 2017; Danieli and Martens, 2018). On the other hand, in microautophagy, lysosomes can directly sequester cytosolic components by invagination of the lysosomal membrane (Ravikumar et al., 2009). It should be noted that the sequestration process in the macro-and micro-types can be either non-selective, which primarily occurs during nutrient starvation, or selective when targeting specific cargoes towards autophagosomes. These cargoes may include protein aggregates (named aggrephagy), damaged organelles (e.g., mitochondria, so-called mitophagy), and invasive pathogens (termed xenophagy) (Mizushima et al., 2008; Glick et al., 2010; Kaushik et al., 2011; Hou et al., 2013; Levine and Klionsky, 2017). In contrast to the former two types, chaperone-mediated autophagy is responsible for selective degradation of specific soluble proteins bearing a particular pentapeptide motif named “KFERQ” (Dice, 2007). This motif is detected by the Hsp70 chaperone complex, which allows the interaction between this substrate protein and the lysosomal receptor. Subsequently, the substrate unfolds and crosses the lysosomal membrane for degradation (Ravikumar et al., 2009; Cuervo, 2011; Kaushik et al., 2011).
5.1.2 Molecular mechanism of autophagy pathway
Our advanced understanding of autophagic machinery and its regulation is attributed to a series of investigations conducted in molecular biology laboratories. These investigations led to the discovery of at least 30 genes named autophagy-related genes (Atg) that are essential for the execution of each step of the autophagic pathway (Ravikumar et al., 2009). The autophagic pathway moves through multiple steps, including the formation of the phagophore, autophagosome, and eventually, autolysosome where degradation of sequestered substances occurs (Yang and Klionsky, 2010; Liu et al., 2017).
The formation of phagophore (also called isolation membrane or autophagosome precursor) occurs primarily at the ER which ultimately elongates and encloses forming the autophagosome structure (Ravikumar et al., 2009). The phagophore, and hence autophagosome formation requires the generation of an initiation complex named Unc51-like kinase 1 (ULK1) complex composed of ULK1, Atg13, FIP200, and Atg101 (Yang et al., 2015; Chen et al., 2016). The ULK1 phosphorylates activating molecule in Beclin1-regulated autophagy protein 1 (AMBRA1) leading to translocation of Bcl-2-interacting myosin-like coiled-coil protein (Beclin-1) towards the ER. The Beclin-1, in turn, forms a complex with vacuolar protein sorting (VPS)-34, VPS15, and Atg14L. In this complex, the lipid kinase, VPS34 phosphorylates phosphatidylinositol into phosphatidylinositol 3-phosphate, which acts as a recruitment signal for multiple proteins that are necessary for the nucleation of phagophore (Karakaş and Gözüaçik, 2014; Lee, 2014; Ryter and Choi, 2015; Mercer et al., 2018; Pavlinov et al., 2020).
The elongation and closure of the autophagosomal membrane are regulated by two ubiquitylation-like systems; the Atg5-Atg12-Atg16L complex and light chain 3 (LC3) (Ravikumar et al., 2009; Karakaş and Gözüaçik, 2014). On the elongating membrane, pro-LC3 is first cleaved to produce the cytosolic form of LC3 (LC3-I). Then, LC3-I is combined with phosphatidylethanolamine to generate the membrane-bound form of LC3 (LC3-II) by the action of many Atgs including Atg7, Atg3, and Atg5-Atg12-Atg16L complex (Ravikumar et al., 2009; Klapan et al., 2022). Upon closure, mature autophagosome engulfs the autophagic cargoes and subsequently fuses with lysosomes. Finally, the autophagic cargoes are metabolized by hydrolytic enzymes of lysosomes, and degraded contents are returned into the cytoplasm through lysosomal efflux permeases for reuse (Jing and Lim, 2012; Jin and Klionsky, 2014; Xu et al., 2020) (Figure 4).

FIGURE 4. Molecular mechanism of the autophagy pathway. (A) the scheme depicts the sequential steps involved in the autophagy process. This process starts with the formation of isolation membrane by the Unc51-like kinase 1 (ULK1) complex primarily at the endoplasmic reticulum (ER) followed by nucleation of this membrane to produce the cup-shaped phagophore. Subsequently, the Atg5–Atg12-Atg16L complex along with processed light chain-II (LC3-II) extended this phagophore to encase autophagic cargo in a double-membraned autophagosome. The loaded autophagosome then merges with the lysosome for breaking down of infused cargo and recycling. Notably, the Atg5–Atg12-Atg16L complex is dissociated from the autophagosomal membrane while LC3-II continues to attach to the mature autophagosomes until vesicle degradation, making it a specific and reliable autophagosomal marker. (B) LC3 processing. Upon activation of autophagy, pro-LC3 is first cleaved by Atg4B to produce LC3-I. This is followed by lipidation of LC3-I with phosphatidylethanolamine (PE) to generate LC3-II with the help of Atg7 and Atg3 as well as the Atg5-Atg12-Atg16L complex.
5.1.3 Regulation of autophagy in mammalian cells
Multiple kinases are responsible for regulation of mammalian autophagy, among them, the mechanistic target of rapamycin (mTOR) and adenosine monophosphate (AMP)-activated protein kinase (AMPK) play important and antagonistic roles (Tamargo-Gómez and Mariño, 2018).
The mTOR is a serine/threonine protein kinase, belonging to the phosphatidylinositol 3-kinase-related kinase family and it is widely known for its negative regulation of the autophagy process (Rabanal-Ruiz et al., 2017; Yuan et al., 2018). The mTOR exists in two complexes called mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) (Hall, 2008). These two complexes vary in their sensitivities to rapamycin, structures, and functions (Yu and Cui, 2016). There is a general consensus that the mTORC1 is the main modulator of autophagy, whereas mTORC2 primarily controls cytoskeleton reorganization (Karakaş and Gözüaçik, 2014).
Under the rich nutrient state, mTORC1 binds to the ULK1 complex and inhibits the initiation of autophagy through inhibitory phosphorylation of ULK1 at Ser-757 (Kim et al., 2011; Jain et al., 2013; Lee, 2014). However, suppression of mTORC1 activity either physiologically by starvation, stress, and certain immunological signals or pharmacologically by rapamycin triggers its dissociation from the ULK1 complex, thus enabling ULK1 to induce autophagy (Ravikumar et al., 2009; Lee, 2014; Liu et al., 2017). It is important to note that, mTOR can directly suppress the activity of Atg13 through inhibitory phosphorylation on Ser-258, thereby preventing association and activation of the ULK1 complex (Jain et al., 2013; Puente et al., 2016). Moreover, it has been found that mTOR may also phosphorylate AMBRA1 to further disrupt the autophagic pathway (Nazio et al., 2013).
Recently, mTOR has also been shown to regulate the autophagy process by controlling the subcellular localization of transcription factor EB (TFEB) (Tong and Song, 2015). The TFEB is widely known for its roles in lipid catabolism, cell metabolism, and lysosomal biogenesis (Soukas and Zhou, 2019). In response to nutrient abundance, mTORC1 phosphorylates TFEB at the lysosomal membrane, thereby inhibiting its migration to the nucleus and promoting its retention in the cytosol in an inactive state (Tong and Song, 2015; Zhitomirsky et al., 2018). In contrast, under conditions of nutrient deprivation, oxidative stress, and lysosomal dysfunction, inhibition of mTOR activity and concurrent activation of calcineurin triggers TFEB dephosphorylation (Medina et al., 2015; Tong and Song, 2015; Napolitano et al., 2018). During these conditions, the lysosome releases Ca2+, which in turn activates the Ca2+-activated phosphatase, calcineurin (Zhang et al., 2020). Upon activation, calcineurin dephosphorylates and stimulates TFEB, which in turn translocates into the nucleus where it binds to the coordinated lysosomal expression and regulation (CLEAR) sequence to upregulate expression of several genes involved in the autophagic pathway (Tong and Song, 2015; Slade and Pulinilkunnil, 2017; Sciarretta et al., 2018).
The AMPK is another important regulator of autophagic machinery (Karakaş and Gözüaçik, 2014). The AMPK has a unique heterotrimeric structure consisting of one catalytic subunit (α) along with two regulatory subunits (β and γ) (Garcia and Shaw, 2017). It is tightly regulated by ATP to AMP ratio which is greatly reduced during glucose deprivation leading to AMPK activation (Jung et al., 2010). During the nutrient deprivation state, AMP molecules bind to the AMPK γ subunit and cause certain conformational changes in the heterotrimeric complex, thereby exposing Thr-172 on the catalytic α subunit for phosphorylation as well as activation by liver kinase B1 (Zaha and Young, 2012). Importantly, AMPK stimulates the autophagic pathway through several mechanisms. Among these mechanisms, AMPK suppresses mTOR signaling either directly via inactivation of regulatory-associated protein of mTOR or indirectly by activation of its downstream negative regulator tuberous sclerosis complex-1 and -2 (Jung et al., 2010). Moreover, AMPK also stimulates autophagy independently of the mTOR signaling pathway through activating phosphorylation of ULK1 (Kim et al., 2011) (Figure 5).
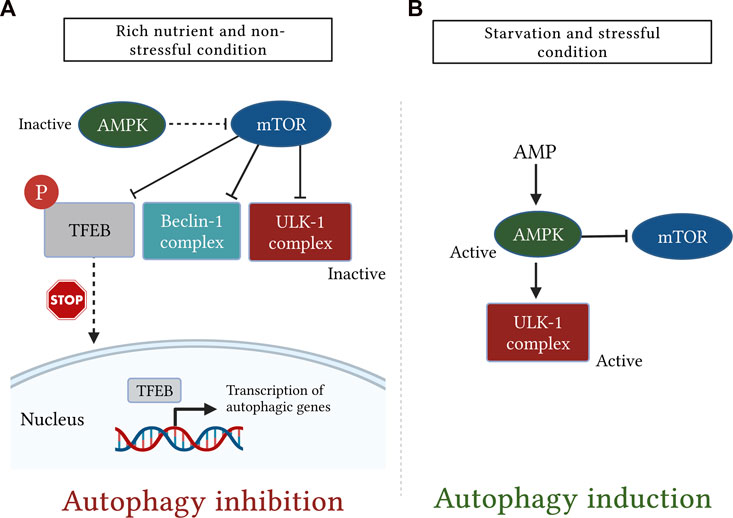
FIGURE 5. A simplistic diagram showing the antagonistic role of the mechanistic target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) in regulation of mammalian autophagy. The cellular energy status is closely monitored by the two kinases, mTOR and AMPK. (A): under rich nutrient state and absence of environmental stress, mTOR is activated and suppresses autophagy by disrupting the activation of Unc51-like kinase (ULK1) and Bcl-2-interacting myosin-like coiled-coil protein (Beclin-1) complexes. mTOR also inhibits autophagy by phosphorylating transcription factor EB (TFEB), which, in turn, sequesters TFEB in the cytosol and prevents it from being translocated into the nucleus. (B): Conversely, following starvation or stress response, AMPK is activated and induces autophagy either by direct phosphorylation of ULK1 or indirectly through downregulation of mTOR signaling.
5.2 Role of enhancing autophagy in CP-induced AKI
There is mounting evidence suggesting that basal autophagic activity in the kidney is clearly required for the maintenance and function of the proximal tubule (Kimura et al., 2011; Havasi and Dong, 2016; Zhu et al., 2020). Recently, autophagy has attracted unprecedented interest due to its importance in several renal diseases including AKI induced by CP (Pallet et al., 2008; Ding et al., 2014; Li T. et al., 2017; Nam et al., 2019; Shen et al., 2021). It is currently known that CP induces oxidative stress and DNA damage leading to autophagy induction (Yang et al., 2008; Takahashi A. et al., 2012). In contrast to CP-induced apoptosis which is going through the preapoptotic lag phase (Kaushal et al., 2001; Cummings and Schnellmann, 2002), it has been demonstrated that treatment of kidney epithelial cells with CP immediately and transiently activated autophagy within a few hours of CP administration (Periyasamy-Thandavan et al., 2008; Yang et al., 2008). Activation of autophagy during the initial phase of CP injury could provide a suitable environment for maintaining cell homeostasis before reaching the CP-induced apoptosis threshold (Herzog et al., 2012). However, importantly, high concentrations (≥50 μM) or prolonged treatment of CP have significantly reduced autophagy, thereby dominating apoptotic cell death in kidney cells (Rovetta et al., 2012; Zhang D. et al., 2017).
The cytoprotective and prosurvival role of autophagy in CP-induced AKI has been proved in many pharmacological and Atg-gene knockout studies (Periyasamy-Thandavan et al., 2008; Jiang et al., 2012; Rovetta et al., 2012; Liu J. et al., 2018). It has been reported that stimulation of autophagy after exposure to CP delayed caspase activation and increased the survival of renal cells (Yang et al., 2008; Liu et al., 2016). It was also found that overexpression of autophagic proteins Atg5 or beclin-1 blocked the activation of caspase-3 and cell death induced by CP (Herzog et al., 2012). In contrast, inhibition of autophagy either by chloroquine or knockout of proximal tubule-specific Atg7 worsens kidney function (Jiang et al., 2012). Similarly, Takahashi A. et al. (2012) found that deficiency of autophagy in proximal tubule cells leads to an increase in tubular injury, p53 activation, and protein aggregation in the CP-induced AKI model. In addition, CP exposure also induced PTEN-induced kinase protein 1 (PINK1) and Parkin, the two mediators of mitochondrial autophagy that were recently involved in CP nephrotoxicity. Knockout of either Parkin or PINK1 suppressed mitochondrial autophagy and aggravated kidney injury (Wang Y. et al., 2018).
The role of autophagy in CP-induced AKI was further proposed in subsequent studies. Periyasamy-Thandavan et al. (2008) and Zhao et al. (2017) pointed out that pharmacological inhibition of autophagy by 3-methyladenine enhanced mitochondrial dysfunction and tubular apoptosis during CP treatment. Furthermore, another study indicated that the nephroprotective effect mediated through TLR-2 is in part attributed to activation of autophagy and knockout of TLR2 reduced expression of autophagy proteins and exacerbated renal dysfunction in the CP-induced AKI model (Andrade-Silva et al., 2018). Likewise, it was also demonstrated that the nephroprotection rendered by hypoxia-inducible factor 1-alpha protein on CP-induced AKI was associated with autophagy upregulation (Liang et al., 2017). Consistently, the study by Tang et al. (2018) showed that inhibition of histone deacetylase 6 attenuated CP-induced AKI partially through activation of autophagy. In line with this, Minocha et al. (2019) found that amniotic fluid stem cells exerted their protective effects against CP-induced AKI by upregulating autophagy. Recently, Lynch et al. (2020) demonstrated that the inhibitory effect of peroxisome proliferator-activated receptor gamma coactivator 1α (PGC1α) on ROS induced by CP was mediated through TFEB-dependent autophagy. More recently, Sears et al. (2022) demonstrated that the loss of neutral ceramidase, an enzyme responsible for the metabolism of sphingolipid prevents the development of CP-indued AKI by enhancing basal autophagic activity in the kidney.
5.2.1 The potential mechanisms underlying autophagy protection
The exact mechanisms behind the cytoprotective role of the autophagy pathway are not yet completely understood. However, multiple mechanisms have been suggested, including the following: i) Through degradation of different cellular components, autophagy allows for the turnover of resulting substrates for energy production and protein formation, which is considered essential for the preservation of cellular bioenergetics during AKI (Jiang et al., 2012). ii) Autophagy eliminates depolarized mitochondria, dysfunctional or damaged organelles, misfolded proteins, and protein aggregates which have toxic effects on kidney tubules. By doing so, it promotes cellular hemostasis and survival. Concerning this matter, previous publications have signified the massive accumulation of impaired mitochondria and protein occlusions in mouse renal cells after deficiency of autophagy (Kimura et al., 2011; Takahashi A. et al., 2012) and that activation of autophagy improved mitochondrial function and protected against CP-induced AKI (Zhu et al., 2020). Moreover, Kimura et al. (2012) found that enhancing autophagic flux by IFN-γ alleviated the accumulation of polyubiquitinated peptides, thereby protecting renal tubular cells from CP-induced AKI. iii) Autophagy may disrupt other mechanisms involved in CP nephrotoxicity, particularly apoptosis and oxidative stress. In this regard, Takahashi A. et al. (2012) showed that knockout of Atg5 enhanced p53 stimulation and mitochondrial ROS formation following CP exposure. Similarly, Jiang et al. (2012) found that knockout of Atg7 augmented activation of the p53 cascade. Additionally, it has been reported that inhibition of autophagy accelerated ROS production and apoptosis induced by CP, while its activation reversed these effects (Zhao et al., 2017). Moreover, activation of AMPK attenuated CP-induced AKI by improving mitochondrial function and suppressing the formation of ROS and apoptosis responses caused by CP, all of which effects were abolished upon AMPK inhibition (Zhang J. et al., 2021). Furthermore, restoration of autophagy flux by polydatin ameliorated CP-induced oxidative stress, inflammation, and cell apoptosis, thereby alleviating CP-induced AKI (Li et al., 2022c). iv) Interestingly, several reports have recently documented the pivotal role of the autophagy process in modulating inflammation and ER stress. In response to elevated ER stress during nephrotoxicity, autophagy is strongly activated to mitigate ER stress and counteract tubular cell apoptosis (Pallet et al., 2008). By activating autophagy, Astragaloside IV prevents activation and assembly of NLRP3 inflammasome and reduces the release of pro-inflammatory cytokines in the liver and kidneys injured by CP (Qu et al., 2019). v) Notably, additional reports have also documented the important role of autophagy in preventing renal fibrosis, the main contributor for CKD progression post-AKI. Activation of autophagy participates in elimination of accumulated collagen, thereby attenuating kidney fibrosis and retard the progression of AKI to CKD (Shi et al., 2016). Similarly in the experimental model of CP-induced CKD, the natural flavonoid, farrerol upregulated mitochondrial autophagy, thereby inhibiting inflammation and renal fibrosis induced by CP and closely contributing to CKD (Ma et al., 2021). Along the same line, Shi et al. (2022) recently demonstrated that deficiency of beclin-1, the key molecule of autophagy, promoted renal fibrosis and consequently delayed recovery following renal injury.
5.2.2 Targeting autophagy in CP-induced AKI
The above observations strongly suggest the cytoprotective role of autophagy against CP-induced AKI, and because activation of autophagy by CP is not stable and is relatively retarded over the course of CP injury (Herzog et al., 2012; Li H. et al., 2017; Zhu et al., 2020). Thus, using autophagy inducers could further augment this activation and provide kidney protection during CP chemotherapy. Table 3 summarizes several natural substances and synthetic agents that have been confirmed to exhibit their renoprotective effects through autophagy/mitophagy upregulation in CP-induced AKI.
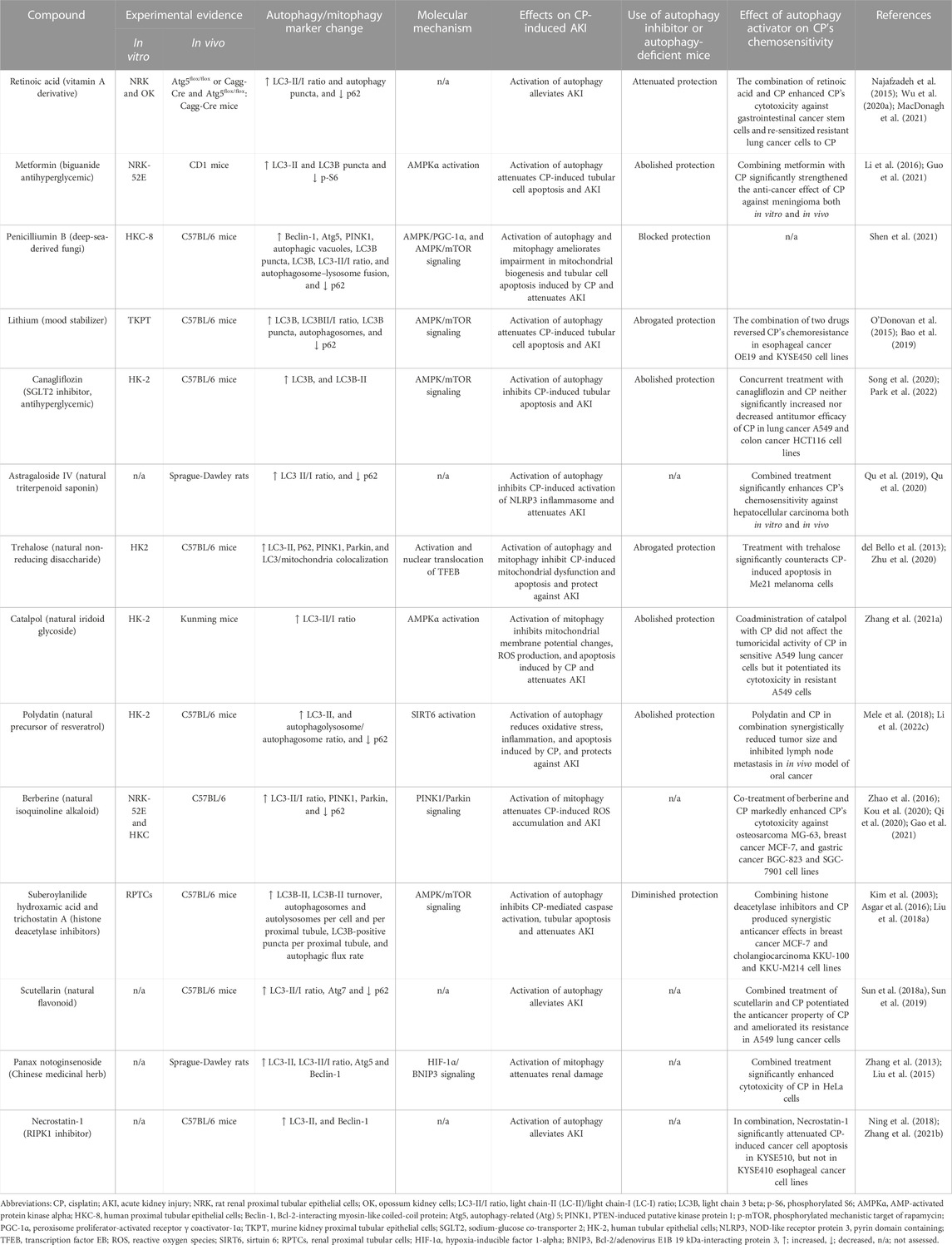
TABLE 3. Summary of renoprotective approaches targeting autophagy/mitophagy in CP-induced AKI.
6 The potential crosstalk between autophagy and necroptosis in CP-induced AKI
Emerging evidence suggests that autophagy and necroptosis can affect each other. To date, only a few studies have provided novel insights into the possible relationship between these two pathways in the kidney. It has been demonstrated that necroptosis molecules may negatively regulate basal autophagy. For example, using the rat CKD model, Shibata et al. (2021) recently suggested that induction of necroptotic signaling in proximal tubular cells may impair the activation of cytoprotective autophagy leading to exacerbation of kidney dysfunction. Moreover, Li R. et al. (2021) showed that RIPK3 inhibits nuclear translocation of TFEB leading to lysosome dysfunction and impairment of autophagic degradation during AKI. Suppression of RIPK3 restored the nuclear translocation of TFEB and attenuated tubular injury in vitro and in vivo. It has been reported that the deficiency of TFEB increased the mitochondrial ROS production and exacerbate tubular injury during CP renal injury (Lynch et al., 2020). Hence, an interesting question that remains unaddressed is whether inhibitors of necroptosis molecules attenuate CP-induced AKI by controlling lysosomal biogenesis and autophagic flux through TFEB? More interestingly, previous studies also reported that Necrostatin-1, the classical inhibitor of necroptosis alleviated renal injury by increasing the activity of renal autophagy and improving disruption in autophagosome elimination in sepsis- and CP-induced AKI (Dong et al., 2018; Ning et al., 2018). However, whether this renal proautophagic activity is mediated through necroptosis inhibition has not been clarified yet. Therefore, further investigations are required to elucidate autophagy-necroptosis crosstalk in the CP-induced AKI model.
On the other hand, this relationship has been much more investigated in several non-renal cells or tissues. A recent study by Wu X. et al. (2020) reported that increased expression of MLKL enhances western diet-induced liver injury by blocking autophagy through impairment of autophagic flux. Similarly, upon necroptotic stimulation, RIPK3 reduced the activity of autophagy by attenuating autophagic flux in intestinal cells (Otsubo et al., 2020). On the opposite hand, previous studies have also suggested that autophagy may protect against cell necroptosis, while its inhibition may activate and potentiate necroptosis (Matsuzawa-Ishimoto et al., 2017; Zhou et al., 2017; Liu S. et al., 2018; Li et al., 2020; Huang et al., 2021). Recently, Abe et al. (2019) further highlighted this reciprocal regulation by showing that the inhibition of mTORC1 by rapamycin treatment significantly inactivates RIPK1 through inhibitory phosphorylation at Ser320. Inactivation of RIPK1, in turn, stimulates autophagy and represses necroptosis through a TFEB-dependent mechanism. The molecular mechanisms through which autophagy regulates necroptosis are not entirely clear. However, this could be attributed to lysosomal dysfunction, since both necroptotic proteins (RIPK1 & RIPK3) may degrade through the lysosome-dependent pathway and therefore inhibition of lysosomal or autophagy function contributes to their accumulation, thereby triggering necroptosis (Liu S. et al., 2018; Lim et al., 2019). Thus, along with inhibiting apoptosis, these data suggest the ability of autophagy to suppress cell necroptosis which may consider an important mechanism for its pro-survival role.
7 Impact of autophagy activation and necroptosis inhibition on anticancer effect of CP
The biggest challenge that remains is to prevent renal injury associated with CP while maintaining or even enhancing its anti-cancer activity (Sun et al., 2019). Therefore, the main question is could autophagy activation or necroptosis inhibition potentially affect the anticancer activity of CP? Current data indicated that this question still cannot be conclusively answered as both autophagy and necroptosis are considered to have a Janus-faced role in tumorigenesis and cancer treatment. Regarding necroptosis, on one hand, RIPK1 plays a prosurvival role that results in chemoresistance of lung cancer cells. Interestingly, RIPK1 reduction sensitized cancer cells to CP and substantially potentiated its cytotoxicity (Wang et al., 2014a; Wang et al., 2014b; Wang et al., 2017). On the other hand, tumor-suppressing effects of necroptosis have been also reported among other cancer types, especially those resistant to apoptosis. Necroptosis was shown to mediate CP’s cytotoxicity, and its downregulation counteracted the anticancer activities of CP in fibrosarcoma and ovarian cancers (Xu et al., 2017; Zheng et al., 2020). In addition, restoring RIPK3 expression was associated with a better prognosis in patients with hepatocellular and esophageal carcinoma treated with CP chemotherapy (Sun Y. et al., 2018; Zhang B. et al., 2019). Similar to necroptosis, autophagy’s role in CP-induced cytotoxicity is extremely complex and remains a matter of debate, as dual effects of pro-metastatic and anti-metastatic have been reported. Activation of autophagy by CP contributes to its chemoresistance in esophageal cancer (O’Donovan et al., 2011), ovarian cancer (Bao et al., 2015), bladder cancer (Lin et al., 2017), and osteosarcoma (Jiang et al., 2017). In contrast, autophagy induction is also shown to mediate CP chemosensitivity in several other reports. In lung cancer, Sirichanchuen et al. (2012) demonstrated that long-term exposure to CP impaired autophagy leading to CP resistance, and interestingly, upregulation of autophagic response under such condition re-sensitized resistant cells to CP-induced cell death. In addition, autophagic degradation of Exo70 reduced CP efflux, thereby increasing its intracellular storage and cytotoxic activity (Zhao et al., 2021). Moreover, it has been reported that induction of autophagy helps to overcome CP resistance and potentiates its cytotoxic activity against lung cancer (Cheng et al., 2019; López-Plana et al., 2020), and oral cancer (Gao et al., 2022). More importantly, under the context of CP-induced AKI, previous studies have shown inconclusive results. Through upregulation of autophagy, scutellarin ameliorated CP-induced AKI and enhanced its antitumor efficiency against lung cancer (Sun C.-Y. et al., 2018; 2019). Conversely, the proautophagic activity of trehalose conferred protection against CP-induced AKI but also antagonized the antitumor effects of CP (del Bello et al., 2013; Zhu et al., 2020). On the other hand, some other novel autophagy activators like retinoic acid and astragaloside IV, which induce autophagic response in the kidney, and therefore protect against CP-induced AKI (Qu et al., 2019; Wu X. et al., 2020), demonstrated a different response in cancer cells, as they suppressed autophagy to enhance CP’s chemosensitivity (Lai et al., 2020; Abbasi et al., 2022). Altogether, while modulation of autophagy and necroptosis signaling pathways protects kidneys from CP injury (Figure 6), it is still difficult to estimate the effect of this modulation on the clinical response of CP, which is expected to vary depending on tumor type, stage, and cell context as well as the specific individual effect of each modulator. Tables 1, 3 showed the effects of different necroptotic inhibitors and autophagic activators on the anticancer activity of CP chemotherapy.
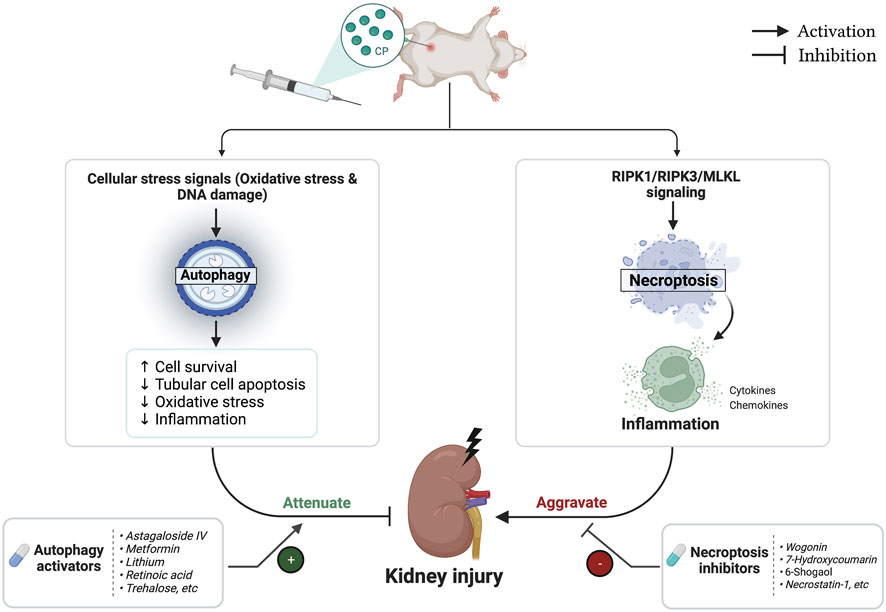
FIGURE 6. Flowchart representing the role of autophagy and necroptosis pathways during cisplatin (CP)-induced acute kidney injury (AKI).
8 Does modulation of autophagy and necroptosis pathways prevent CP-induced toxicities in other organs?
Besides CP-induced nephrotoxicity, administration of CP may induce serious injury in other normal tissues such as the ear, liver, heart, and others (Qi et al., 2019) (Figure 2). Interestingly, several recent publications have indicated the implicated role of necroptosis in CP-mediated ototoxicity (Choi et al., 2019; Ruhl et al., 2019). In Choi et al.’s (2019) study, pretreatment with necroptotic inhibitor, Necrostatin-1 markedly suppressed CP-induced auditory cell death, while the treatment of apoptosis inhibitor, ZVAD did not. Likewise, the protective effect of the autophagy pathway was also reported in CP-induced damage to the liver cells (Qu et al., 2019; Nashar et al., 2021), and cochlear cells (Fang and Xiao, 2014; Yang Q. et al., 2018; Liu et al., 2019; Pang et al., 2019). More Interestingly, trehalose which alleviated CP-induced AKI by activating autophagy (Zhu et al., 2020), recently, its pro-autophagic activity has made it also a potential treatment for CP-induced ototoxicity (Li et al., 2022b). Similarly, finding from other studies demonstrated the ability of the autophagy activator, metformin to attenuate CP-induced ototoxicity, cardiotoxicity, and neurotoxicity primarily through AMPKα activation (Guo et al., 2021; Liang et al., 2021; Nageeb et al., 2022). These data indicate that modulation of autophagy and necroptosis pathways during CP treatment may not only ameliorates CP injury in the kidney but also in other tissues and organs.
9 Conclusion and future directions
Nephrotoxicity, especially AKI, is the main serious problem that affects cancer patients treated with CP chemotherapy and often requires cessation of therapy. Therefore, understanding the principal mechanisms underlying this injury would be extremely helpful for the development of effective therapeutic strategies that could substantially help cancer patients to take full efficacy of CP, meanwhile reducing the potential of AKI episodes. In recent years, both autophagy and necroptosis are extensively investigated and their importance in the pathogenesis of CP-induced AKI is increasing at a remarkable pace. Recent in vitro and in vivo studies have given compelling evidence that modulation of these pathways could provide significant protection against CP-induced AKI. The pharmaceutical industry so far has made significant investments in the development of autophagy and necroptosis modulators, however, none of these agents successfully paved the way for clinical investigation. In the coming years, additional investigations in this area will help the future development of promising modulators with greater efficacy, plasma stability, and specificity. Moreover, given the contribution of various cellular processes in the pathogenesis of CP-induced AKI, using a combination of several modulators or identifying agents that can simultaneously modulate multiple targets may serve as an important strategy for developing future treatments. Importantly, before the clinical translation of any of these modulators, their effects should be adequately examined in tumor-bearing animals to make sure that their renoprotective effects are not compromising the anticancer activity of CP.
Author contributions
NA and HA conceptualized the idea of this research. NA wrote the original draft and created the figures. HA revised and edited the manuscript. All authors have read and approved the final version of this review.
Acknowledgments
All images included in this review article were created using biorender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abbasi, A., Hosseinpourfeizi, M., and Safaralizadeh, R. (2022). All-trans retinoic acid-mediated miR-30a up-regulation suppresses autophagy and sensitizes gastric cancer cells to cisplatin. Life Sci. 307, 120884. doi:10.1016/j.lfs.2022.120884
Abdel-Daim, M. M., Abushouk, A. I., Donia, T., Alarifi, S., Alkahtani, S., Aleya, L., et al. (2019). The nephroprotective effects of allicin and ascorbic acid against cisplatin-induced toxicity in rats. Environ. Sci. Pollut. Res. 26, 13502–13509. doi:10.1007/s11356-019-04780-4
Abe, K., Yano, T., Tanno, M., Miki, T., Kuno, A., Sato, T., et al. (2019). mTORC1 inhibition attenuates necroptosis through RIP1 inhibition-mediated TFEB activation. Biochimica Biophysica Acta (BBA) - Mol. Basis Dis. 1865, 165552. doi:10.1016/j.bbadis.2019.165552
Al-Salam, S., Jagadeesh, G. S., Sudhadevi, M., Tageldeen, H., and Yasin, J. (2021). Galectin-3 possesses anti-necroptotic and anti-apoptotic effects in cisplatin-induced acute tubular necrosis. Cell Physiol. Biochem. 55, 344–363. doi:10.33594/000000381
Ali, F. E. M., Hassanein, E. H. M., El-Bahrawy, A. H., Omar, Z. M. M., Rashwan, E. K., Abdel-Wahab, B. A., et al. (2021). Nephroprotective effect of umbelliferone against cisplatin-induced kidney damage is mediated by regulation of NRF2, cytoglobin, SIRT1/FOXO-3, and NF- kB-p65 signaling pathways. J. Biochem. Mol. Toxicol. 35, e22738. doi:10.1002/jbt.22738
Amaravadi, R. K., Yu, D., Lum, J. J., Bui, T., Christophorou, M. A., Evan, G. I., et al. (2007). Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Invest 117, 326–336. doi:10.1172/JCI28833
Anders, H.-J. (2018). Necroptosis in acute kidney injury. Nephron 139, 342–348. doi:10.1159/000489940
Andrade-Silva, M., Cenedeze, M. A., Perandini, L. A., Felizardo, R. J. F., Watanabe, I. K. M., Agudelo, J. S. H., et al. (2018). TLR2 and TLR4 play opposite role in autophagy associated with cisplatin-induced acute kidney injury. Clin. Sci. 132, 1725–1739. doi:10.1042/CS20170262
Andrea, D., and Reddy, M. A. (2018). Current concepts in the use of cisplatin: A historical perspective and its recent advances in the treatment of non-small cell lung cancer. J. Drug Des. Res. 5, 1073.
Asgar, A., Senawong, G., Sripa, B., and Senawong, T. (2016). Synergistic anticancer effects of cisplatin and histone deacetylase inhibitors (SAHA and TSA) on cholangiocarcinoma cell lines. Int. J. Oncol. 48, 409–420. doi:10.3892/ijo.2015.3240
Baek, J.-S., Na, Y.-G., and Cho, C.-W. (2018). Sustained cytotoxicity of wogonin on breast cancer cells by encapsulation in solid lipid nanoparticles. Nanomater. (Basel) 8, 159. doi:10.3390/nano8030159
Bao, H., Zhang, Q., Liu, X., Song, Y., Li, X., Wang, Z., et al. (2019). Lithium targeting of AMPK protects against cisplatin-induced acute kidney injury by enhancing autophagy in renal proximal tubular epithelial cells. FASEB J. 33, 14370–14381. doi:10.1096/fj.201901712R
Bao, L., Jaramillo, M. C., Zhang, Z., Zheng, Y., Yao, M., Zhang, D. D., et al. (2015). Induction of autophagy contributes to cisplatin resistance in human ovarian cancer cells. Mol. Med. Rep. 11, 91–98. doi:10.3892/mmr.2014.2671
Beck, K. F., and Pfeilschifter, J. (2022). The pathophysiology of H2S in renal glomerular diseases. Biomolecules 12, 207. doi:10.3390/biom12020207
Benyamini, Y., McClain, C. S., Leventhal, E. A., and Leventhal, H. (2003). Living with the worry of cancer: Health perceptions and behaviors of elderly people with self, vicarious, or no history of cancer. Psychooncology 12, 161–172. doi:10.1002/pon.637
Biton, S., and Ashkenazi, A. (2011). NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-α feedforward signaling. Cell 145, 92–103. doi:10.1016/j.cell.2011.02.023
Bonnet, M. C., Preukschat, D., Welz, P.-S., van Loo, G., Ermolaeva, M. A., Bloch, W., et al. (2011). The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity 35, 572–582. doi:10.1016/j.immuni.2011.08.014
Cao, X., Xiong, S., Zhou, Y., Wu, Z., Ding, L., Zhu, Y., et al. (2018). Renal protective effect of hydrogen sulfide in cisplatin-induced nephrotoxicity. Antioxid. Redox Signal 29, 455–470. doi:10.1089/ars.2017.7157
Chen, H., Fang, Y., Wu, J., Chen, H., Zou, Z., Zhang, X., et al. (2018). RIPK3-MLKL-mediated necroinflammation contributes to AKI progression to CKD. Cell Death Dis. 9, 878. doi:10.1038/s41419-018-0936-8
Chen, S.-H., and Chang, J.-Y. (2019). New insights into mechanisms of cisplatin resistance: From tumor cell to microenvironment. Int. J. Mol. Sci. 20, 4136. doi:10.3390/ijms20174136
Chen, S., Wang, C., Yeo, S., Liang, C.-C., Okamoto, T., Sun, S., et al. (2016). Distinct roles of autophagy-dependent and -independent functions of FIP200 revealed by generation and analysis of a mutant knock-in mouse model. Genes Dev. 30, 856–869. doi:10.1101/gad.276428.115
Cheng, H., Yang, Z. T., Bai, Y. Q., Cai, Y. F., and Zhao, J. P. (2019). Overexpression of Ulk2 inhibits proliferation and enhances chemosensitivity to cisplatin in non-small cell lung cancer. Oncol. Lett. 17, 79–86. doi:10.3892/ol.2018.9604
Chirino, Y. I., Sánchez-González, D. J., Martínez-Martínez, C. M., Cruz, C., and Pedraza-Chaverri, J. (2008). Protective effects of apocynin against cisplatin-induced oxidative stress and nephrotoxicity. Toxicology 245, 18–23. doi:10.1016/j.tox.2007.12.007
Cho, Y. S., Challa, S., Moquin, D., Genga, R., Ray, T. D., Guildford, M., et al. (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123. doi:10.1016/j.cell.2009.05.037
Cho, Y. S. (2018). The role of necroptosis in the treatment of diseases. BMB Rep. 51, 219–224. doi:10.5483/bmbrep.2018.51.5.074
Choi, M. J., Kang, H., Lee, Y. Y., Choo, O. S., Jang, J. H., Park, S. H., et al. (2019). Cisplatin-induced ototoxicity in rats is driven by RIP3-dependent necroptosis. Cells 8, 409. doi:10.3390/cells8050409
Coornaert, I., Hofmans, S., Devisscher, L., Augustyns, K., van der Veken, P., de Meyer, G. R. Y., et al. (2018). Novel drug discovery strategies for atherosclerosis that target necrosis and necroptosis. Expert Opin. Drug Discov. 13, 477–488. doi:10.1080/17460441.2018.1457644
Cuervo, A. M. (2011). Chaperone-mediated autophagy: Dice’s “wild” idea about lysosomal selectivity. Nat. Rev. Mol. Cell Biol. 12, 535–541. doi:10.1038/nrm3150
Cummings, B. S., and Schnellmann, R. G. (2002). Cisplatin-induced renal cell apoptosis: Caspase 3-dependent and -independent pathways. J. Pharmacol. Exp. Ther. 302, 8–17. doi:10.1124/jpet.302.1.8
Danieli, A., and Martens, S. (2018). p62-mediated phase separation at the intersection of the ubiquitin-proteasome system and autophagy. J. Cell Sci. 131, jcs214304. doi:10.1242/jcs.214304
Dasari, S., and Bernard Tchounwou, P. (2014). Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 740, 364–378. doi:10.1016/j.ejphar.2014.07.025
Degterev, A., Hitomi, J., Germscheid, M., Ch’en, I. L., Korkina, O., Teng, X., et al. (2008). Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321. doi:10.1371/journal.pone.0057236
del Bello, B., Toscano, M., Moretti, D., and Maellaro, E. (2013). Cisplatin-induced apoptosis inhibits autophagy, which acts as a pro-survival mechanism in human melanoma cells. PLoS One 8, e57236. doi:10.1371/journal.pone.0057236
Deng, F., Zheng, X., Sharma, I., Dai, Y., Wang, Y., and Kanwar, Y. S. (2021). Regulated cell death in cisplatin-induced AKI: Relevance of myo-inositol metabolism. Am. J. Physiology-Renal Physiology 320, F578–F595. doi:10.1152/ajprenal.00016.2021
Deter, R. L., and de Duve, C. (1967). Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J. Cell Biol. 33, 437–449. doi:10.1083/jcb.33.2.437
Dhuriya, Y. K., and Sharma, D. (2018). Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflammation 15, 199. doi:10.1186/s12974-018-1235-0
Ding, Y., Kim, S. L, Lee, S.Y., Koo, J. K., Wang, Z., and Choi, M. E. (2014). Autophagy regulates TGF-β expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J. Am. Soc. Nephrol. 25, 2835–2846. doi:10.1681/ASN.2013101068
Dong, W., Li, Z., Chen, Y., Zhang, L., Ye, Z., Liang, H., et al. (2018). Necrostatin-1 attenuates sepsis-associated acute kidney injury by promoting autophagosome elimination in renal tubular epithelial cells. Mol. Med. Rep. 17, 3194–3199. doi:10.3892/mmr.2017.8214
Dugbartey, G. J., Peppone, L. J., and de Graaf, I. A. M. (2016). An integrative view of cisplatin-induced renal and cardiac toxicities: Molecular mechanisms, current treatment challenges and potential protective measures. Toxicology 371, 58–66. doi:10.1016/j.tox.2016.10.001
Fan, Y., Lu, J., Yu, Z., Qu, X., and Guan, S. (2022). 1,3-Dichloro-2-propanol-Induced renal tubular cell necroptosis through the ROS/RIPK3/MLKL pathway. J. Agric. Food Chem. 70, 10847–10857. doi:10.1021/acs.jafc.2c02619
Fang, B., and Xiao, H. (2014). Rapamycin alleviates cisplatin-induced ototoxicity in vivo. Biochem. Biophys. Res. Commun. 448, 443–447. doi:10.1016/j.bbrc.2014.04.123
Feng, Y., He, D., Yao, Z., and Klionsky, D. J. (2014). The machinery of macroautophagy. Cell Res. 24, 24–41. doi:10.1038/cr.2013.168
Fernandez-Gil, B. I., Guerra-Librero, A., Shen, Y.-Q., Florido, J., Martínez-Ruiz, L., García-López, S., et al. (2019). Melatonin enhances cisplatin and radiation cytotoxicity in head and neck squamous cell carcinoma by stimulating mitochondrial ROS generation, apoptosis, and autophagy. Oxid. Med. Cell Longev. 2019, 7187128. doi:10.1155/2019/7187128
Fulda, S. (2013). The mechanism of necroptosis in normal and cancer cells. Cancer Biol. Ther. 14, 999–1004. doi:10.4161/cbt.26428
Gao, L., Liu, M.-M., Zang, H., Ma, Q.-Y., Yang, Q., Jiang, L., et al. (2018). Restoration of E-cadherin by PPBICA protects against cisplatin-induced acute kidney injury by attenuating inflammation and programmed cell death. Lab. Investig. 98, 911–923. doi:10.1038/s41374-018-0052-5
Gao, L., Wu, W.-F., Dong, L., Ren, G.-L., Li, H.-D., Yang, Q., et al. (2016). Protocatechuic aldehyde attenuates cisplatin-induced acute kidney injury by suppressing Nox-mediated oxidative stress and renal inflammation. Front. Pharmacol. 7, 479. doi:10.3389/fphar.2016.00479
Gao, L., Zhang, Q., Li, S., Zheng, J., Ren, W., and Zhi, K. (2022). Circ-PKD2 promotes Atg13-mediated autophagy by inhibiting miR-646 to increase the sensitivity of cisplatin in oral squamous cell carcinomas. Cell Death Dis. 13, 192. doi:10.1038/s41419-021-04497-8
Gao, X., Zhang, C., Wang, Y., Zhang, P., Zhang, J., and Hong, T. (2021). Berberine and cisplatin exhibit synergistic anticancer effects on osteosarcoma MG-63 cells by inhibiting the MAPK pathway. Molecules 26, 1666. doi:10.3390/molecules26061666
Garcia, D., and Shaw, R. J. (2017). AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 66, 789–800. doi:10.1016/j.molcel.2017.05.032
Gautheron, J., Vucur, M., Reisinger, F., Cardenas, D. V., Roderburg, C., Koppe, C., et al. (2014). A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol. Med. 6, 1062–1074. doi:10.15252/emmm.201403856
Ghosh, S. (2019). Cisplatin: The first metal based anticancer drug. Bioorg Chem. 88, 102925. doi:10.1016/j.bioorg.2019.102925
Glick, D., Barth, S., and Macleod, K. F. (2010). Autophagy: Cellular and molecular mechanisms. J. Pathology 221, 3–12. doi:10.1002/path.2697
Gómez-Sierra, T., Eugenio-Pérez, D., Sánchez-Chinchillas, A., and Pedraza-Chaverri, J. (2018). Role of food-derived antioxidants against cisplatin induced-nephrotoxicity. Food Chem. Toxicol. 120, 230–242. doi:10.1016/j.fct.2018.07.018
Guo, L., Cui, J., Wang, H., Medina, R., Zhang, S., Zhang, X., et al. (2021). Metformin enhances anti-cancer effects of cisplatin in meningioma through AMPK-mTOR signaling pathways. Mol. Ther. Oncolytics 20, 119–131. doi:10.1016/j.omto.2020.11.004
Gwon, M. G., Gu, H., Leem, J., and Park, K. K. (2021). Protective effects of 6-shogaol, an active compound of ginger, in a murine model of cisplatin-induced acute kidney injury. Molecules 26, 5931. doi:10.3390/molecules26195931
Hägerström, E., Lindberg, L., Bentzen, J., Brødbæk, K., Zerahn, B., and Kristensen, B. (2019). The nephroprotective effect of mannitol in head and neck cancer patients receiving cisplatin therapy. Clin. Med. Insights Oncol. 13, 1179554918821320. doi:10.1177/1179554918821320
Hall, M. N. (2008). mTOR—what does it do? Transpl. Proc. 40, S5–S8. doi:10.1016/j.transproceed.2008.10.009
Hamroun, A., Lenain, R., Bigna, J. J., Speyer, E., Bui, L., Chamley, P., et al. (2019). Prevention of cisplatin-induced acute kidney injury: A systematic review and meta-analysis. Drugs 79, 1567–1582. doi:10.1007/s40265-019-01182-1
Havasi, A., and Dong, Z. (2016). Autophagy and tubular cell death in the kidney. Semin. Nephrol. 36, 174–188. doi:10.1016/j.semnephrol.2016.03.005
Herzog, C., Yang, C., Holmes, A., and Kaushal, G. P. (2012). zVAD-fmk prevents cisplatin-induced cleavage of autophagy proteins but impairs autophagic flux and worsens renal function. Am. J. Physiol. Ren. Physiol. 303, F1239–F1250. doi:10.1152/ajprenal.00659.2011
Hill, N. R., Cook, H. T., Pusey, C. D., and Tarzi, R. M. (2018). RIPK3-deficient mice were not protected from nephrotoxic nephritis. BMC Nephrol. 19, 61. doi:10.1186/s12882-018-0850-4
Holditch, S. J., Brown, C. N., Lombardi, A. M., Nguyen, K. N., and Edelstein, C. L. (2019). Recent advances in models, mechanisms, biomarkers, and interventions in cisplatin-induced acute kidney injury. Int. J. Mol. Sci. 20, 3011. doi:10.3390/ijms20123011
Hou, J., Ju, J., Zhang, Z., Zhao, C., Li, Z., Zheng, J., et al. (2019). Discovery of potent necroptosis inhibitors targeting RIPK1 kinase activity for the treatment of inflammatory disorder and cancer metastasis. Cell Death Dis. 10, 493. doi:10.1038/s41419-019-1735-6
Hou, W., Zhang, Q., Yan, Z., Chen, R., Zeh, H. J., Kang, R., et al. (2013). Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell Death Dis. 4, e966. doi:10.1038/cddis.2013.493
Hu, X., Ma, Z., Wen, L., Li, S., and Dong, Z. (2021). Autophagy in cisplatin nephrotoxicity during cancer therapy. Cancers (Basel) 13, 5618. doi:10.3390/cancers13225618
Huang, Y., Feng, Y., Cui, L., Yang, L., Zhang, Q., Zhang, J., et al. (2021). Autophagy-related LC3 accumulation interacted directly with LIR containing RIPK1 and RIPK3, stimulating necroptosis in hypoxic cardiomyocytes. Front. Cell Dev. Biol. 9, 679637. doi:10.3389/fcell.2021.679637
Jain, M. v., Paczulla, A. M., Klonisch, T., Dimgba, F. N., Rao, S. B., Roberg, K., et al. (2013). Interconnections between apoptotic, autophagic and necrotic pathways: Implications for cancer therapy development. J. Cell Mol. Med. 17, 12–29. doi:10.1111/jcmm.12001
Jantas, D., and Lasoń, W. (2021). Preclinical evidence for the interplay between oxidative stress and RIP1-dependent cell death in neurodegeneration: State of the art and possible therapeutic implications. Antioxidants 10, 1518. doi:10.3390/antiox10101518
Jiang, K., Zhang, C., Yu, B., Chen, B., Liu, Z., Hou, C., et al. (2017). Autophagic degradation of FOXO3a represses the expression of PUMA to block cell apoptosis in cisplatin-resistant osteosarcoma cells. Am. J. Cancer Res. 7, 1407–1422.
Jiang, M., Wei, Q., Dong, G., Komatsu, M., Su, Y., and Dong, Z. (2012). Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 82, 1271–1283. doi:10.1038/ki.2012.261
Jiang, M., Yi, X., Hsu, S., Wang, C.-Y., and Dong, Z. (2004). Role of p53 in cisplatin-induced tubular cell apoptosis: Dependence on p53 transcriptional activity. Am. J. Physiology-Renal Physiology 287, F1140–F1147. doi:10.1152/ajprenal.00262.2004
Jiang, Y. J., Jin, J., Nan, Q. Y., Ding, J., Cui, S., Xuan, M. Y., et al. (2022). Coenzyme Q10 attenuates renal fibrosis by inhibiting RIP1-RIP3-MLKL-mediated necroinflammation via Wnt3α/β-catenin/GSK-3β signaling in unilateral ureteral obstruction. Int. Immunopharmacol. 108, 108868. doi:10.1016/j.intimp.2022.108868
Jin, M., and Klionsky, D. J. (2014). Regulation of autophagy: Modulation of the size and number of autophagosomes. FEBS Lett. 588, 2457–2463. doi:10.1016/j.febslet.2014.06.015
Jing, K., and Lim, K. (2012). Why is autophagy important in human diseases? Exp. Mol. Med. 44, 69–72. doi:10.3858/emm.2012.44.2.028
Jung, C. H., Ro, S.-H., Cao, J., Otto, N. M., and Kim, D.-H. (2010). mTOR regulation of autophagy. FEBS Lett. 584, 1287–1295. doi:10.1016/j.febslet.2010.01.017
Kaczmarek, A., Vandenabeele, P., and Krysko, D. v. (2013). Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 38, 209–223. doi:10.1016/j.immuni.2013.02.003
Karakaş, H. E., and Gözüaçik, D. (2014). Autophagy and cancer. Turkish J. Biol. 38, 720–739. doi:10.3906/biy-1408-16
Karasawa, T., and Steyger, P. S. (2015). An integrated view of cisplatin-induced nephrotoxicity and ototoxicity. Toxicol. Lett. 237, 219–227. doi:10.1016/j.toxlet.2015.06.012
Kaushal, G. P. (2012). Autophagy protects proximal tubular cells from injury and apoptosis. Kidney Int. 82, 1250–1253. doi:10.1038/ki.2012.337
Kaushal, G. P., Kaushal, V., Hong, X., and Shah, S. V. (2001). Role and regulation of activation of caspases in cisplatin-induced injury to renal tubular epithelial cells. Kidney Int. 60, 1726–1736. doi:10.1046/j.1523-1755.2001.00026.x
Kaushik, S., Bandyopadhyay, U., Sridhar, S., Kiffin, R., Martinez-Vicente, M., Kon, M., et al. (2011). Chaperone-mediated autophagy at a glance. J. Cell Sci. 124, 495–499. doi:10.1242/jcs.073874
Kim, J.-Y., Jo, J., Leem, J., and Park, K.-K. (2020). Kahweol ameliorates cisplatin-induced acute kidney injury through pleiotropic effects in mice. Biomedicines 8, 572. doi:10.3390/biomedicines8120572
Kim, J., Kundu, M., Viollet, B., and Guan, K.-L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi:10.1038/ncb2152
Kim, J., Long, K. E., Tang, K., and Padanilam, B. J. (2012). Poly(ADP-ribose) polymerase 1 activation is required for cisplatin nephrotoxicity. Kidney Int. 82, 193–203. doi:10.1038/ki.2012.64
Kim, J. W., Jo, J., Kim, J.-Y., Choe, M., Leem, J., and Park, J.-H. (2019). Melatonin attenuates cisplatin-induced acute kidney injury through dual suppression of apoptosis and necroptosis. Biol. (Basel) 8, 64. doi:10.3390/biology8030064
Kim, M. S., Blake, M., Baek, J. H., Kohlhagen, G., Pommier, Y., and Carrier, F. (2003). Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res. 63, 7291–7300.
Kimura, A., Ishida, Y., Inagaki, M., Nakamura, Y., Sanke, T., Mukaida, N., et al. (2012). Interferon-γ is protective in cisplatin-induced renal injury by enhancing autophagic flux. Kidney Int. 82, 1093–1104. doi:10.1038/ki.2012.240
Kimura, T., Takabatake, Y., Takahashi, A., Kaimori, J., Matsui, I., Namba, T., et al. (2011). Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J. Am. Soc. Nephrol. 22, 902–913. doi:10.1681/ASN.2010070705
Klapan, K., Simon, D., Karaulov, A., Gomzikova, M., Rizvanov, A., Yousefi, S., et al. (2022). Autophagy and skin diseases. Front. Pharmacol. 13, 844756. doi:10.3389/fphar.2022.844756
Klionsky, D. J. (2008). Autophagy revisited: A conversation with christian de Duve. Autophagy 4, 740–743. doi:10.4161/auto.6398
Kocarnik, J. M., Compton, K., Dean, F. E., Fu, W., Gaw, B. L., Harvey, J. D., et al. (2022). Cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life years for 29 cancer groups from 2010 to 2019: A systematic analysis for the global burden of disease study 2019. JAMA Oncol. 8, 420–444. doi:10.1001/jamaoncol.2021.6987
Kociba, R. J., and Sleight, S. D. (1971). Acute toxicologic and pathologic effects of cis-diamminedichloroplatinum (NSC-119875) in the male rat. Cancer Chemother. Rep. 55, 1–8.
Kou, Y., Tong, B., Wu, W., Liao, X., and Zhao, M. (2020). Berberine improves chemo-sensitivity to cisplatin by enhancing cell apoptosis and repressing PI3K/AKT/mTOR signaling pathway in gastric cancer. Front. Pharmacol. 11. doi:10.3389/fphar.2020.616251
Lai, S. T., Wang, Y., and Peng, F. (2020). Astragaloside IV sensitizes non-small cell lung cancer cells to cisplatin by suppressing endoplasmic reticulum stress and autophagy. J. Thorac. Dis. 12, 3715–3724. doi:10.21037/jtd-20-2098
Landau, S. I., Guo, X., Velazquez, H., Torres, R., Olson, E., Garcia-Milian, R., et al. (2019). Regulated necrosis and failed repair in cisplatin-induced chronic kidney disease. Kidney Int. 95, 797–814. doi:10.1016/j.kint.2018.11.042
Lau, A. H. (1999). Apoptosis induced by cisplatin nephrotoxic injury. Kidney Int. 56, 1295–1298. doi:10.1046/j.1523-1755.1999.00687.x
Lau, A., Wang, S., Jiang, J., Haig, A., Pavlosky, A., Linkermann, A., et al. (2013). RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. Am. J. Transplant. 13, 2805–2818. doi:10.1111/ajt.12447
Lee, M.-S. (2014). Role of islet β cell autophagy in the pathogenesis of diabetes. Trends Endocrinol. Metabolism 25, 620–627. doi:10.1016/j.tem.2014.08.005
Lee, S. H., Kwon, J. Y, Moon, J., Choi, J., Jhun, J., Jung, K., et al. (2020). Inhibition of RIPK3 pathway attenuates intestinal inflammation and cell death of inflammatory bowel disease and suppresses necroptosis in peripheral mononuclear cells of ulcerative colitis patients. Immune Netw. 20. doi:10.4110/in.2020.20.e16
Leeper, N. J. (2016). The role of necroptosis in atherosclerotic disease. JACC Basic Transl. Sci. 1, 548–550. doi:10.1016/j.jacbts.2016.08.002
Levine, B., and Klionsky, D. J. (2017). Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: Breakthroughs in baker’s yeast fuel advances in biomedical research. Proc. Natl. Acad. Sci. 114, 201–205. doi:10.1073/pnas.1619876114
Levine, B., and Yuan, J. (2005). Autophagy in cell death: An innocent convict? J. Clin. Invest 115, 2679–2688. doi:10.1172/JCI26390
Lewington, A. J. P., Cerdá, J., and Mehta, R. L. (2013). Raising awareness of acute kidney injury: A global perspective of a silent killer. Kidney Int. 84, 457–467. doi:10.1038/ki.2013.153
Li, C., Chen, Q., He, Y., Liu, Y., Meng, X., and Liu, M. (2022a). Discovery of a chalcone derivative as potent necroptosis inhibitor for the treatment of acute kidney injury. Clin. Exp. Pharmacol. Physiol. 49, 824–835. doi:10.1111/1440-1681.13670
Li, C., Mu, N., Gu, C., Liu, M., Yang, Z., Yin, Y., et al. (2020). Metformin mediates cardioprotection against aging-induced ischemic necroptosis. Aging Cell 19, e13096. doi:10.1111/acel.13096
Li, H., Tang, Y., Wen, L., Kong, X., Chen, X., Liu, P., et al. (2017a). Neferine reduces cisplatin-induced nephrotoxicity by enhancing autophagy via the AMPK/mTOR signaling pathway. Biochem. Biophys. Res. Commun. 484, 694–701. doi:10.1016/j.bbrc.2017.01.180
Li, J., Gui, Y., Ren, J., Liu, X., Feng, Y., Zeng, Z., et al. (2016). Metformin protects against cisplatin-induced tubular cell apoptosis and acute kidney injury via AMPKα-regulated autophagy induction. Sci. Rep. 6, 23975. doi:10.1038/srep23975
Li, L., Tong, A., Zhang, Q., Wei, Y., and Wei, X. (2021a). The molecular mechanisms of MLKL-dependent and MLKL-independent necrosis. J. Mol. Cell Biol. 13, 3–14. doi:10.1093/jmcb/mjaa055
Li, R., Zhao, X., Zhang, S., Dong, W., Zhang, L., Chen, Y., et al. (2021b). RIP3 impedes transcription factor EB to suppress autophagic degradation in septic acute kidney injury. Cell Death Dis. 12, 593. doi:10.1038/s41419-021-03865-8
Li, T., Liu, Y., Zhao, J., Miao, S., Xu, Y., Liu, K., et al. (2017b). Aggravation of acute kidney injury by mPGES-2 down regulation is associated with autophagy inhibition and enhanced apoptosis. Sci. Rep. 7, 10247. doi:10.1038/s41598-017-10271-8
Li, Z., Yao, Q., Tian, Y., Jiang, Y., Xu, M., Wang, H., et al. (2022b). Trehalose protects against cisplatin-induced cochlear hair cell damage by activating TFEB-mediated autophagy. Biochem. Pharmacol. 197, 114904. doi:10.1016/j.bcp.2021.114904
Li, Z., Zhou, L., Du, Y., Li, H., Feng, L., Li, X., et al. (2022c). Polydatin attenuates cisplatin-induced acute kidney injury via SIRT6-mediated autophagy activation. Oxid. Med. Cell Longev. 2022, 9035547. doi:10.1155/2022/9035547
Liang, H., Zhang, Z., He, L., and Wang, Y. (2016). CXCL16 regulates cisplatin-induced acute kidney injury. Oncotarget 7, 31652–31662. doi:10.18632/oncotarget.9386
Liang, X., Yang, Y., Huang, Z., Zhou, J., Li, Y., and Zhong, X. (2017). Panax notoginseng saponins mitigate cisplatin induced nephrotoxicity by inducing mitophagy via HIF-1α. Oncotarget 8, 102989–103003. doi:10.18632/oncotarget.19900
Liang, Z., Zhang, T., Zhan, T., Cheng, G., Zhang, W., Jia, H., et al. (2021). Metformin alleviates cisplatin-induced ototoxicity by autophagy induction possibly via the AMPK/FOXO3a pathway. J. Neurophysiol. 125, 1202–1212. doi:10.1152/jn.00417.2020
Lim, J., Park, H., Heisler, J., Maculins, T., Roose-Girma, M., Xu, M., et al. (2019). Autophagy regulates inflammatory programmed cell death via turnover of RHIM-domain proteins. Elife 8, e44452. doi:10.7554/eLife.44452
Lin, J. F., Lin, Y. C., Tsai, T. F., Chen, H. E., Chou, K. Y., and Hwang, T. I. S. (2017). Cisplatin induces protective autophagy through activation of BECN1 in human bladder cancer cells. Drug Des. Devel Ther. 11, 1517–1533. doi:10.2147/DDDT.S126464
Lin, J., Li, H., Yang, M., Ren, J., Huang, Z., Han, F., et al. (2013). A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep. 3, 200–210. doi:10.1016/j.celrep.2012.12.012
Lin, Y., Choksi, S., Shen, H.-M., Yang, Q.-F., Hur, G. M., Kim, Y. S., et al. (2004). Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J. Biol. Chem. 279, 10822–10828. doi:10.1074/jbc.M313141200
Linkermann, A., Bräsen, J. H., Darding, M., Jin, M. K., Sanz, A. B., Heller, J.-O., et al. (2013). Two independent pathways of regulated necrosis mediate ischemia–reperfusion injury. Proc. Natl. Acad. Sci. 110, 12024–12029. doi:10.1073/pnas.1305538110
Linkermann, A., de Zen, F., Weinberg, J., Kunzendorf, U., and Krautwald, S. (2012). Programmed necrosis in acute kidney injury. Nephrol. Dial. Transplant. 27, 3412–3419. doi:10.1093/ndt/gfs373
Linkermann, A. (2016). Nonapoptotic cell death in acute kidney injury and transplantation. Kidney Int. 89, 46–57. doi:10.1016/j.kint.2015.10.008
Linkermann, A., Stockwell, B. R., Krautwald, S., and Anders, H.-J. (2014). Regulated cell death and inflammation: An auto-amplification loop causes organ failure. Nat. Rev. Immunol. 14, 759–767. doi:10.1038/nri3743
Liu, H., Gu, L., Tu, Y., Hu, H., Huang, Y., and Sun, W. (2016). Emodin ameliorates cisplatin-induced apoptosis of rat renal tubular cells in vitro by activating autophagy. Acta Pharmacol. Sin. 37, 235–245. doi:10.1038/aps.2015.114
Liu, J., Livingston, M. J., Dong, G., Tang, C., Su, Y., Wu, G., et al. (2018a). Histone deacetylase inhibitors protect against cisplatin-induced acute kidney injury by activating autophagy in proximal tubular cells. Cell Death Dis. 9, 322. doi:10.1038/s41419-018-0374-7
Liu, S., Li, Y., Choi, H. M. C., Sarkar, C., Koh, E. Y., Wu, J., et al. (2018b). Lysosomal damage after spinal cord injury causes accumulation of RIPK1 and RIPK3 proteins and potentiation of necroptosis. Cell Death Dis. 9, 476. doi:10.1038/s41419-018-0469-1
Liu, T., Zong, S., Luo, P., Qu, Y., Wen, Y., Du, P., et al. (2019). Enhancing autophagy by down-regulating GSK-3β alleviates cisplatin-induced ototoxicity in vivo and in vitro. Toxicol. Lett. 313, 11–18. doi:10.1016/j.toxlet.2019.05.025
Liu, X., Huang, Z., Zou, X., Yang, Y., Qiu, Y., and Wen, Y. (2015). Possible mechanism of PNS protection against cisplatin-induced nephrotoxicity in rat models. Toxicol. Mech. Methods 25, 347–354. doi:10.3109/15376516.2015.1006492
Liu, X., Liu, M., Jiang, L., Gao, L., Zhang, Y., Huang, Y., et al. (2022). A novel small molecule Hsp90 inhibitor, C-316-1, attenuates acute kidney injury by suppressing RIPK1-mediated inflammation and necroptosis. Int. Immunopharmacol. 108, 108849. doi:10.1016/j.intimp.2022.108849
Liu, X., Xie, X., Ren, Y., Shao, Z., Zhang, N., Li, L., et al. (2021). The role of necroptosis in disease and treatment. MedComm (Beijing) 2, 730–755. doi:10.1002/mco2.108
Liu, Z., Li, Z., Chen, Z., Li, C., Lei, L., Wu, X., et al. (2020). Numb ameliorates necrosis and inflammation in acute kidney injury induced by cisplatin. Chem. Biol. Interact. 330, 109251. doi:10.1016/j.cbi.2020.109251
Liu, Z., Xiao, Y., Torresilla, C., Rassart, E., and Barbeau, B. (2017). Implication of different HIV-1 genes in the modulation of autophagy. Viruses 9, E389. doi:10.3390/v9120389
López-Plana, A., Fernández-Nogueira, P., Muñoz-Guardiola, P., Solé-Sánchez, S., Megías-Roda, E., Pérez-Montoyo, H., et al. (2020). The novel proautophagy anticancer drug ABTL0812 potentiates chemotherapy in adenocarcinoma and squamous nonsmall cell lung cancer. Int. J. Cancer 147, 1163–1179. doi:10.1002/ijc.32865
Lynch, M. R., Tran, M. T., Ralto, K. M., Zsengeller, Z. K., Raman, V., Bhasin, S. S., et al. (2020). TFEB-driven lysosomal biogenesis is pivotal for PGC1α-dependent renal stress resistance. JCI Insight 4, e126749. doi:10.1172/jci.insight.126749
Ma, N., wei, Z., Hu, J., Gu, W., and Ci, X. (2021). Farrerol ameliorated cisplatin-induced chronic kidney disease through mitophagy induction via Nrf2/PINK1 pathway. Front. Pharmacol. 12, 768700. doi:10.3389/fphar.2021.768700
MacDonagh, L., Santiago, R. M., Gray, S. G., Breen, E., Cuffe, S., Finn, S. P., et al. (2021). Exploitation of the vitamin A/retinoic acid axis depletes ALDH1-positive cancer stem cells and re-sensitises resistant non-small cell lung cancer cells to cisplatin. Transl. Oncol. 14, 101025. doi:10.1016/j.tranon.2021.101025
Manohar, S., and Leung, N. (2018). Cisplatin nephrotoxicity: A review of the literature. J. Nephrol. 31, 15–25. doi:10.1007/s40620-017-0392-z
Massey, A., Kiffin, R., and Cuervo, A. M. (2004). Pathophysiology of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 36, 2420–2434. doi:10.1016/j.biocel.2004.04.010
Matsuzawa-Ishimoto, Y., Shono, Y., Gomez, L. E., Hubbard-Lucey, V. M., Cammer, M., Neil, J., et al. (2017). Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J. Exp. Med. 214, 3687–3705. doi:10.1084/jem.20170558
Medina, D. L., di Paola, S., Peluso, I., Armani, A., de Stefani, D., Venditti, R., et al. (2015). Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 17, 288–299. doi:10.1038/ncb3114
Mele, L., Paino, F., Papaccio, F., Regad, T., Boocock, D., Stiuso, P., et al. (2018). A new inhibitor of glucose-6-phosphate dehydrogenase blocks pentose phosphate pathway and suppresses malignant proliferation and metastasis in vivo. Cell Death Dis. 9, 572. doi:10.1038/s41419-018-0635-5
Meng, X.-M., Li, H.-D., Wu, W.-F., Ming-Kuen Tang, P., Ren, G.-L., Gao, L., et al. (2018). Wogonin protects against cisplatin-induced acute kidney injury by targeting RIPK1-mediated necroptosis. Lab. Investig. 98, 79–94. doi:10.1038/labinvest.2017.115
Mercer, T. J., Gubas, A., and Tooze, S. A. (2018). A molecular perspective of mammalian autophagosome biogenesis. J. Biol. Chem. 293, 5386–5395. doi:10.1074/jbc.R117.810366
Mihaly, S. R., Ninomiya-Tsuji, J., and Morioka, S. (2014). TAK1 control of cell death. Cell Death Differ. 21, 1667–1676. doi:10.1038/cdd.2014.123
Miller, R. P., Tadagavadi, R. K., Ramesh, G., and Reeves, W. B. (2010). Mechanisms of cisplatin nephrotoxicity. Toxins (Basel) 2, 2490–2518. doi:10.3390/toxins2112490
Minocha, E., Sinha, R. A., Jain, M., Chaturvedi, C. P., and Nityanand, S. (2019). Amniotic fluid stem cells ameliorate cisplatin-induced acute renal failure through induction of autophagy and inhibition of apoptosis. Stem Cell Res. Ther. 10, 370. doi:10.1186/s13287-019-1476-6
Mizushima, N., Levine, B., Cuervo, A. M., and Klionsky, D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. doi:10.1038/nature06639
Molitoris, B. A., Dagher, P. C., Sandoval, R. M., Campos, S. B., Ashush, H., Fridman, E., et al. (2009). siRNA targeted to p53 attenuates ischemic and cisplatin-induced acute kidney injury. J. Am. Soc. Nephrol. 20, 1754–1764. doi:10.1681/ASN.2008111204
Mulay, S. R., Desai, J., Kumar, S. v., Eberhard, J. N., Thomasova, D., Romoli, S., et al. (2016a). Cytotoxicity of crystals involves RIPK3-MLKL-mediated necroptosis. Nat. Commun. 7, 10274. doi:10.1038/ncomms10274
Mulay, S. R., Linkermann, A., and Anders, H.-J. (2016b). Necroinflammation in kidney disease. J. Am. Soc. Nephrol. 27, 27–39. doi:10.1681/ASN.2015040405
Nageeb, M. M., Saadawy, S. F., and Attia, S. H. (2022). Breast milk mesenchymal stem cells abate cisplatin-induced cardiotoxicity in adult male albino rats via modulating the AMPK pathway. Sci. Rep. 12, 17554. doi:10.1038/s41598-022-22095-2
Nailwal, H., and Chan, F. K.-M. (2019). Necroptosis in anti-viral inflammation. Cell Death Differ. 26, 4–13. doi:10.1038/s41418-018-0172-x
Najafzadeh, N., Mazani, M., Abbasi, A., Farassati, F., and Amani, M. (2015). Low-dose all-trans retinoic acid enhances cytotoxicity of cisplatin and 5-fluorouracil on CD44+ cancer stem cells. Biomed. Pharmacother. 74, 243–251. doi:10.1016/j.biopha.2015.08.019
Nam, S. A., Kim, W.-Y., Kim, J. W., Park, S. H., Kim, H. L., Lee, M.-S., et al. (2019). Autophagy attenuates tubulointerstital fibrosis through regulating transforming growth factor-β and NLRP3 inflammasome signaling pathway. Cell Death Dis. 10, 78. doi:10.1038/s41419-019-1356-0
Napolitano, G., Esposito, A., Choi, H., Matarese, M., Benedetti, V., di Malta, C., et al. (2018). mTOR-dependent phosphorylation controls TFEB nuclear export. Nat. Commun. 9, 3312. doi:10.1038/s41467-018-05862-6
Napolitano, G., Johnson, J. L., He, J., Rocca, C. J., Monfregola, J., Pestonjamasp, K., et al. (2015). Impairment of chaperone-mediated autophagy leads to selective lysosomal degradation defects in the lysosomal storage disease cystinosis. EMBO Mol. Med. 7, 158–174. doi:10.15252/emmm.201404223
Nashar, E. M. el, Alghamdi, M. A., Alasmari, W. A., Hussein, M. M. A., Hamza, E., Taha, R. I., et al. (2021). Autophagy promotes the survival of adipose mesenchymal stem/stromal cells and enhances their therapeutic effects in cisplatin-induced liver injury via modulating TGF-β1/Smad and PI3K/AKT signaling pathways. Cells 10, 2475. doi:10.3390/cells10092475
Nazio, F., Strappazzon, F., Antonioli, M., Bielli, P., Cianfanelli, V., Bordi, M., et al. (2013). mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 15, 406–416. doi:10.1038/ncb2708
Nguyen, L., Nguyen Vo, T.-H., Trinh, Q. H., Nguyen, B. H., Nguyen-Hoang, P.-U., Le, L., et al. (2022). iANP-EC: Identifying anticancer natural products using ensemble learning incorporated with evolutionary computation. J. Chem. Inf. Model 62, 5080–5089. doi:10.1021/acs.jcim.1c00920
Nikoletopoulou, V., Markaki, M., Palikaras, K., and Tavernarakis, N. (2013). Crosstalk between apoptosis, necrosis and autophagy. Biochimica Biophysica Acta (BBA) - Mol. Cell Res. 1833, 3448–3459. doi:10.1016/j.bbamcr.2013.06.001
Ning, Y., Shi, Y., Chen, J., Song, N., Cai, J., Fang, Y., et al. (2018). Necrostatin-1 attenuates cisplatin-induced nephrotoxicity through suppression of apoptosis and oxidative stress and retains klotho expression. Front. Pharmacol. 9, 384. doi:10.3389/fphar.2018.00384
O’Donovan, T. R., O’Sullivan, G. C., and McKenna, S. L. (2011). Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 7, 509–524. doi:10.4161/auto.7.6.15066
O’Donovan, T. R., Rajendran, S., O’Reilly, S., O’Sullivan, G. C., and McKenna, S. L. (2015). Lithium modulates autophagy in esophageal and colorectal cancer cells and enhances the efficacy of therapeutic agents in vitro and in vivo. PLoS One 10, e0134676. doi:10.1371/journal.pone.0134676
Oh, C. J., Ha, C.-M., Choi, Y.-K., Park, S., Choe, M. S., Jeoung, N. H., et al. (2017). Pyruvate dehydrogenase kinase 4 deficiency attenuates cisplatin-induced acute kidney injury. Kidney Int. 91, 880–895. doi:10.1016/j.kint.2016.10.011
Oh, G. S., Kim, H. J., Shen, A. H., Lee, S. B, Khadka, D., Pandit, A., et al. (2014). Cisplatin-induced kidney dysfunction and perspectives on improving treatment strategies. Electrolyte Blood Press. 12, 55–65. doi:10.5049/EBP.2014.12.2.55
Oliveira, S. R., Amaral, J. D., and Rodrigues, C. M. P. (2018). Mechanism and disease implications of necroptosis and neuronal inflammation. Cell Death Dis. 9, 903. doi:10.1038/s41419-018-0872-7
Ostermann, M., and Joannidis, M. (2016). Acute kidney injury 2016: Diagnosis and diagnostic workup. Crit. Care 20, 299. doi:10.1186/s13054-016-1478-z
Otsubo, K., Maeyashiki, C., Nibe, Y., Tamura, A., Aonuma, E., Matsuda, H., et al. (2020). Receptor-Interacting Protein Kinase 3 (RIPK3) inhibits autophagic flux during necroptosis in intestinal epithelial cells. FEBS Lett. 594, 1586–1595. doi:10.1002/1873-3468.13748
Ozkok, A., and Edelstein, C. L. (2014). Pathophysiology of cisplatin-induced acute kidney injury. Biomed. Res. Int. 2014, 967826. doi:10.1155/2014/967826
Ozkok, A., Ravichandran, K., Wang, Q., Ljubanovic, D., and Edelstein, C. L. (2016). NF-κB transcriptional inhibition ameliorates cisplatin-induced acute kidney injury (AKI). Toxicol. Lett. 240, 105–113. doi:10.1016/j.toxlet.2015.10.028
Pabla, N., Dong, G., Jiang, M., Huang, S., Kumar, M. V., Messing, R. O., et al. (2011). Inhibition of PKCδ reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J. Clin. Invest 121, 2709–2722. doi:10.1172/JCI45586
Pabla, N., and Dong, Z. (2008). Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 73, 994–1007. doi:10.1038/sj.ki.5002786
Pallet, N., Bouvier, N., Legendre, C., Gilleron, J., Codogno, P., Beaune, P., et al. (2008). Autophagy protects renal tubular cells against cyclosporine toxicity. Autophagy 4, 783–791. doi:10.4161/auto.6477
Pang, J., Xiong, H., Zhan, T., Cheng, G., Jia, H., Ye, Y., et al. (2019). Sirtuin 1 and autophagy attenuate cisplatin-induced hair cell death in the mouse cochlea and zebrafish lateral line. Front. Cell Neurosci. 12. doi:10.3389/fncel.2018.00515
Park, C. H., Lee, B., Han, M., Rhee, W. J., Kwak, M. S., Yoo, T.-H., et al. (2022). Canagliflozin protects against cisplatin-induced acute kidney injury by AMPK-mediated autophagy in renal proximal tubular cells. Cell Death Discov. 8, 12. doi:10.1038/s41420-021-00801-9
Pavlinov, I., Salkovski, M., and Aldrich, L. N. (2020). Beclin 1–ATG14L protein–protein interaction inhibitor selectively inhibits autophagy through disruption of VPS34 complex I. J. Am. Chem. Soc. 142, 8174–8182. doi:10.1021/jacs.9b12705
Peres, L. A., and da Cunha, A. D. (2013). Acute nephrotoxicity of cisplatin: Molecular mechanisms. J. Bras. Nefrol. 35, 332–340. doi:10.5935/0101-2800.20130052
Periyasamy-Thandavan, S., Jiang, M., Wei, Q., Smith, R., Yin, X.-M., and Dong, Z. (2008). Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int. 74, 631–640. doi:10.1038/ki.2008.214
Puente, C., Hendrickson, R. C., and Jiang, X. (2016). Nutrient-regulated phosphorylation of ATG13 inhibits starvation-induced autophagy. J. Biol. Chem. 291, 6026–6035. doi:10.1074/jbc.M115.689646
Qi, J., Xue, Q., Kuang, L., Xie, L., Luo, R., and Nie, X. (2020). Berberine alleviates cisplatin-induced acute kidney injury by regulating mitophagy via PINK 1/Parkin pathway. Transl. Androl. Urol. 9, 1712–1724. doi:10.21037/tau-20-1129
Qi, L., Luo, Q., Zhang, Y., Jia, F., Zhao, Y., and Wang, F. (2019). Advances in toxicological research of the anticancer drug cisplatin. Chem. Res. Toxicol. 32, 1469–1486. doi:10.1021/acs.chemrestox.9b00204
Qu, X., Gao, H., Tao, L., Zhang, Y., Zhai, J., Sun, J., et al. (2019). Astragaloside IV protects against cisplatin-induced liver and kidney injury via autophagy-mediated inhibition of NLRP3 in rats. J. Toxicol. Sci. 44, 167–175. doi:10.2131/jts.44.167
Qu, X., Gao, H., Zhai, J., Sun, J., Tao, L., Zhang, Y., et al. (2020). Astragaloside IV enhances cisplatin chemosensitivity in hepatocellular carcinoma by suppressing MRP2. Eur. J. Pharm. Sci. 148, 105325. doi:10.1016/j.ejps.2020.105325
Rabanal-Ruiz, Y., Otten, E. G., and Korolchuk, V. I. (2017). mTORC1 as the main gateway to autophagy. Essays Biochem. 61, 565–584. doi:10.1042/EBC20170027
Ramachandran, A., McGill, M. R., Xie, Y., Ni, H.-M., Ding, W.-X., and Jaeschke, H. (2013). Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology 58, 2099–2108. doi:10.1002/hep.26547
Ravikumar, B., Futter, M., Jahreiss, L., Korolchuk, V. I., Lichtenberg, M., Luo, S., et al. (2009). Mammalian macroautophagy at a glance. J. Cell Sci. 122, 1707–1711. doi:10.1242/jcs.031773
Rosenberg, B., van Camp, L., and Krigas, T. (1965). Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature 205, 698–699. doi:10.1038/205698a0
Rosenberg, B., Vancamp, L., Trosko, J. E., and Mansour, V. H. (1969). Platinum compounds: A new class of potent antitumour agents. Nature 222, 385–386. doi:10.1038/222385a0
Rovetta, F., Stacchiotti, A., Consiglio, A., Cadei, M., Grigolato, P. G., Lavazza, A., et al. (2012). ER signaling regulation drives the switch between autophagy and apoptosis in NRK-52E cells exposed to cisplatin. Exp. Cell Res. 318, 238–250. doi:10.1016/j.yexcr.2011.11.008
Ruhl, D., Du, T.-T., Wagner, E. L., Choi, J. H., Li, S., Reed, R., et al. (2019). Necroptosis and apoptosis contribute to cisplatin and aminoglycoside ototoxicity. J. Neurosci. 39, 2951–2964. doi:10.1523/JNEUROSCI.1384-18.2019
Ryter, S. W., and Choi, A. M. K. (2015). Autophagy in lung disease pathogenesis and therapeutics. Redox Biol. 4, 215–225. doi:10.1016/j.redox.2014.12.010
Sciarretta, S., Forte, M., Frati, G., and Sadoshima, J. (2018). New insights into the role of mTOR signaling in the cardiovascular system. Circ. Res. 122, 489–505. doi:10.1161/CIRCRESAHA.117.311147
Sears, S. M., Dupre, T. v., Shah, P. P., Davis, D. L., Doll, M. A., Sharp, C. N., et al. (2022). Neutral ceramidase deficiency protects against cisplatin-induced acute kidney injury. J. Lipid Res. 63, 100179. doi:10.1016/j.jlr.2022.100179
Seifert, L., and Miller, G. (2017). Molecular pathways: The necrosome—a target for cancer therapy. Clin. Cancer Res. 23, 1132–1136. doi:10.1158/1078-0432.CCR-16-0968
Seo, J., Nam, Y. W., Kim, S., Oh, D.-B., and Song, J. (2021). Necroptosis molecular mechanisms: Recent findings regarding novel necroptosis regulators. Exp. Mol. Med. 53, 1007–1017. doi:10.1038/s12276-021-00634-7
Shan, B., Pan, H., Najafov, A., and Yuan, J. (2018). Necroptosis in development and diseases. Genes Dev. 32, 327–340. doi:10.1101/gad.312561.118
Sharp, C. N., and Siskind, L. J. (2017). Developing better mouse models to study cisplatin-induced kidney injury. Am. J. Physiology-Renal Physiology 313, F835–F841. doi:10.1152/ajprenal.00285.2017
Shen, W., Jia, N., Miao, J., Chen, S., Zhou, S., Meng, P., et al. (2021). Penicilliumin B protects against cisplatin-induced renal tubular cell apoptosis through activation of AMPK-induced autophagy and mitochondrial biogenesis. Kidney Dis. 7, 278–292. doi:10.1159/000514657
Shi, M., Flores, B., Gillings, N., Bian, A., Cho, H. J., Yan, S., et al. (2016). αKlotho mitigates progression of AKI to CKD through activation of autophagy. J. Am. Soc. Nephrol. 27, 2331–2345. doi:10.1681/ASN.2015060613
Shi, M., Maique, J., Shepard, S., Li, P., Seli, O., Moe, O. W., et al. (2022). In vivo evidence for therapeutic applications of beclin 1 to promote recovery and inhibit fibrosis after acute kidney injury. Kidney Int. 101, 63–78. doi:10.1016/j.kint.2021.09.030
Shibata, S., Moniwa, N., Kuno, A., Kimura, A., Ohwada, W., Sugawara, H., et al. (2021). Involvement of necroptosis in contrast-induced nephropathy in a rat CKD model. Clin. Exp. Nephrol. 25, 708–717. doi:10.1007/s10157-021-02048-1
Shiraishi, F., Curtis, L. M., Truong, L., Poss, K., Visner, G. A., Madsen, K., et al. (2000). Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. Am. J. Physiology-Renal Physiology 278, F726–F736. doi:10.1152/ajprenal.2000.278.5.F726
Sirichanchuen, B., Pengsuparp, T., and Chanvorachote, P. (2012). Long-term cisplatin exposure impairs autophagy and causes cisplatin resistance in human lung cancer cells. Mol. Cell Biochem. 364, 11–18. doi:10.1007/s11010-011-1199-1
Slade, L., and Pulinilkunnil, T. (2017). The MiTF/TFE family of transcription factors: Master regulators of organelle signaling, metabolism, and stress adaptation. Mol. Cancer Res. 15, 1637–1643. doi:10.1158/1541-7786.MCR-17-0320
Song, Z., Zhu, J., Wei, Q., Dong, G., and Dong, Z. (2020). Canagliflozin reduces cisplatin uptake and activates Akt to protect against cisplatin-induced nephrotoxicity. Am. J. Physiology-Renal Physiology 318, F1041–F1052. doi:10.1152/ajprenal.00512.2019
Soukas, A. A., and Zhou, B. (2019). Surviving starvation simply without TFEB. PLoS Biol. 17, e3000285. doi:10.1371/journal.pbio.3000285
Sridevi, P., Nhiayi, M. K., and Wang, J. Y. J. (2013). Genetic disruption of Abl nuclear import reduces renal apoptosis in a mouse model of cisplatin-induced nephrotoxicity. Cell Death Differ. 20, 953–962. doi:10.1038/cdd.2013.42
Sun, C.-Y., Nie, J., Zheng, Z.-L., Zhao, J., Wu, L.-M., Zhu, Y., et al. (2019). Renoprotective effect of scutellarin on cisplatin-induced renal injury in mice: Impact on inflammation, apoptosis, and autophagy. Biomed. Pharmacother. 112, 108647. doi:10.1016/j.biopha.2019.108647
Sun, C.-Y., Zhu, Y., Li, X.-F., Wang, X.-Q., Tang, L.-P., Su, Z.-Q., et al. (2018a). Scutellarin increases cisplatin-induced apoptosis and autophagy to overcome cisplatin resistance in non-small cell lung cancer via ERK/p53 and c-met/AKT signaling pathways. Front. Pharmacol. 9, 92. doi:10.3389/fphar.2018.00092
Sun, Y., Zhai, L., Ma, S., Zhang, C., Zhao, L., Li, N., et al. (2018b). Down-regulation of RIP3 potentiates cisplatin chemoresistance by triggering HSP90-ERK pathway mediated DNA repair in esophageal squamous cell carcinoma. Cancer Lett. 418, 97–108. doi:10.1016/j.canlet.2018.01.022
Takahashi, A., Kimura, T., Takabatake, Y., Namba, T., Kaimori, J., Kitamura, H., et al. (2012a). Autophagy guards against cisplatin-induced acute kidney injury. Am. J. Pathol. 180, 517–525. doi:10.1016/j.ajpath.2011.11.001
Takahashi, N., Duprez, L., Grootjans, S., Cauwels, A., Nerinckx, W., DuHadaway, J. B., et al. (2012b). Necrostatin-1 analogues: Critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 3, e437–e438. doi:10.1038/cddis.2012.176
Tamargo-Gómez, I., and Mariño, G. (2018). AMPK: Regulation of metabolic dynamics in the context of autophagy. Int. J. Mol. Sci. 19, 3812. doi:10.3390/ijms19123812
Tang, J., Shi, Y., Liu, N., Xu, L., Zang, X., Li, P., et al. (2018). Blockade of histone deacetylase 6 protects against cisplatin-induced acute kidney injury. Clin. Sci. 132, 339–359. doi:10.1042/CS20171417
Tong, Y., and Song, F. (2015). Intracellular calcium signaling regulates autophagy via calcineurin-mediated TFEB dephosphorylation. Autophagy 11, 1192–1195. doi:10.1080/15548627.2015.1054594
Tristão, V. R., Gonçalves, P. F., Dalboni, M. A., Batista, M. C., Durão, M. de S., and Monte, J. C. M. (2012). Nec-1 protects against nonapoptotic cell death in cisplatin-induced kidney injury. Ren. Fail 34, 373–377. doi:10.3109/0886022X.2011.647343
Tristão, V. R., Pessoa, E. A., Nakamichi, R., Reis, L. A., Batista, M. C., de Souza Durão Junior, M., et al. (2016). Synergistic effect of apoptosis and necroptosis inhibitors in cisplatin-induced nephrotoxicity. Apoptosis 21, 51–59. doi:10.1007/s10495-015-1190-5
Vanden Berghe, T., Hassannia, B., and Vandenabeele, P. (2016). An outline of necrosome triggers. Cell. Mol. Life Sci. 73, 2137–2152. doi:10.1007/s00018-016-2189-y
Vandenabeele, P., Galluzzi, L., vanden Berghe, T., and Kroemer, G. (2010). Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714. doi:10.1038/nrm2970
Wang, F., Wang, J., He, X., Suo, X., Li, C., Ni, W., et al. (2022a). Stratifin promotes renal dysfunction in ischemic and nephrotoxic AKI mouse models via enhancing RIPK3-mediated necroptosis. Acta Pharmacol. Sin. 43, 330–341. doi:10.1038/s41401-021-00649-w
Wang, G., Qu, F. Z., Li, L., Lv, J. C., and Sun, B. (2016a). Necroptosis: A potential, promising target and switch in acute pancreatitis. Apoptosis 21, 121–129. doi:10.1007/s10495-015-1192-3
Wang, J., Liu, M., Wang, F., Wei, B., Yang, Q., Cai, Y., et al. (2019). RIPK1 inhibitor Cpd-71 attenuates renal dysfunction in cisplatin-treated mice via attenuating necroptosis, inflammation and oxidative stress. Clin. Sci. 133, 1609–1627. doi:10.1042/CS20190599
Wang, Q., Chen, W., Bai, L., Chen, W., Padilla, M. T., Lin, A. S., et al. (2014a). Receptor-interacting protein 1 increases chemoresistance by maintaining inhibitor of apoptosis protein levels and reducing reactive oxygen species through a microRNA-146a-mediated catalase pathway. J. Biol. Chem. 289, 5654–5663. doi:10.1074/jbc.M113.526152
Wang, Q., Shi, S., He, W., Padilla, M. T., Zhang, L., Wang, X., et al. (2014b). Retaining MKP1 expression and attenuating JNK-mediated apoptosis by RIP1 for cisplatin resistance through miR-940 inhibition. Oncotarget 5, 1304–1314. doi:10.18632/oncotarget.1798
Wang, R., Zheng, X., Zhang, L., Zhou, B., Hu, H., Li, Z., et al. (2017). Histone H4 expression is cooperatively maintained by IKKβ and Akt1 which attenuates cisplatin-induced apoptosis through the DNA-PK/RIP1/IAPs signaling cascade. Sci. Rep. 7, 41715. doi:10.1038/srep41715
Wang, S., Liu, X., and Liu, Y. (2022b). Hydrogen sulfide protects from acute kidney injury via attenuating inflammation activated by necroptosis in dogs. J. Vet. Sci. 23, e72. doi:10.4142/jvs.22064
Wang, S., Zhang, C., Hu, L., and Yang, C. (2016b). Necroptosis in acute kidney injury: A shedding light. Cell Death Dis. 7, e2125. doi:10.1038/cddis.2016.37
Wang, X., Yousefi, S., and Simon, H.-U. (2018a). Necroptosis and neutrophil-associated disorders. Cell Death Dis. 9, 111. doi:10.1038/s41419-017-0058-8
Wang, Y., Tang, C., Cai, J., Chen, G., Zhang, D., Zhang, Z., et al. (2018b). PINK1/Parkin-mediated mitophagy is activated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis. 9, 1113. doi:10.1038/s41419-018-1152-2
Wei, Q., Dong, G., Yang, T., Megyesi, J., Price, P. M., and Dong, Z. (2007). Activation and involvement of p53 in cisplatin-induced nephrotoxicity. Am. J. Physiology-Renal Physiology 293, F1282–F1291. doi:10.1152/ajprenal.00230.2007
Weinlich, R., Oberst, A., Beere, H. M., and Green, D. R. (2017). Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 18, 127–136. doi:10.1038/nrm.2016.149
Welz, P.-S., Wullaert, A., Vlantis, K., Kondylis, V., Fernández-Majada, V., Ermolaeva, M., et al. (2011). FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 477, 330–334. doi:10.1038/nature10273
Wu, J., Huang, Z., Ren, J., Zhang, Z., He, P., Li, Y., et al. (2013). Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 23, 994–1006. doi:10.1038/cr.2013.91
Wu, J., Zheng, C., Wan, X., Shi, M., McMillan, K., Maique, J., et al. (2020a). Retinoic acid alleviates cisplatin-induced acute kidney injury through activation of autophagy. Front. Pharmacol. 11, 987. doi:10.3389/fphar.2020.00987
Wu, W.-F., Wang, J.-N., Li, Z., Wei, B., Jin, J., Gao, L., et al. (2020b). 7-Hydroxycoumarin protects against cisplatin-induced acute kidney injury by inhibiting necroptosis and promoting Sox9-mediated tubular epithelial cell proliferation. Phytomedicine 69, 153202. doi:10.1016/j.phymed.2020.153202
Wu, X.-N., Yang, Z.-H., Wang, X.-K., Zhang, Y., Wan, H., Song, Y., et al. (2014). Distinct roles of RIP1–RIP3 hetero- and RIP3–RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 21, 1709–1720. doi:10.1038/cdd.2014.77
Wu, X., Poulsen, K. L., Sanz-Garcia, C., Huang, E., McMullen, M. R., Roychowdhury, S., et al. (2020c). MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcohol-associated fatty liver and steatohepatitis. J. Hepatol. 73, 616–627. doi:10.1016/j.jhep.2020.03.023
Xiao, X., Du, C., Yan, Z., Shi, Y., Duan, H., and Ren, Y. (2017). Inhibition of necroptosis attenuates kidney inflammation and interstitial fibrosis induced by unilateral ureteral obstruction. Am. J. Nephrol. 46, 131–138. doi:10.1159/000478746
Xu, J., Zhang, X.-Q., and Zhang, Z. (2020). Transcription factor EB agonists from natural products for treating human diseases with impaired autophagy-lysosome pathway. Chin. Med. 15, 123. doi:10.1186/s13020-020-00402-1
Xu, Y., Ma, H., Fang, Y., Zhang, Z., Shao, J., Hong, M., et al. (2017). Cisplatin-induced necroptosis in TNFα dependent and independent pathways. Cell Signal 31, 112–123. doi:10.1016/j.cellsig.2017.01.004
Xu, Y., Ma, H., Shao, J., Wu, J., Zhou, L., Zhang, Z., et al. (2015). A role for tubular necroptosis in cisplatin-induced AKI. J. Am. Soc. Nephrol. 26, 2647–2658. doi:10.1681/ASN.2014080741
Xuan, M. Y., Piao, S. G., Ding, J., Nan, Q. Y., Piao, M. H., Jiang, Y. J., et al. (2022). Dapagliflozin alleviates renal fibrosis by inhibiting RIP1-RIP3-MLKL-mediated necroinflammation in unilateral ureteral obstruction. Front. Pharmacol. 12, 798381. doi:10.3389/fphar.2021.798381
Yang, C., Kaushal, V., Shah, S. v., and Kaushal, G. P. (2008). Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am. J. Physiology-Renal Physiology 294, F777–F787. doi:10.1152/ajprenal.00590.2007
Yang, Q., Gao, L., Hu, X., Wang, J., Zhang, Y., Dong, Y., et al. (2021). Smad3-targeted therapy protects against cisplatin-induced AKI by attenuating programmed cell death and inflammation via a NOX4-dependent mechanism. Kidney Dis. 7, 372–390. doi:10.1159/000512986
Yang, Q., Ren, G., Wei, B., Jin, J., Huang, X. R., Shao, W., et al. (2019). Conditional knockout of TGF-βRII/Smad2 signals protects against acute renal injury by alleviating cell necroptosis, apoptosis and inflammation. Theranostics 9, 8277–8293. doi:10.7150/thno.35686
Yang, Q., Sun, G., Yin, H., Li, H., Cao, Z., Wang, J., et al. (2018a). PINK1 protects auditory hair cells and spiral ganglion neurons from cisplatin-induced ototoxicity via inducing autophagy and inhibiting JNK signaling pathway. Free Radic. Biol. Med. 120, 342–355. doi:10.1016/j.freeradbiomed.2018.02.025
Yang, X., Yu, D.-D., Yan, F., Jing, Y.-Y., Han, Z.-P., Sun, K., et al. (2015). The role of autophagy induced by tumor microenvironment in different cells and stages of cancer. Cell Biosci. 5, 14. doi:10.1186/s13578-015-0005-2
Yang, Z., and Klionsky, D. J. (2010). Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 22, 124–131. doi:10.1016/j.ceb.2009.11.014
Yang, Z., Wang, Y., Zhang, Y., He, X., Zhong, C.-Q., Ni, H., et al. (2018b). RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat. Cell Biol. 20, 186–197. doi:10.1038/s41556-017-0022-y
Yoon, S., and Kim, J. (2018). Exogenous spermidine ameliorates tubular necrosis during cisplatin nephrotoxicity. Anat. Cell Biol. 51, 189–199. doi:10.5115/acb.2018.51.3.189
Yu, J. S. L., and Cui, W. (2016). Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 143, 3050–3060. doi:10.1242/dev.137075
Yuan, J., Zhao, X., Hu, Y., Sun, H., Gong, G., Huang, X., et al. (2018). Autophagy regulates the degeneration of the auditory cortex through the AMPK-mTOR-ULK1 signaling pathway. Int. J. Mol. Med. 41, 2086–2098. doi:10.3892/ijmm.2018.3393
Zaha, V. G., and Young, L. H. (2012). AMP-activated protein kinase regulation and biological actions in the heart. Circ. Res. 111, 800–814. doi:10.1161/CIRCRESAHA.111.255505
Zhang, B., Cao, K., Liu, Z., Shan, W., Wen, Q., and Wang, R. (2019a). Receptor interacting protein kinase 3 promotes cisplatin-induced necroptosis in apoptosis-resistant HepG2/DDP cells. Neoplasma 66, 694–703. doi:10.4149/neo_2018_180710N466
Zhang, C., Tong, X., Qi, B., Yu, X., Dong, S., Zhang, S., et al. (2013). Components of Panax notoginseng saponins enhance the cytotoxicity of cisplatin via their effects on gap junctions. Mol. Med. Rep. 8, 897–902. doi:10.3892/mmr.2013.1597
Zhang, D.-W., Shao, J., Lin, J., Zhang, N., Lu, B.-J., Lin, S.-C., et al. (2009). RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336. doi:10.1126/science.1172308
Zhang, D., Pan, J., Xiang, X., Liu, Y., Dong, G., Livingston, M. J., et al. (2017a). Protein kinase cδ suppresses autophagy to induce kidney cell apoptosis in cisplatin nephrotoxicity. J. Am. Soc. Nephrol. 28, 1131–1144. doi:10.1681/ASN.2016030337
Zhang, J., Yang, Y., He, W., and Sun, L. (2016). Necrosome core machinery: Mlkl. Cell. Mol. Life Sci. 73, 2153–2163. doi:10.1007/s00018-016-2190-5
Zhang, J., Zhao, T., Wang, C., Meng, Q., Huo, X., Wang, C., et al. (2021a). Catalpol-induced AMPK activation alleviates cisplatin-induced nephrotoxicity through the mitochondrial-dependent pathway without compromising its anticancer properties. Oxid. Med. Cell Longev. 2021, 7467156. doi:10.1155/2021/7467156
Zhang, L., and Hanigan, M. H. (2003). Role of cysteine S-conjugate β-lyase in the metabolism of cisplatin. J. Pharmacol. Exp. Ther. 306, 988–994. doi:10.1124/jpet.103.052225
Zhang, S., Li, R., Dong, W., Yang, H., Zhang, L., Chen, Y., et al. (2019b). RIPK3 mediates renal tubular epithelial cell apoptosis in endotoxin-induced acute kidney injury. Mol. Med. Rep. 20, 1613–1620. doi:10.3892/mmr.2019.10416
Zhang, W., Li, X., Wang, S., Chen, Y., and Liu, H. (2020). Regulation of TFEB activity and its potential as a therapeutic target against kidney diseases. Cell Death Discov. 6, 32. doi:10.1038/s41420-020-0265-4
Zhang, Y., Du, J., Duan, X., Peng, W., Lv, L., Chen, Z., et al. (2021b). RIPK1 contributes to cisplatin-induced apoptosis of esophageal squamous cell carcinoma cells via activation of JNK pathway. Life Sci. 269, 119064. doi:10.1016/j.lfs.2021.119064
Zhang, Y., Su, S. S., Zhao, S., Yang, Z., Zhong, C.-Q., Chen, X., et al. (2017b). RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 8, 14329. doi:10.1038/ncomms14329
Zhao, C., Chen, Z., Qi, J., Duan, S., Huang, Z., Zhang, C., et al. (2017). Drp1-dependent mitophagy protects against cisplatin-induced apoptosis of renal tubular epithelial cells by improving mitochondrial function. Oncotarget 8, 20988–21000. doi:10.18632/oncotarget.15470
Zhao, Y., Hong, X., Chen, X., Hu, C., Lu, W., Xie, B., et al. (2021). Deregulation of Exo70 facilitates innate and acquired cisplatin resistance in epithelial ovarian cancer by promoting cisplatin efflux. Cancers (Basel) 13, 3467. doi:10.3390/cancers13143467
Zhao, Y., Jing, Z., Li, Y., and Mao, W. (2016). Berberine in combination with cisplatin suppresses breast cancer cell growth through induction of DNA breaks and caspase-3-dependent apoptosis. Oncol. Rep. 36, 567–572. doi:10.3892/or.2016.4785
Zheng, X., Yang, J., Wang, Y., Li, Q., Song, Y., Su, M., et al. (2020). RIP1 promotes proliferation through G2/M checkpoint progression and mediates cisplatin-induced apoptosis and necroptosis in human ovarian cancer cells. Acta Pharmacol. Sin. 41, 1223–1233. doi:10.1038/s41401-019-0340-7
Zhitomirsky, B., Yunaev, A., Kreiserman, R., Kaplan, A., Stark, M., and Assaraf, Y. G. (2018). Lysosomotropic drugs activate TFEB via lysosomal membrane fluidization and consequent inhibition of mTORC1 activity. Cell Death Dis. 9, 1191. doi:10.1038/s41419-018-1227-0
Zhou, X., Xie, L., Xia, L., Bergmann, F., Büchler, M. W., Kroemer, G., et al. (2017). RIP3 attenuates the pancreatic damage induced by deletion of ATG7. Cell Death Dis. 8, e2918. doi:10.1038/cddis.2017.313
Zhu, L., Yuan, Y., Yuan, L., Li, L., Liu, F., Liu, J., et al. (2020). Activation of TFEB-mediated autophagy by trehalose attenuates mitochondrial dysfunction in cisplatin-induced acute kidney injury. Theranostics 10, 5829–5844. doi:10.7150/thno.44051
Zhu, P., Hu, S., Jin, Q., Li, D., Tian, F., Toan, S., et al. (2018). Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: A mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 16, 157–168. doi:10.1016/j.redox.2018.02.019
Keywords: cisplatin, nephrotoxicity, acute kidney injury, necroptosis, necroinflammation, autophagy, macroautophagy
Citation: Alassaf N and Attia H (2023) Autophagy and necroptosis in cisplatin-induced acute kidney injury: Recent advances regarding their role and therapeutic potential. Front. Pharmacol. 14:1103062. doi: 10.3389/fphar.2023.1103062
Received: 19 November 2022; Accepted: 16 January 2023;
Published: 30 January 2023.
Edited by:
Norberto Perico, Mario Negri Institute for Pharmacological Research (IRCCS), ItalyReviewed by:
Man Livingston, Augusta University, United StatesRobert Louis Safirstein, Yale University, United States
Copyright © 2023 Alassaf and Attia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Noha Alassaf, noha-006@hotmail.com