 Elyse C. Page1,2,3
Elyse C. Page1,2,3 Susan L. Heatley1,3
Susan L. Heatley1,3 Jacqueline Rehn1,3
Jacqueline Rehn1,3 Paul Q. Thomas3,4David T. Yeung1,3,5,6,7
Paul Q. Thomas3,4David T. Yeung1,3,5,6,7 Deborah L. White1,2,3,5,8*
Deborah L. White1,2,3,5,8*- 1Blood Cancer Program, Precision Cancer Medicine Theme, South Australian Health and Medical Research Institute, Adelaide, SA, Australia
- 2School of Biological Sciences, Faculty of Sciences, Engineering, and Technology, University of Adelaide, Adelaide, SA, Australia
- 3Adelaide Medical School, Faculty of Health and Medical Science, University of Adelaide, Adelaide, SA, Australia
- 4SA Gene Editing Program, Lifelong Health Theme, South Australian Health and Medical Research Institute, Adelaide, SA, Australia
- 5Australasian Leukaemia and Lymphoma Group, Melbourne, VIC, Australia
- 6Department of Hematology, Royal Adelaide Hospital and SA Pathology, Adelaide, SA, Australia
- 7School of Pharmacy and Medical Science, University of South Australia, Adelaide, SA, Australia
- 8Australian and New Zealand Children’s Hematology/Oncology Group (ANZCHOG), Clayton, VIC, Australia
Acute lymphoblastic leukemia (ALL) patients with a gain of chromosome 21, intrachromosomal amplification of chromosome 21 (iAMP21), or Down syndrome (DS), have increased expression of genes in the DS critical region (DSCR) of chromosome 21, including the high-mobility group nucleosome-binding protein 1, HMGN1. Children with DS are predisposed to develop hematologic malignancies, providing insight into the role of chromosome 21 in the development of leukemias. A 320-kb deletion in the pseudoautosomal region of the X/Y chromosome in leukemic cells, resulting in a gene fusion between the purinergic receptor and cytokine receptor-like factor-2 (P2Y Receptor Family Member 8 (P2RY8)::CRLF2), is a common feature in ~60% of DS-ALL and ~40% of iAMP21 patients, suggesting a link between chromosome 21 and P2RY8::CRLF2. In an Australian cohort of pediatric B-ALL patients with P2RY8::CRLF2 (n = 38), eight patients harbored gain of chromosome 21 (+21), and two patients had iAMP21, resulting in a significantly increased HMGN1 expression. An inducible CRISPR/Cas9 system was used to model P2RY8::CRLF2 and investigate its cooperation with HMGN1. This model was then used to validate HMGN1 as an influencing factor for P2RY8::CRLF2 development. Using Cas9 to cleave the DNA at the pseudoautosomal region without directed repair, cells expressing HMGN1 favored repair, resulting in P2RY8::CRLF2 generation, compared with cells without HMGN1. CRISPR/Cas9 P2RY8::CRLF2 cells expressing HMGN1 exhibit increased proliferation, thymic stromal lymphopoietin receptor (TSLPR) expression, and JAK/STAT signaling, consistent with cells from patients with P2RY8::CRLF2. Our patient expression data and unique CRISPR/Cas9 modeling, when taken together, suggest that HMGN1 increases the propensity for P2RY8::CRLF2 development. This has important implications for patients with DS, +21, or iAMP21.
1 Introduction
Gain of chromosome 21 is the most common whole chromosome copy number variation (CNV) that occurs in hematological malignancies (1, 2), with the highest frequency in acute lymphoblastic leukemia (ALL) at ~15% (3). Children with Down syndrome (DS) and constitutional trisomy 21 have a 20-fold increased risk of developing ALL (4), a 150-fold increased risk of developing acute myeloid leukemia (AML), and are 400–600 times more likely to develop acute megakaryoblastic leukemia (AMKL) (5, 6). The purinergic receptor and cytokine receptor-like factor-2 (P2Y Receptor Family Member 8 (P2RY8)::CRLF2) gene fusion has been identified in ~60% of DS-ALL (+21) patients and ~40% of patients with intrachromosomal amplification of chromosome 21 (iAMP21), compared with only 5%–16% of pediatric ALL patients without +21 (7, 8). The genomic basis for the predisposition in DS-ALL has been investigated (9), but the role of chromosome 21 remains unknown.
Chromosome 21 harbors over 30 candidate genes that may contribute to leukemogenesis (10), including the high-mobility group nucleosome-binding protein 1 (HMGN1) (10). Many genes in the Down syndrome critical region (DSCR) of chromosome 21 have been implicated in various hematological malignancies due to their roles in cancer-associated or gene activation pathways (10, 11). These genes have not yet been linked with the high proportion of DS-ALL patients with P2RY8::CRLF2. Genes including the dual-specificity tyrosine phosphorylated and regulated kinase 1A (DYRK1A), ETS-related gene (ERG), ETS variant transcription factor 6 (ETV6), EBF transcription factor 1 (EBF1), and RUNX family transcription factor 1 (RUNX1) have been studied in ALL (12–18), while many more have been characterized in the context of AML, including the GATA-binding factor 1 (GATA1) (2), ubiquitin-specific peptidase 16 (USP16) (19), chromatin assembly factor 1 (CHAF1B) (20), and the microRNAs (mir99A and mir125b) (21, 22).
Interest in the potential involvement of HMGN1 in leukemia development has arisen due to its demethylase activity associated with enhanced transcriptional activation (11). While ALL patients with CRLF2 alterations are considered at high risk of treatment failure, there are no current effective targeted therapies for this cohort (23). CRISPR/Cas9 facilitates the modeling of individual patient genomic variants (24) with reduced off-target effects and higher efficacy than older gene editing technologies (25) and has been used to model AML fusions (26), but not ALL fusion genes. As there are no cell lines endogenously expressing P2RY8::CRLF2, it is necessary to establish a pre-leukemic cell model to investigate HMGN1 and its effect on P2RY8::CRLF2 fusion generation. We hypothesize that the expression of HMGN1 prior to a Cas9-induced DNA break will lead to increased P2RY8::CRLF2 development in cell lines compared with cells that have low HMGN1 expression.
2 Methods
2.1 Cell lines and maintenance
HEK293T cells (ATCC, Manassas, VA, USA) were maintained in Dulbecco’s modified Eagle’s medium (MEM) and utilized for lentiviral transduction, and Jurkat cells (ATCC, Manassas, VA, USA), which were maintained in Roswell Park Memorial Institute (RPMI), were supplemented with 10% fetal calf serum (FCS), 200 mM of L-glutamine (SAFC Biosciences), 5,000 U/mL of penicillin, and 5,000 µg/mL of streptomycin sulfate.
2.2 Constructing the FgH1tUTG gRNA vector
The Benchling gRNA design tool (Biology Software, 2019, https://benchling.com) was used to design gRNAs targeting the intron following the first non-coding exon of P2RY8 and preceding the first exon of CRLF2 with 5′ Esp3I restriction sites (Key Resources Table). The FUCas9Cherry and FgH1tUTG plasmids were a gift from Marco Herold (Addgene #70182 and #70183) (27). The FgH1tUTG vector was digested with Esp3I [New England Biolabs (NEB) #R0734L] and rSAP (NEB #M0371L) for 1 h at 37°C. The complementary gRNAs were phosphorylated at a final concentration of 10 µM using T4 PNK (NEB #M0201L) and then diluted 1:125 with nuclease-free water. Moreover, 5 ng/µL of FgH1tUTG vector was digested with 0.8 pmol of diluted gRNA and ligated with T4 ligase overnight (NEB #M0204L) at 4°C. The ligation was transformed overnight into competent DH5α E. coli (NEB #C2987H) on ampicillin-containing Luria–Bertani agar plates. Single colonies were isolated and cultured for plasmid purification using the Qiagen QIAprep spin miniprep kit (#27104).
2.3 Lentiviral transduction
Lentivirus was produced by transfecting 5.5 µg of the FuCas9mCherry vector or FgH1tUTG gRNA vector, with packaging constructs pMD2.G (2.25 µg), pMDL-PRRE (3.375 µg), and pRSV-REV (1.575 µg), with 30 µL of lipofectamine added into 1 × 106 HEK293T cells in a T25 culture flask in 5-mL media. Viral supernatant was harvested 48 h later and passed through a 0.45-µm filter. Jurkat cells at a concentration of 5 × 105/mL were centrifuged at 1,800 rpm for 1 h with 30 µg/mL of polybrene in 4 mL of viral supernatant in a six-well plate (28).
2.4 Flow cytometry cell sorting
Jurkat cells transduced with FuCas9mCherry and FgH1tUTG were resuspended in 1 mL of RPMI with 2% FCS at a concentration of 1 × 107 cells. This suspension was sorted on a BD FACSAria™ for GFP and mCherry double-positive cells. Pure populations were resuspended in 1 mL of RPMI with 2% FCS and sorted into single cells in a 96-well plate with 100 µL of RPMI with 20% FCS on a BD FACS Melody™. The clones were subcultured into 1 mL of media in a 24-well plate 3 weeks after sorting.
2.5 Genome-targeting efficiency assay
Jurkat cells transduced with Cas9 and gRNA vectors (Key Resources Table) were exposed to 1 µg/mL of doxycycline hyclate (Sigma-Aldrich, St. Louis, MO, USA) in milli-Q water for 72 h to induce the 320-kb deletion. gDNA was isolated from transduced cells by phenol-chloroform extraction, and the P2RY8::CRLF2 fusion breakpoint was amplified via PCR using the Phusion kit (NEB, Notting Hill, VIC, Australia). The primer sequences are outlined in the Key Resources Table. Heteroduplexes were formed by denaturing the PCR products at 95°C for 5 min and reannealing the breakpoint amplification PCR product by slowly ramping down the temperature to room temperature. The reannealed PCR products were digested with 1 µL of T7 endonuclease I (NEB) for 1 h at 37°C. The resulting products were gel-purified (Qiagen, Venlo, The Netherlands) and Sanger-sequenced.
2.6 Surface flow cytometry
Transduced Jurkat cells were stained with 5 µL of TSLPR-APC or isotype control IgG2a (Invitrogen, Carlsbad, CA, USA) for 30 min in 100 µL of RPMI with 10% FCS on ice. Approximately 5 × 106 cells were washed with 1 mL of RPMI with 10% FCS and resuspended in 200 µL of 1× PBS and read on a BD FACS Fortessa™ analyzer.
2.7 Intracellular flow cytometry
Jurkat cells were fixed with a final concentration of 1.6% paraformaldehyde for 10 min, washed in 1× PBS, and then permeabilized with 80% methanol overnight at -80°C (28). The cells were washed in 1× PBS followed by 1× PBS containing 1% bovine serum albumin (BSA). All intracellular staining procedures were carried out in the dark, on ice, for 60 min at room temperature in 1× PBS/1% BSA with the antibodies outlined in the Key Resources Table. The cells were washed in 1× PBS before reading on a BD FACSCanto™ analyzer.
2.8 Real-time PCR analysis
RNA was isolated from transduced Jurkat cells using TRIzol® (Invitrogen), and cDNA was synthesized using Quantitect reverse transcriptase (Qiagen). SYBR Green reagents (Qiagen) were used with 10 µM of CRLF2 or HMGN1 primers as outlined in the Key Resources Table.
2.9 Proliferation assay
Jurkat cells were seeded at 390 cells/mL in duplicate in a 24-well plate. On days 0, 2, 4, and 6, 20 µL of CellTiter-Glo 2.0® reagent (Promega, Fitchburg, WI, USA) was added to 20 µL of cell suspension. Following 30 min of incubation in the dark, luminescence was measured on a Perkin Elmer Victor X5 luminometer set to luminescence at 0.1 s.
2.10 Development of a patient-derived xenograft murine model
NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice (The Jackson Laboratory, Bar Harbor, ME, USA) were treated subcutaneously with 0.1 mg of Baytril in 0.9% sodium chloride per 10 g body weight prior to sublethal gamma irradiation at 200 cGy. Spleen- or bone marrow (BM)-derived ALL patient (P2RY8::CRLF2 or BCR::ABL1) blasts (1 × 106 cells) were injected into the tail vein of NSG mice. Engraftment and disease progression were monitored by fortnightly blood sampling from the tail vein and flow cytometric analysis of hCD45+. The animals were monitored daily and were humanely killed when they displayed clinical signs of leukemia (e.g., weight loss, reduced activity, ruffled fur). At the end of the experiment, cardiac bleeding and complete blood count (CBC) were performed, and the spleen, liver, and BM were harvested. Flow cytometric immunophenotyping was performed on peripheral blood and/or BM (28). All experiments were performed on protocols approved by the Institutional Animal Ethics Committee.
2.11 mRNA sequencing
mRNA sequencing was performed on the blast cells of pediatric and adolescent/young adult ALL patients (n = 508) using the Universal Plus mRNA seq with NuQuant kit as per the manufacturer’s instructions, with 1 µg of high-quality total RNA, and sequenced on the Illumina NextSeq 500 platforms (29). A read depth of 70 million reads was achieved for most samples. FusionCatcher, SOAPfuse, and JAFFA software were used to identify fusion transcripts from the mRNA sequencing data. Variant calling on the mRNA-seq data was based on the Broad Institute GATK best practices. Gene expression data were generated from the STAR (v2.7.3a)-aligned BAM file using featureCounts and normalized with edgeR. Sub-types were assigned according to identified gene fusions, single nucleotide variants, and gene expression profiles. Gene deletions were detected by multiplex ligation-dependent probe amplification (MLPA). Age-matched ALL patients assessed for in-depth analysis included P2RY8::CRLF2 (n = 38) and BCR::ABL1 (n = 38). Of the P2RY8::CRLF2 patients, eight harbored +21, two of which were +21c. Four patients harboring BCR::ABL1 also harbored +21, one of which was +21c (Supplementary Table S1).
2.12 Multiplex ligation-dependent probe amplification
SALSA MLPA assays #P202, #P335, and #P327 (MRC Holland, Amsterdam, The Netherlands) were performed according to the manufacturer’s instructions using 100 ng of patient DNA and run on a SeqStudio Genetic Analyzer (Applied Biosystems, Waltham, MA, USA).
2.13 Quantification and statistical analysis
GraphPad Prism software Version 8.4.0© (GraphPad Software Inc.) and FlowJo software (FlowJo LLC) were used for the analyses. Graphs represent the median value or mean with SEM error bars as indicated in the figure legends. Student’s t-test and Welch’s ANOVA were used to determine differences between experimental groups as indicated. Differences were considered statistically significant when the p-value was <0.05. Experiments were carried out a minimum of three times (n = 3) unless otherwise stated.
3 Results
We have assessed a cohort of 580 pediatric and adolescent/young adult B-ALL patients for HMGN1/2 expression levels. The expression of HMGN1 varied significantly in ALL patients studied (Figure 1A; p < 0.001). From these samples, an age-matched cohort of P2RY8::CRLF2 (n = 38) and BCR::ABL1 (n = 38) patients were compared for chromosome 21 alterations and the impact on HMGN1 expression (Supplementary Table S1). A +21 cytogenetic aberration was observed in eight patients with the P2RY8::CRLF2 gene fusion, two of which were +21c, compared with four patients with +21 who harbored BCR::ABL1, one of which was +21c. Two patients with P2RY8::CRLF2 also harbored iAMP21, whereas no iAMP21 was observed in the BCR::ABL1 cohort. Significantly higher HMGN1 expression was identified in patients harboring P2RY8::CRLF2 compared with the age-matched control BCR::ABL1+ ALL patients (Figure 1B; p < 0.0001). BCR::ABL1+ patients were chosen as the control due to their similar gene signature and the number of aged-matched patients to the P2RY8::CRLF2 group. There was no difference in the expression of the control genes HMGN2 (p = 0.7881) or JAK2 (p = 0.1171) between the cohort of ALL patients or specifically between P2RY8::CRLF2 and BCR::ABL1+ patients (Figures 1C, D, Supplementary Figure S1). Furthermore, 21% (8/38) of pediatric P2RY8::CRLF2 ALL patients harbored +21 (n = 2 + 21c), resulting in a significantly increased HMGN1 expression, and 5% (2/38) harbored iAMP21, also with a significantly higher HMGN1 expression (Figure 1E; p = 0.0075).
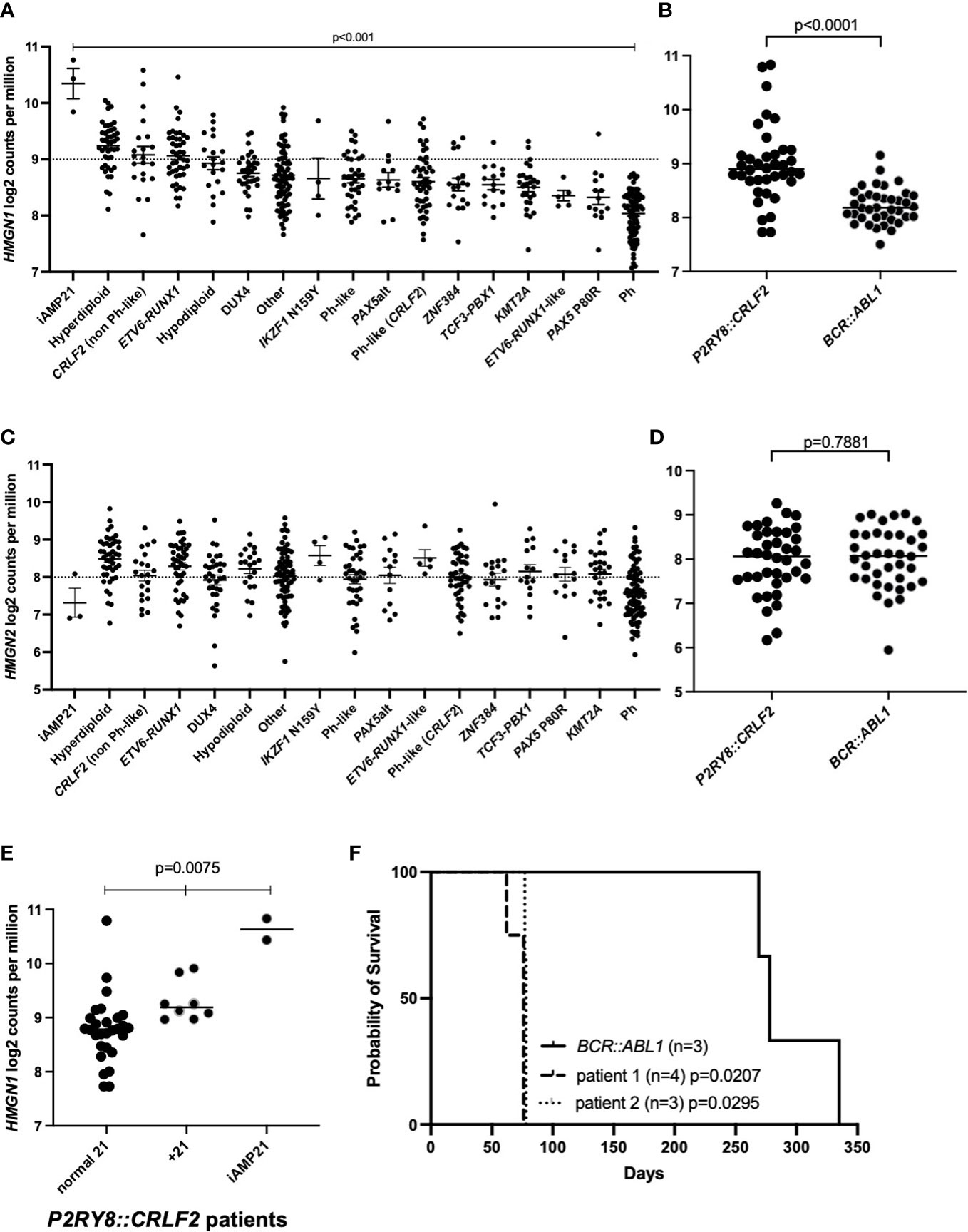
Figure 1 Acute lymphoblastic leukemia (ALL) cohort analysis of high-mobility group nucleosome-binding protein 1 (HMGN1) expression. (A, C) Gene expression analysis of HMGN1 or HMGN2 divided into distinct ALL subtypes. (B, D) HMGN1 or HMGN2 RNA expression data from 38 age-matched pediatric and adolescent/young adult patients in the purinergic receptor and cytokine receptor-like factor-2 (P2RY8::CRLF2) and BCR::ABL1 control cohorts. Welch’s ANOVA was used to determine significance. (E) HMGN1 expression of P2RY8::CRLF2 patients divided into normal chromosome 21, + 21, or intrachromosomal amplification of chromosome 21 (iAMP21) groups. (F) Kaplan–Meier curve of sub-lethally irradiated NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice engrafted with P2RY8::CRLF2 or BCR::ABL1 leukemia patient cells, analyzed using a log-rank test.
To investigate the latency of patient blasts harboring P2RY8::CRLF2 and high HMGN1 expression, NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice were engrafted with blasts from two ALL patients with P2RY8::CRLF2 and high HMGN1 expression (+21). The patient-derived xenograft (PDX) mice succumbed to their leukemia at a median of 76 and 78 days (Figure 1F; p = 0.0207 and p = 0.0295, respectively, compared with the BCR::ABL1 control with a median survival of 278 days). In addition to P2RY8::CRLF2, patient 1 also harbored an activating JAK2 p.F694L mutation, and patient 2 harbored additional lesions including deletions of CDKN2A/B and IKZF1 exons 4–6 and a KRAS p.G12S mutation; however, both patient samples engrafted into NSG mice at the same rate.
To further explore the increased HMGN1 expression and decreased survival in PDX mice, a cell model endogenously expressing P2RY8::CRLF2 was created with CRISPR/Cas9 to delete 320 kb in the pseudoautosomal region (PAR1) of the X/Y chromosome (Figure 2A, Supplementary Figure S2). Prior to the induction of P2RY8 and CRLF2 gRNAs, HMGN1 was overexpressed (1.5-fold) in control Cas9-only expressing cells to represent a trisomic level of expression (Figure 2B; p = 0.019).
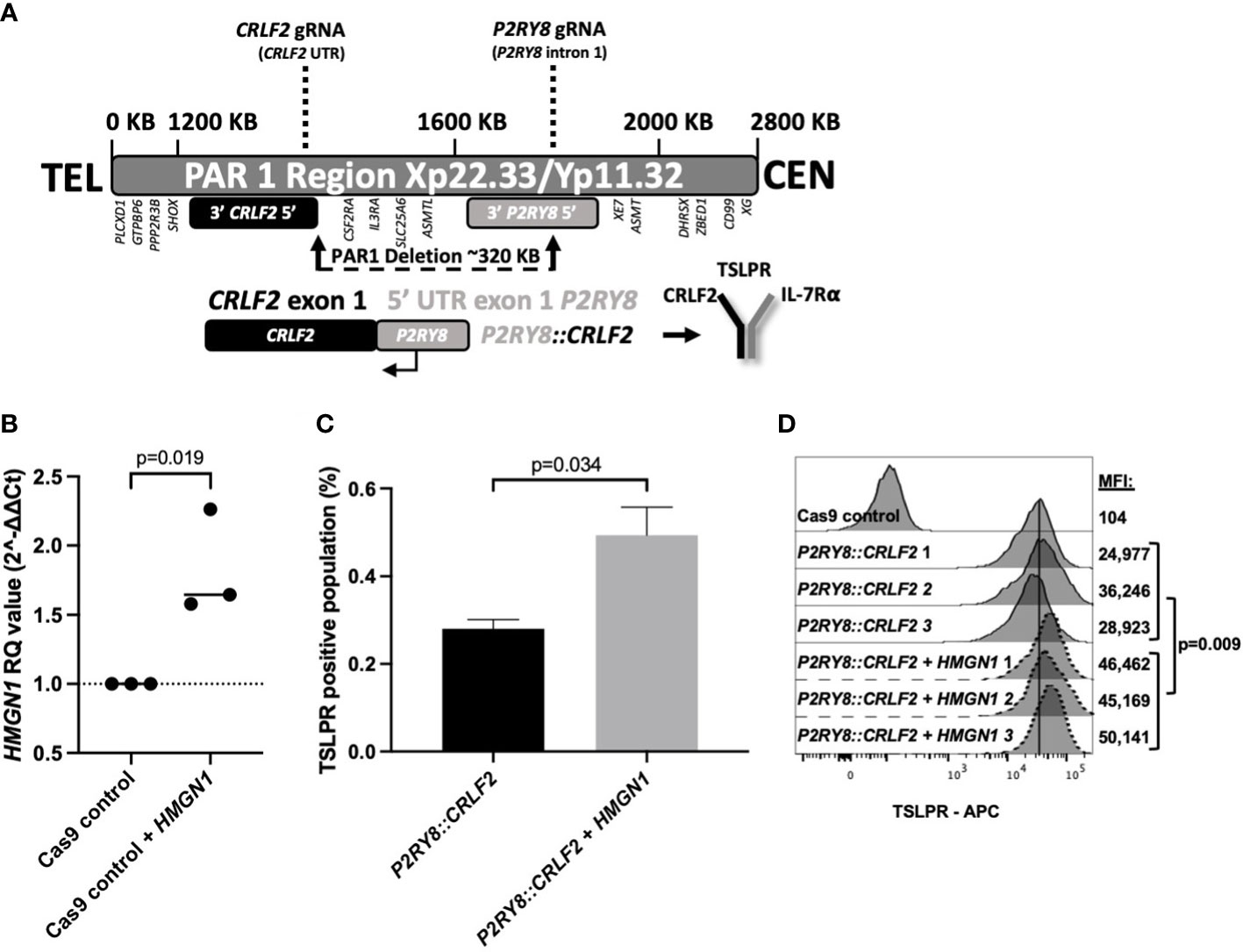
Figure 2 Generating CRISPR/Cas9-edited purinergic receptor and cytokine receptor-like factor-2 (P2RY8::CRLF2)-expressing cells and evaluation of functional changes. (A) Schematic display of gRNAs, designed using Benchling, targeting the intron after the first non-coding exon of P2RY8 and the 5′ untranslated region (UTR) of CRLF2 to create the P2RY8::CRLF2 breakpoint found in patients. (B) Using qRT-PCR to measure high-mobility group nucleosome-binding protein 1 (HMGN1) mRNA expression in Jurkat CRISPR/Cas9 cell lines before gRNA transduction. Relative quantification (RQ) values were determined using the housekeeping actin expression and normalized to the parental Cas9 control cells. Student’s t-test was used between the HMGN1 line compared with control Cas9 cells to determine significance. (C) Jurkat P2RY8::CRLF2 cells with or without HMGN1 expression were stained with thymic stromal lymphopoietin receptor (TSLPR) for flow cytometry after 3 days of gRNA induction to assess the efficiency of P2RY8::CRLF2 generation and favored repair outcomes. (D) TSLPR expression of single-cell clones of Jurkat CRISPR/Cas9-edited P2RY8::CRLF2 cells measured by flow cytometry.
Cells with or without HMGN1 expression were subjected to the same gRNAs/Cas9 without directed repair to determine the favored repair outcome after a DNA break, using TSLPR surface expression as a first readout of successful P2RY8::CRLF2 fusion creation. HMGN1-expressing cells favored P2RY8::CRLF2 generation as demonstrated by upregulated TSLPR on the cell surface from 0.28% in P2RY8::CRLF2 cells to 0.49% in cells expressing P2RY8::CRLF2 + HMGN1 (Figure 2C; p = 0.034). This finding suggests that a higher HMGN1 expression increases the likelihood of P2RY8::CRLF2 development after a DNA break compared with cells that do not express HMGN1-favoring repair for WT CRLF2. Upon induction of Cas9 cleavage at P2RY8 intron 1 and the 5′ UTR of CRLF2, cells with and without HMGN1 expression demonstrated TSLPR surface expression; however, P2RY8::CRLF2 + HMGN1 clones had a significantly higher TSLPR expression (MFI: 47,247 ± 1,489; compared with P2RY8::CRLF2 MFI: 30,049 ± 3,301; p = 0.009; Figure 2D). The breakpoint PCR of genomic DNA confirmed the presence of the P2RY8::CRLF2 fusion in a polyclonal pool and single-cell clones of CRISPR/Cas9-edited TSLPR+ cells, consistent with the independent development of P2RY8::CRLF2 (Supplementary Figure S2).
HMGN1 may assist in the repair of the double-strand DNA breaks to promote P2RY8::CRLF2 formation. To confirm this in vitro, Cas9 gene editing activity was quantified with or without HMGN1 expression using a T7-endonuclease assay. The P2RY8::CRLF2 line resulted in only one isoform after T7-endonuclease digestion, whereas the co-expressing P2RY8::CRLF2 + HMGN1 line resulted in increased gene editing with three bands present in the population (Figure 3A). Sequencing of these isoforms revealed the canonical breakpoint, intron retention, and partial CRLF2 exon 1 deletion.
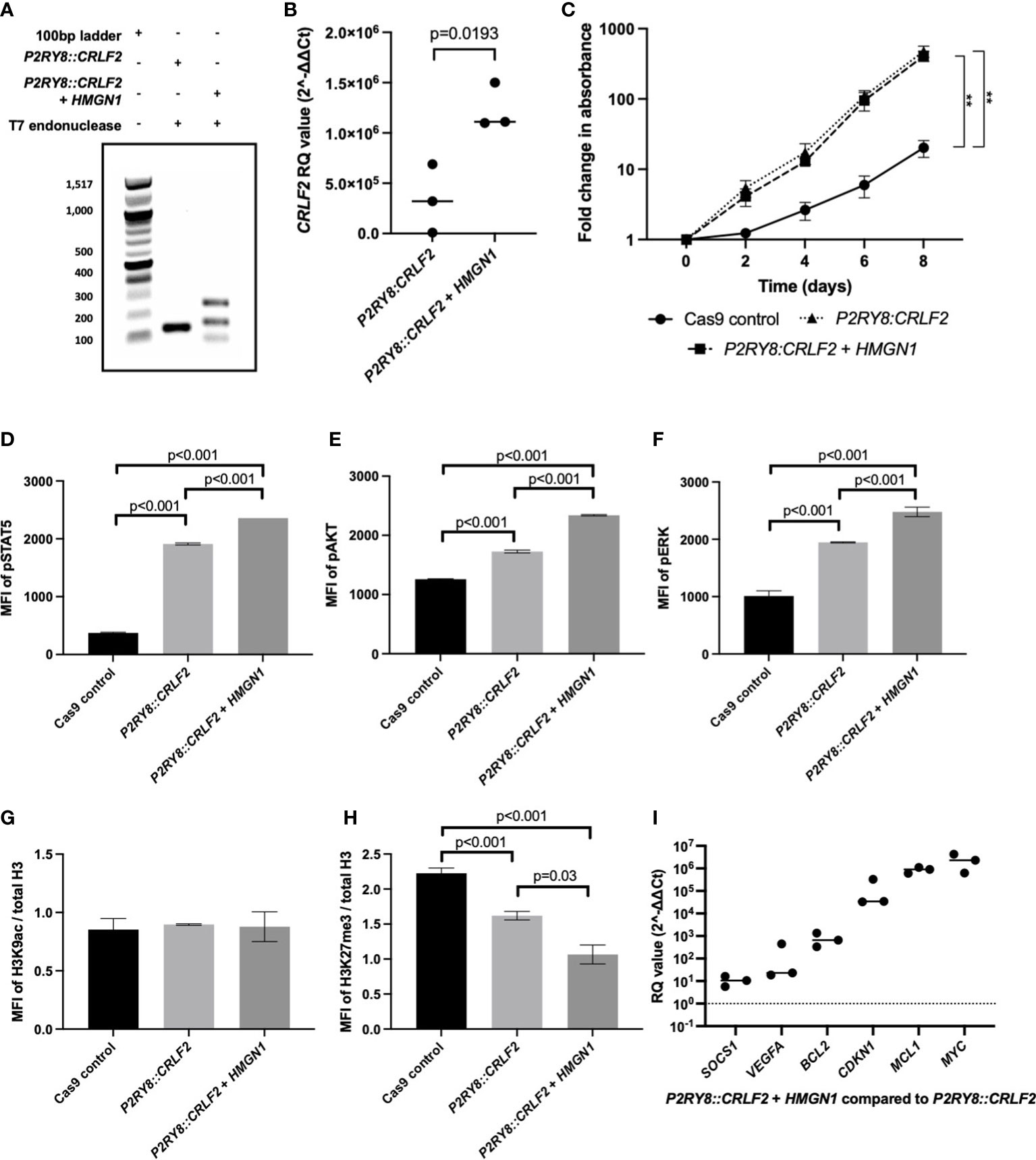
Figure 3 Assessing the effect of high-mobility group nucleosome-binding protein 1 (HMGN1) expression on CRISPR/Cas9-edited purinergic receptor and cytokine receptor-like factor-2 (P2RY8::CRLF2) cells. (A) T7 endonuclease gene editing analysis identifies additional P2RY8::CRLF2 breakpoint PCR products present in HMGN1-expressing cells. (B) Using qRT-PCR to measure CRLF2 mRNA expression in Jurkat CRISPR/Cas9-edited P2RY8::CRLF2 cell lines. Relative quantification (RQ) values were determined using the housekeeping actin expression and normalized to the parental Cas9 control cells. (C) The fold change in the proliferation of Jurkat CRISPR/Cass9-edited P2RY8::CRLF2 cells was measured over a period of 6 days. Phosphorylation levels of STAT5 (D), AKT (E), ERK (F), or H3K9ac (G) and H3K27me3 (H) of CRISPR/Cas9-edited P2RY8::CRLF2 cells with or without high HMGN1 expression measured by flow cytometry. (I) Measuring the expression of genes downstream of STAT5 by qRT-PCR in Jurkat CRISPR/Cas9-edited P2RY8::CRLF2 cell lines. RQ values were determined using the housekeeping actin expression and normalized to the parental Cas9 control cells. The graphs represent the mean of biological replicates of n = 3 with SEM error bars, and a Student’s t-test was used between each P2RY8::CRLF2 cell line compared with control Cas9 cells to determine significance (**p < 0.01).
Consistent with increased P2RY8::CRLF2 generation in the +HMGN1 line, an increase in CRLF2 mRNA expression was also identified via qRT-PCR. P2RY8::CRLF2 cells had higher CRLF2 expression [relative quantification (RQ): 3.4 × 105 ± 1.9 × 105] compared with the control cells (RQ: 47 ± 10). Importantly, a significant increase in CRLF2 expression was observed in P2RY8::CRLF2 + HMGN1 cells (RQ: 1.2 × 106 ± 1.3 × 105; p < 0.001; Figure 3B). The P2RY8::CRLF2 cell pool grew at a seven-fold higher rate than the Cas9 control cells (Figure 3C; p < 0.001). This was also observed in the single-cell clones (#1: p = 0.005; #2: p = 0.015; Supplementary Figure S3A). Interestingly, no difference in proliferation was observed between P2RY8::CRLF2 populations with or without HMGN1 expression despite the increase in CRLF2 expression, suggesting that the role of HMGN1 is epigenetic rather than a direct effect on cell proliferation.
To determine the effect of HMGN1 on cell signaling, the phosphorylation (p) levels of STAT5, AKT, and ERK were assessed. Interestingly, a stepwise increase in phosphorylation of all three proteins was observed between the Cas9 control cells (MFI of pSTAT5: 373 ± 7.4; pAKT: 1,258 ± 5; pERK: 1,011 ± 52), the P2RY8::CRLF2 cells (MFI of pSTAT5: 1,910 ± 10.2; pAKT: 1,727 ± 13.5; pERK: 1,946 ± 6.3), and the P2RY8::CRLF2 + HMGN1 cells (MFI of pSTAT5: 2,359 ± 1; pAKT: 2,339.6 ± 6.3; pERK: 2,478 ± 47.5; Figures 3D–F; all p < 0.001). This signaling profile is consistent with the reported phenotype of P2RY8::CRLF2 patients.
As HMGN1 is a demethylase, the acetylation of H3K9 and trimethylation of H3K27 were assessed. No change in gene activation was identified (Figure 3G); however, a stepwise decrease in H3K27me3 was identified from Cas9 control cells (Figure 3H; H3K27me3 MFI: 2.2 ± 0.04) to P2RY8::CRLF2 cells (H3K27me3 MFI: 1.6 ± 0.03; p < 0.001), with a further reduction in P2RY8::CRLF2 + HMGN1 cells (H3K27me3 MFI: 1 ± 0.07; p = 0.03). This reduction in H3K27me3 may indicate that previously silenced genes have become active in this line.
Assessment of pSTAT5-mediated transcriptional activation indicated higher expression levels of BCL2, CDKN1, and particularly MCL1 and MYC in cells co-expressing HMGN1 and P2RY8::CRLF2 compared with Cas9 control cells (Figure 3I). This finding suggests the potential leukemic survival mechanisms in ALL patients with an increased expression of HMGN1 and the P2RY8::CRLF2 fusion.
4 Discussion
We have evaluated an Australian cohort of pediatric/adolescent B-ALL patients and identified a significantly higher HMGN1 expression in P2RY8::CRLF2 ALL patients compared with a control subgroup. In particular, a significantly higher HMGN1 expression was observed in P2RY8::CRLF2 patients with +21 or iAMP21. In a PDX model of two separate patients with P2RY8::CRLF2 and high HMGN1 expression, the mice succumbed to the disease at the same rate, indicating an aggressive disease burden despite the additional lesions in each patient’s blasts. This was compared with the engraftment of blasts from a patient with BCR::ABL1 which had a latency 3.6 times slower than the P2RY8::CRLF2 blasts. To test the hypothesis that HMGN1 is an influencing factor for P2RY8::CRLF2 development, a cell line in a state before P2RY8::CRLF2 development was required.
Modeling loss-of-function tumor suppressors or gain-of-function oncogenes is fundamental to studying cancer, and CRISPR/Cas9 technology has streamlined this process with high efficiency. Recurrent chromosomal alterations and novel gene fusions have been and continue to be identified in ALL patients (30). To understand the implications of these alterations, they need to be modeled using in vitro and in vivo systems. Subsequently, mechanistic assays and drug panels can be used to identify therapeutic candidates to rationally target the leukemic cells harboring these lesions. The current modeling of ALL gene fusions involves cloning, which can be complex with repetitive sequences or very large transcripts. CRISPR/Cas9 presents a solution to overcome these difficulties and has been used to create chromosomal alterations found in other diseases (24, 26, 31) but has not been previously attempted in ALL. We have utilized this technology to generate an inducible endogenous cell model of P2RY8::CRLF2 that can be employed to determine co-occurring factors in leukemogenesis.
The P2RY8::CRLF2 fusion alone is not sufficient for leukemic transformation and frequently co-occurs with mutations in Janus kinase 2 (JAK2) (7). P2RY8::CRLF2 is increased in DS-ALL patients with a frequency of ~60% (8); however, these patients do not harbor JAK mutations as frequently as non-DS-ALL patients (32). Therefore, the “double hit” is likely to occur first on chromosome 21 and remains to be identified. We have previously demonstrated that a frameshift mutation in NF1 can cooperate with P2RY8::CRLF2 as a mechanism of leukemic relapse (33). Therefore, patients with P2RY8::CRLF2 who do not harbor a JAK or Ras pathway mutation need to be carefully assessed to determine the cooperating lesions for leukemic transformation. Previous reports have demonstrated that increased HMGN1 expression results in a B-cell progenitor phenotype due to its role in lineage determination (11) and may cooperate with P2RY8::CRLF2.
The CRISPR/Cas9 model generated here allowed a pre-leukemic state to be modeled. Increasing the HMGN1 expression before inducing P2RY8 and CRLF2 gRNAs favored fusion development. A potential mechanism via increased DNA double-strand break repair was demonstrated. Consistent with this role of DNA repair, previous reports have identified that the loss of HMGN1 leads to an impaired DNA damage response (34). Therefore, in a trisomy 21 cell with increased HMGN1, an increased chance of repairing double-strand breaks to create P2RY8::CRLF2 is likely. This finding indicates that HMGN1 expression may increase the susceptibility of P2RY8::CRLF2 development.
The P2RY8::CRLF2 and HMGN1 co-expressing cells demonstrated increased proliferation and TSLPR expression and had the most clinically relevant trends in cell signaling compared with previous reports of CRLF2 patient cell signaling (35). Therefore, using CRISPR/Cas9, a role for HMGN1 in cooperation with P2RY8::CRLF2 was demonstrated. Interestingly, P2RY8::CRLF2 + HMGN1 cells had increased expression of BCL2, CDKN1A, MCL1, and MYC. Furthermore, a global decrease in H3K27me3 was associated with an increase in transcriptional activation, consistent with previous reports (11). Increased MYC expression has been previously demonstrated in HMGN1-overexpressing cells (11) and trisomy 21 B-ALL cells (36). BCL2 and MCL1 are anti-apoptotic and novel targets in ALL that warrant further investigation in DS-ALL patients.
The P2RY8::CRLF2 gene fusion is prevalent in DS, +21, and iAMP21 ALL patients. Here we demonstrate that P2RY8::CRLF2 is associated with a high expression of HMGN1 (chr21) in ALL patient cells. Using CRISPR/Cas9 in an in vitro model, we demonstrate that forced high expression of HMGN1 alters the DSB repair mechanism, favoring PAR1 deletion and the subsequent formation of the P2RY8::CRLF2 gene fusion (7), with associated higher expression of STAT5 target genes. Furthermore, this was achieved by the first reported CRISPR/Cas9 320-kb deletion resulting in a clinically relevant fusion gene found in ALL. Importantly, this model will be valuable to advance the ALL field by investigating leukemia-initiating events as the inducible gRNAs allow recapitulation of a pre-leukemic state. Understanding the role of HMGN1 in the disproportionate number of DS–ALL patients who are diagnosed with P2RY8::CRLF2 ALL has the potential to lead to novel therapeutic interventions in this high-risk group of patients where effective therapeutic options are currently limited.
The limitations of this study include the use of viral vectors to deliver the Cas9 machinery into the cell of interest rather than a transient expression system. This was necessary to overcome the low efficiency of transfection in leukemic cells; however, we recognize that this could potentially lead to the disruption of oncogenes at any given locus. Additionally, the Cas9 system has been reported to sporadically induce large deletions in a chromosome (37). In this case, it was a large deletion that was directed by the Cas9 machinery to result in the P2RY8::CRLF2 gene fusion but this does not infer that other deletions did not occur. RNA sequencing could be utilized to determine if any other chromosomal regions were disrupted. To validate the specificity of HMGN1 susceptibility to P2RY8::CRLF2, this study could be repeated to model another gene fusion with the same approach, such as ETV6::RUNX1 which is present in ~10% of DS–ALL patients (38).
Data availability statement
Additional data and requests for resources should be directed to the lead contact, Deborah White (deborah.white@sahmri.com). Materials can be obtained via material transfer agreement from authors’ institutions upon reasonable request to corresponding authors.
Ethics statement
Studies involving human participants were reviewed and approved by each relevant institutional Human Research Ethics Committee. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Animal studies were reviewed and approved by the South Australian Health and Medical Research Institute Ethics Committee.
Author contributions
EP, SH, and DW conceived of and designed the experiments. PT, DY, and DW provided all study materials. ECP collected, assembled, and analyzed the data and wrote the manuscript. JR performed the bioinformatics analysis. DW, SH, PT, DY, and JR critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The funding for this study was provided by the National Health and Medical Research Council, Australia (APP1057746 and APP1044884), the Beat Cancer Project, and the Leukaemia Foundation, Australia.
Acknowledgments
Flow cytometry analysis and cell sorting were performed at the South Australian Health Medical Research Institute (SAHMRI) in the ACRF Cellular Imaging and Cytometry Core Facility. The facility is generously supported by the Detmold Hoopman Group, the Australian Cancer Research Foundation, and the Australian Government through the Zero Childhood Cancer Program.
Conflict of interest
DW receives research support from BMS and honoraria from BMS and AMGEN. DY receives research support from BMS and Novartis and honoraria from BMS, Novartis, Pfizer, and AMGEN. None of these companies had a role in the preparation of this manuscript.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1177871/full#supplementary-material
References
1. Laurent AP, Kotecha RS, Malinge S. Gain of chromosome 21 in hematological malignancies: lessons from studying leukemia in children with down syndrome. Leukemia (2020) 34(8):1984–99. doi: 10.1038/s41375-020-0854-5
2. Izraeli S, Rainis L, Hertzberg L, Smooha G, Birger Y. Trisomy of chromosome 21 in leukemogenesis. Blood Cells Mol Dis (2007) 39(2):156–9. doi: 10.1016/j.bcmd.2007.04.004
3. Mitelman F, Heim S, Mandahl N. Trisomy 21 in neoplastic cells. Am J Med Genet Suppl (1990) 7:262–6. doi: 10.1002/ajmg.1320370752
4. Malinge S, Izraeli S, Crispino JD. Insights into the manifestations, outcomes, and mechanisms of leukemogenesis in down syndrome. Blood (2009) 113(12):2619–28. doi: 10.1182/blood-2008-11-163501
5. Roberts I, Izraeli S. Haematopoietic development and leukaemia in down syndrome. Br J Haematol (2014) 167(5):587–99. doi: 10.1111/bjh.13096
6. Marlow EC, Ducore J, Kwan ML, Cheng SY, Bowles EJA, Greenlee RT, et al. Leukemia risk in a cohort of 3.9 million children with and without down syndrome. J Pediatr (2021) 234:172–80.e3. doi: 10.1016/j.jpeds.2021.03.001
7. Mullighan CG, Collins-Underwood JR, Phillips LA, Loudin MG, Liu W, Zhang J, et al. Rearrangement of CRLF2 in b-progenitor- and down syndrome-associated acute lymphoblastic leukemia. Nat Genet (2009) 41(11):1243–6. doi: 10.1038/ng.469
8. Hertzberg L, Vendramini E, Ganmore I, Cazzaniga G, Schmitz M, Chalker J, et al. Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: a report from the international BFM study group. Blood (2010) 115(5):1006–17. doi: 10.1182/blood-2009-08-235408
9. Brown AL, de Smith AJ, Gant VU, Yang W, Scheurer ME, Walsh KM, et al. Inherited genetic susceptibility to acute lymphoblastic leukemia in down syndrome. Blood (2019) 134(15):1227–37. doi: 10.1182/blood.2018890764
10. Lane AA, Chapuy B, Lin CY, Tivey T, Li H, Townsend EC, et al. Triplication of a 21q22 region contributes to b cell transformation through HMGN1 overexpression and loss of histone H3 Lys27 trimethylation. Nat Genet (2014) 46(6):618–23. doi: 10.1038/ng.2949
11. Mowery CT, Reyes JM, Cabal-Hierro L, Higby KJ, Karlin KL, Wang JH, et al. Trisomy of a down syndrome critical region globally amplifies transcription via HMGN1 overexpression. Cell Rep (2018) 25(7):1898–911.e5. doi: 10.1016/j.celrep.2018.10.061
12. Li S, Sok P, Xu K, Muskens IS, Elliott N, Myint SS, et al. Epigenome-wide association study of acute lymphoblastic leukemia in children with down syndrome. Blood Adv (2022) 6(14):4132–6. doi: 10.1182/bloodadvances.2022007098
13. Lee P, Bhansali R, Izraeli S, Hijiya N, Crispino JD. The biology, pathogenesis and clinical aspects of acute lymphoblastic leukemia in children with down syndrome. Leukemia (2016) 30(9):1816–23. doi: 10.1038/leu.2016.164
14. Malinge S, Bliss-Moreau M, Kirsammer G, Diebold L, Chlon T, Gurbuxani S, et al. Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic leukemia in a murine model of down syndrome. J Clin Invest (2012) 122(3):948–62. doi: 10.1172/JCI60455
15. Zhang J, McCastlain K, Yoshihara H, Xu B, Chang Y, Churchman ML, et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet (2016) 48(12):1481–9. doi: 10.1038/ng.3691
16. Chatterton Z, Morenos L, Mechinaud F, Ashley DM, Craig JM, Sexton-Oates A, et al. Epigenetic deregulation in pediatric acute lymphoblastic leukemia. Epigenetics (2014) 9(3):459–67. doi: 10.4161/epi.27585
17. Izraeli S, Vora A, Zwaan CM, Whitlock J. How I treat ALL in down's syndrome: pathobiology and management. Blood (2014) 123(1):35–40. doi: 10.1182/blood-2013-07-453480
18. Kubota Y, Uryu K, Ito T, Seki M, Kawai T, Isobe T, et al. Integrated genetic and epigenetic analysis revealed heterogeneity of acute lymphoblastic leukemia in down syndrome. Cancer Sci (2019) 110(10):3358–67. doi: 10.1111/cas.14160
19. Adorno M, Sikandar S, Mitra SS, Kuo A, Nicolis Di Robilant B, Haro-Acosta V, et al. Usp16 contributes to somatic stem-cell defects in down's syndrome. Nature (2013) 501(7467):380–4. doi: 10.1038/nature12530
20. Volk A, Liang K, Suraneni P, Li X, Zhao J, Bulic M, et al. A CHAF1B-dependent molecular switch in hematopoiesis and leukemia pathogenesis. Cancer Cell (2018) 34(5):707–23.e7. doi: 10.1016/j.ccell.2018.10.004
21. Boucher AC, Caldwell KJ, Crispino JD, Flerlage JE. Clinical and biological aspects of myeloid leukemia in down syndrome. Leukemia (2021) 35(12):3352–60. doi: 10.1038/s41375-021-01414-y
22. Alejo-Valle O, Weigert K, Bhayadia R, Ng M, Issa H, Beyer C, et al. The megakaryocytic transcription factor ARID3A suppresses leukemia pathogenesis. Blood (2022) 139(5):651–65. doi: 10.1182/blood.2021012231
23. Meyr F, Escherich G, Mann G, Klingebiel T, Kulozik A, Rossig C, et al. Outcomes of treatment for relapsed acute lymphoblastic leukaemia in children with down syndrome. Br J Haematol (2013) 162(1):98–106. doi: 10.1111/bjh.12348
24. García-Tuñón I, Hernández-Sánchez M, Ordoñez JL, Alonso-Pérez V, Álamo-Quijada M, Benito R, et al. The CRISPR/Cas9 system efficiently reverts the tumorigenic ability of BCR/ABL in vitro and in a xenograft model of chronic myeloid leukemia. Oncotarget (2017) 8(16):26027–40. doi: 10.18632/oncotarget.15215
25. Sayin VI, Papagiannakopoulos T. Application of CRISPR-mediated genome engineering in cancer research. Cancer Lett (2017) 387:10–7. doi: 10.1016/j.canlet.2016.03.029
26. Sarrou E, Richmond L, Carmody RJ, Gibson B, Keeshan K. CRISPR gene editing of murine blood stem and progenitor cells induces MLL-AF9 chromosomal translocation and MLL-AF9 leukaemogenesis. Int J Mol Sci (2020) 21(12). doi: 10.3390/ijms21124266
27. Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS, O'Connor L, Milla L, et al. An inducible lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations in vivo. Cell Rep (2015) 10(8):1422–32. doi: 10.1016/j.celrep.2015.02.002
28. Page EC, Heatley SL, Eadie LN, McClure BJ, de Bock CE, Omari S, et al. HMGN1 plays a significant role in CRLF2 driven down syndrome leukemia and provides a potential therapeutic target in this high-risk cohort. Oncogene (2021) 41:797–808. doi: 10.1038/s41388-021-02126-4
29. Rehn J, Mayoh C, Heatley SL, McClure BJ, Eadie LN, Schutz C, et al. RaScALL: rapid (Ra) screening (Sc) of RNA-seq data for prognostically significant genomic alterations in acute lymphoblastic leukaemia (ALL). PloS Genet (2022) 18(10):e1010300. doi: 10.1371/journal.pgen.1010300
30. Iacobucci I, Mullighan CG. Genetic basis of acute lymphoblastic leukemia. J Clin Oncol (2017) 35(9):975–83. doi: 10.1200/JCO.2016.70.7836
31. Vanoli F, Tomishima M, Feng W, Lamribet K, Babin L, Brunet E, et al. CRISPR-Cas9-guided oncogenic chromosomal translocations with conditional fusion protein expression in human mesenchymal cells. Proc Natl Acad Sci U S A (2017) 114(14):3696–701. doi: 10.1073/pnas.1700622114
32. Nikolaev SI, Garieri M, Santoni F, Falconnet E, Ribaux P, Guipponi M, et al. Frequent cases of RAS-mutated down syndrome acute lymphoblastic leukaemia lack JAK2 mutations. Nat Commun (2014) 5:4654. doi: 10.1038/ncomms5654
33. Heatley SL, Page EC, Eadie LN, McClure BJ, Rehn J, Yeung DT, et al. Case report: precision medicine target revealed by. Front Oncol (2022) 12:851572. doi: 10.3389/fonc.2022.851572
34. Rochman M, Taher L, Kurahashi T, Cherukuri S, Uversky VN, Landsman D, et al. Effects of HMGN variants on the cellular transcription profile. Nucleic Acids Res (2011) 39(10):4076–87. doi: 10.1093/nar/gkq1343
35. Tasian SK, Doral MY, Borowitz MJ, Wood BL, Chen IM, Harvey RC, et al. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged b-precursor acute lymphoblastic leukemia. Blood (2012) 120(4):833–42. doi: 10.1182/blood-2011-12-389932
36. Laurent AP, Siret A, Ignacimouttou C, Panchal K, Diop M, Jenni S, et al. Constitutive activation of RAS/MAPK pathway cooperates with trisomy 21 and is therapeutically exploitable in down syndrome b-cell leukemia. Clin Cancer Res (2020) 26(13):3307–18. doi: 10.1158/1078-0432.CCR-19-3519
37. Adikusuma F, Piltz S, Corbett MA, Turvey M, McColl SR, Helbig KJ, et al. Large Deletions induced by Cas9 cleavage. Nature (2018) 560(7717):E8–9. doi: 10.1038/s41586-018-0380-z
Keywords: leukemia, gene expression, HMGN1, P2RY8::CRLF2, CRISPR/Cas9
Citation: Page EC, Heatley SL, Rehn J, Thomas PQ, Yeung DT and White DL (2023) Gain of chromosome 21 increases the propensity for P2RY8::CRLF2 acute lymphoblastic leukemia via increased HMGN1 expression. Front. Oncol. 13:1177871. doi: 10.3389/fonc.2023.1177871
Received: 06 March 2023; Accepted: 15 June 2023;
Published: 06 July 2023.
Edited by:
Carlo V. Bruschi, University of Salzburg, AustriaReviewed by:
Jan-Henning Klusmann, Martin Luther University of Halle-Wittenberg, GermanyMaria S. Pombo-de-Oliveira, National Cancer Institute (INCA), Brazil
Copyright © 2023 Page, Heatley, Rehn, Thomas, Yeung and White. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deborah L. White, Deborah.white@sahmri.com