 Ida Micaily
Ida Micaily Megan Roche1
Megan Roche1 Ubaldo Martinez-Outschoorn
Ubaldo Martinez-Outschoorn Atrayee Basu Mallick
Atrayee Basu Mallick- 1Department of Medical Oncology, Thomas Jefferson University, Philadelphia, PA, United States
- 2Saint Francis Medical Center, Seton Hall University, Trenton, NJ, United States
Chondrosarcomas are the second most common primary bone malignancy. Chondrosarcomas are characterized by the production of cartilaginous matrix and are generally resistant to radiation and chemotherapy and the outcomes are overall poor. Hence, there is strong interest in determining mechanisms of cancer aggressiveness and therapeutic resistance in chondrosarcomas. There are metabolic alterations in chondrosarcoma that are linked to the epigenetic state and tumor microenvironment that drive treatment resistance. This review focuses on metabolic changes in chondrosarcoma, and the relationship between signaling via isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2), hedgehog, PI3K-mTOR-AKT, and SRC, as well as histone acetylation and angiogenesis. Also, potential treatment strategies targeting metabolism will be discussed including potential synergy with immunotherapies.
Introduction
Chondrosarcomas encompass a heterogeneous group of malignant cartilaginous matrix- producing tumors of the bone and are the second most common primary bone malignancy in humans, representing 25% of bone neoplasms (1). Histological grading and staging of chondrosarcomas are the most important factors to aid in prognostication. 85% of chondrosarcomas are histologically classified as conventional chondrosarcomas and can be subcategorized as central or peripherally located (1–3). The other 15% of chondrosarcoma histological subtypes include dedifferentiated, mesenchymal, clear cell and myxoid. Chondrosarcomas are graded from 1 to 3, or low, intermediate and high grade, based on cellularity, nuclear size, nuclear atypia, mitotic activity and matrix alterations (3). The majority of non-conventional chondrosarcomas include low and intermediate grade tumors that are slowly growing with low metastatic potential. However, 5-10% of chondrosarcomas are high-grade and this includes mesenchymal and dedifferentiated chondrosarcomas as well as a subset of conventional chondrosarcomas, which have a high metastatic potential and carry a poor prognosis (4).
The histological grade, stage, and tumor location determine treatment of chondrosarcomas. It is unclear if anticancer agents are active in chondrosarcomas emphasizing the need to understand drivers of cancer aggressiveness that may be the basis of novel therapeutics (2). Surgical management for localized disease is the mainstay of treatment but the specific surgical approach depends on the size, histological type and grade of chondrosarcoma. Small, atypical cartilaginous tumors/grade 1 chondrosarcoma are treated with intralesional curettage, and local high-grade chondrosarcoma are resected en-block (4). Conventional chondrosarcoma that is inoperable or widespread is routinely treated with chemotherapy due to its aggressive nature although it is unclear if any agents are active since there have been no high quality randomized controlled clinical trials in this disease. Chemotherapy was associated with improved overall survival for unresectable conventional chondrosarcoma in a retrospective study from the Rizzoli Institute in Bologna, Italy and Leiden University Medical Center in the Netherlands (3). Specifically, there was a survival advantage when being treated with chemotherapy at 3 years (OS of 26% with chemotherapy vs. 8% without chemotherapy, p<0.05) and the median overall survival was only 11 months for those patients that did not receive chemotherapy (3). However, it is unknown if selection bias contributed significantly to the differences in survival in this retrospective study. Another multi-institutional retrospective analysis with a 180 patients confirmed the poor outcomes for patients with unresectable chondrosarcoma with a median PFS of 4.7 months for first-line chemotherapy (4). The most commonly used chemotherapy regimens for chondrosarcoma are anthracycline-based regimens. However, there are no preferred regimens since there is a lack of positive randomized controlled trials and these tumors are generally chemo-resistant, with the exception of dedifferentiated and mesenchymal which may be partially chemo-sensitive (5).
Chondrosarcoma clinical research is focused on novel therapeutic approaches for inoperable, chemotherapy-refractory or metastatic chondrosarcomas. Although most cancer subtypes have multiple novel therapeutic options, chondrosarcoma lacks effective therapies with the exception of surgery and continues to be associated with poor clinical outcomes (4). Altered metabolism is a hallmark of cancers (6). A deeper understanding of chondrosarcoma metabolism, including glycolytic and TCA enzymes, metabolites, as well as signaling pathways, and tumor suppressor genes and oncogenes that modulate metabolism may identify vulnerabilities that could be exploited therapeutically (7). Genetic changes that affect mitochondrial metabolism are of particular interest in chondrosarcomas, since the most common mutations in these cancers are in the isocitrate dehydrogenase (IDH) gene, and the encoded mutant enzyme modulates mitochondrial metabolism (8). Specifically, mutations in IDH1 or IDH2 isoforms are identified in approximately 60% of chondrosarcomas (2). Also, glutaminolysis affects mitochondrial metabolism in a similar fashion to IDH mutations and is increased in chondrosarcoma (2, 6).
Targeting Isocitrate Dehydrogenase (IDH) Mutations in Chondrosarcoma
IDH enzymes are the most commonly mutated genes in chondrosarcoma and the IDH1 isoform is more commonly mutated than IDH2 (8, 9). Mutant IDH-mediated epigenetic dysregulation leads to chondrocyte differentiation of mesenchymal stem cells and downregulation of osteogenic markers, which may explain the high and low prevalence of IDH mutations in chondrosarcoma and osteosarcoma respectively (10). Post-natal IDH1 R172Q mutations in chondrocytes in mice leads to enchondromas, which is a precursor lesion for chondrosarcoma (11). Knockout of both the mutated and wild type IDH1 via CRISPR–Cas9 from chondrosarcoma cell lines impaired anchorage-independent cell growth and migration with downregulation of integrins, which implicated mutant IDH1 in chondrosarcoma in the epithelial-to-mesenchymal transition with reduced growth rates in vivo and this phenotype was not rescued with restoration experiments with wild type IDH1 (12). Interestingly, mutations in IDH in high grade chondrosarcomas have been associated with CDKN2A/2B and TP53 alterations, prolonged relapse-free and metastasis-free survival (9). In contrast, a meta-analysis revealed that IDH mutations in chondrosarcoma are associated with poor outcomes (13).
IDH mutations are being targeted therapeutically in many cancer subtypes (8). IDH catalyzes the conversion of isocitrate to alpha-ketoglutarate (a-KG) and CO2 in the Krebs or TCA cycle (8). Three isoforms of IDH exist: IDH1 (cytoplasmic), IDH2 (mitochondrial-TCA cycle) and IDH3 (mitochondrial-TCA cycle). IDH1 and IDH2 are NADP-dependent enzymes that are mutated in 38-70% of conventional and dedifferentiated chondrosarcoma cases and induce a neomorphic enzymatic activity (6–13). The mutant forms of IDH1 and IDH2 require a-KG in order to produce a novel metabolite, D-2-hydroxyglutarate (D-2-HG), which becomes a competitive inhibitor in a-KG dependent enzymes and an oncometabolite, which activates hypoxia inducible factor (HIF) (11). HIF-1α expression is highly expressed in the majority of chondrsarcomas and is associated with poor outcomes (14). Also, HIF-2α is upregulated in human high-grade chondrosarcoma biopsies compared to low grade samples and gene amplification is associated with poor prognosis in chondrosarcoma patients (15). HIF-2α promotes chondrosarcoma tumor growth, invasion and metastasis using xenograft models in vivo and pharmacological inhibition of HIF-2α induces apoptosis (15). HIF-1α and HIF-2α are degraded in the proteosome via von-Hippel Lindau (VHL) and VHL disease is due to inactivating mutations in the VHL gene (pVHL), which leads to HIF-1α and HIF-2α stabilization (16). Chondrosarcomas have been described in patients with VHL disease (17–19). Also, low VHL expression is associated with higher grade, higher stage and worse outcomes in chondrosarcoma (20). In sum, IDH and HIF signaling is frequently altered in chondrosarcoma.
IDH1 and IDH2 mutated chondrosarcomas have a distinct metabolomic profile with increased lactate, the TCA intermediates succinate, fumarate, and malate, amino acids broadly and acylcarnitines compared to non-mutated cancer cells based on patient derived xenografts (21). This metabolomic profile of IDH mutated chondrosarcoma is consistent with increased glycolysis, anaplerotic flux into the TCA cycle and fatty acid oxidation (21) and is consistent with other studies that have revealed that there is HIF activation, high rates of glutaminolysis and lipid metabolism in chondrosarcomas (7, 15, 22, 23). Chondrosarcomas with high mRNA expression of glycolytic, TCA, glutamine catabolism and fatty acid oxidation genes were associated with poor outcomes in patients with chondrosarcoma (21).
Pharmacological inhibition of IDH1 reduced colony formation, migration, proliferation, and induced apoptosis in chondrosarcoma cells in vitro (24). However, a different group of investigators using other chondrosarcoma cell lines in vitro did not find that a mutant IDH1 inhibitor had an effect on proliferation and migration (25). IDH1 and 2 mutated chondrosarcoma cells have been shown to up-regulate glutaminolysis and glycolysis for the production of a-KG (25–27). High-grade chondrosarcomas have higher glutaminase expression although no significant differences were observed on the basis of IDH status (14). A subset of chondrosarcoma cell lines are sensitive to drugs targeting glutaminolysis and Bovee et al. studied the glutaminase inhibitor CB-839, the glutamate dehydrogenase inhibitor chloroquine as well as metformin and phenformin, which are inhibitors of mitochondrial oxidative phosphorylation (OXPHOS) and c-MYC and downstream of glutaminolysis (22). Interestingly, this study demonstrated that chloroquine increased apoptosis by increasing caspase 3/7 activity in three out of five cell lines (22). Metformin and phenformin reduced mTOR signaling in these chondrosarcoma cells by decreasing phosphorylated S6 (22) as has been previously demonstrated in other cancers (6). Metformin modulated autophagy with a decrease in LC3B-II levels. Metformin did not affect chondrosarcoma cell viability despite its known effects on mTOR signaling and mitochondrial respiration or OXPHOS. IDH2 mutations in chondrosarcoma cells are known to modulate mesenchymal differentiation through epigenetic dysregulation and treatment with the hypomethylating agent 5-azacytidine is a promising therapeutic strategy (15).
IDH1 inhibition in preclinical and clinical studies has had mixed results (12, 15, 16). A phase I multicenter clinical trial investigating the mutant IDH1 inhibitor ivosidenib (AG-120) in twenty-one patients with advanced chondrosarcoma demonstrated safety with minimal toxicity and a median progression free survival (PFS) of 5.6 months with a PFS rate of 39.5% at 6 months (28). 52% of the 21 patients had stable disease (28). There are ongoing phase I/II clinical trials of novel IDH inhibitors including AG-881 (NCT02481154) and AG-120 (NCT02073994). Aphase I/II clinical trial of metformin and chloroquine in IDH1/2-mutated solid tumors including three patients with chondrosarcoma (NCT02496741) did not find clinical activity (29). A clinical trial of the IDH-inhibitor AG-221 in chondrosarcoma is pending results (NCT02273739) (Table 1). In sum, mutant IDH causes metabolic reprogramming with production of D-2-HG that results in dysregulation of gene expression, differentiation status, DNA damage repair, inflammation, intracellular trafficking, ageing and cell death programs in cancer cells (8). Further research on mutant IDH inhibition is needed to determine its therapeutic value in chondrosarcoma.

Table 1 Clinical trials utilizing IDH and Hh pathway inhibitors in Chondrosarcoma.
Targeting Lipid Metabolism in Chondrosarcoma
Lipids in cancer are thought to be protumorigenic due to their effects on epigenetic reprogramming, maintainance of redox balance by generating NADPH, reduction of ER stress/unfolded protein response, reduction of ferroptosis, effects as second messengers, pro-inflammatory effects, substrates for biomass production, and serving as catabolites to generate ATP (30). Also, increased amounts of cholesterol in the tumor microenvironment induce exhaustion in CD8+ T cells (31). Specifically, high levels of phospholipids and cholesterol are associated with aggressive cancer (32, 33). High cholesterol levels transform the lipid membrane and promote the activity of multidrug efflux pumps driving a multidrug resistant phenotype (33). Cholesterol acting as a second messenger activates mTOR and Hedgehog signaling (34). Conversely, mTOR activation induces denovo lipid and cholesterol synthesis (30, 35).
Inhibition of intracellular cholesterol biogenesis by deletion of sterol regulatory element–binding protein cleavage-activating protein (SCAP) reduces Hedgehog signaling and alters chondrocyte development (36). Statin drugs, which inhibit cholesterol synthesis, restore chondrocyte development and morphology (37, 38). There is upregulation of genes that are activated in cholesterol biosynthesis in IDH1 mutant chondrocytes (23). Consistent with increased biosynthesis, cholesterol levels are higher in IDH1 mutated chondrosarcomas (23). From a therapeutic standpoint, statins reduce cholesterol synthesis and administration of the statin lovastatin reduced chondrosarcoma progression in vivo with reduced proliferation and increased apoptosis rates (23). Hypoxia and starvation, as well as gene mutations in PI3K/AKT, RAS, and MYC have been implicated in upregulation of de novo lipogenesis, cholesterol synthesis and fatty acid desaturation (39). Autophagy is also related to lipid metabolism and there is upregulation in chondrosarcomas (40, 41).
The role of cholesterol and lipid homeostasis in chondrosarcoma is not elucidated. IDH1 mutations are known to cause metabolic reprogramming and regulate intracellular cholesterol biosynthesis. A mouse model utilizing IDH1 mutated chondrosarcoma cells identified an activation of cholesterol biosynthesis (21). IDH1/2 mutated chondrosarcoma cells treated with lovastatin reduced viability (42). These preclinical study suggests that targeting cholesterol metabolism may be promising in chondrosarcoma.
Targeting Glucose Metabolism in Chondrosarcoma
Metabolism of chondrocytes using cell lines has been studied (17, 18). One of the earliest studies of metabolism of UDP-sugar metabolism in chondrocytes utilized Swarm rat chondrosarcoma chondrocytes to better understand the role of glycosylation in cartilage glycosaminoglycan and proteoglycan synthesis. This model demonstrated that stimulation through insulin or fetal calf-serum could increase biosynthetic activity of chondrocytes that can increase the demand of UDP-sugars in cartilage synthesis (43).
Glucose catabolism to C02 and water in the mitochondria occurs in the majority of cells, however under anaerobic conditions catabolism of glucose occurs exclusively in the cytosol and the end product of glycolysis is lactate. Aerobic glycolysis occurs at high rates in cancer cells and has been termed the Warburg Effect. Also, there is a greater appreciation of other cells that are glycolytic within the tumor microenvironment. The Reverse Warburg Effect describes when glycolysis in the cancer-associated stroma metabolically supports adjacent cancer cells. This catabolite transfer, which induces stromal-cancer metabolic coupling, allows cancer cells to generate ATP, increase proliferation, and reduce cell death (44).
Studies have been performed with inhibition of glycolysis in order to treat disease and overcome resistance to other anticancer agents. Multiple nodes in glucose metabolism and reactive oxygen species have been targeted in conjunction with conventional cytotoxic chemotherapy including doxorubicin, cisplatin and paclitaxel (19–23). Expression of lactate dehydrogenase- A (LDHA), which is a key glycolytic enzyme, in cancer cells has been associated with chemotherapy and radiotherapy resistance (2). Additionally, glycolysis can be suppressed by utilizing LDHA inhibitors such as oxamate and FX11 (2). Early studies focused on targeting glycolysis as a mechanism to overcome drug resistance were focused on LDHA. In vitro, paclitaxel resistance was overcome by utilizing the LDHA inhibitor oxamate which induced apoptosis (45). Given the high activity of LDHA and glycolysis in chondrosarcoma cells, LDHA may be a therapeutic target in chondrosarcoma. A cell line exposed to the LDHA inhibitor oxamate can overcome doxorubicin-resistance in chondrosarcoma (46). Another study demonstrated that the addition of oxamate to cisplatin helped overcome chemo-resistance in a chondrosarcoma mouse model (47). Also, the LDHA inhibitor, 1-(phenylseleno)-4-(trifluouromethyl) benzene (PSTMB) has been studied in models of colon cancer, lung cancer, breast cancer, hepatocellular carcinoma and has shown anticancer activity (48). Glucose metabolism may be a future therapeutic target in chondrosarcoma; although to our knowledge, no clinical trials are testing this therapeutic strategy.
Targeting the Hedgehog Pathways in Metabolism of Chondrosarcoma
The hedgehog (Hh) protein regulatory pathway has been implicated in metabolic control, homeostasis and development (49, 50–52). The Hh pathway, which exerts potent metabolic effects, requires the appropriate control of Hh ligand production, processing, secretion and transport (49). A pathological role of the Hh pathway is associated with constitutive activation of transcriptional responses. Binding of Hh to the negative patched regulator (PTCH1) depressed positive smoothened regulator (SMO) in the cilia and phosphorylation and the cytoplasmic end, ultimately mediating downstream signal transduction (49). Glioma zinc finger transcription factors are activated by SMO, which translocate the transcription factors into the nucleus and bind to the promoters of several oncogenes. The disruption of this pathway drives tumorigenesis and glycolysis (49). Chondrosarcomas express high levels of hedgehog (Hh) proteins, and inhibition of the hedgehog pathway in vitro decreased proliferation of chondrosarcoma cells (49) and other solid tumors with down-regulation of P-glycoprotein (50). Additionally, the Indian Hedgehog (IHH)/parathyroid hormone related peptide pathway has been implicated in the pathogenesis of chondrosarcoma. IPI-926 (saridegib), a potent oral- Hh inhibitor, has been assessed in chondrosarcoma cell lines and has shown activity (51). Thereafter, another human chondrosarcoma cell line was also studied with Hh inhibition revealing decreased proliferation, invasion and migratory capacity (52, 53). IPI-926 was tested in a phase II study in chondrosarcoma that was stopped to accrual due to lack of efficacy (51). Another phase II, multicenter clinical trial was conducted on 45 patients with advanced, progressive chondrosarcoma utilizing the Hh pathway inhibitor GDC-0449 (vismodegib). The primary end point was a clinical benefit rate (CBR). The goal CBR was 40% at 6 months. However, the primary end point was not met, as the CBR at 6 months was 25.6% (95% CI 13-42%). Despite the negative results of this clinical trial, clinical benefit was observed in a subset of patients. A possible subpopulation of patients with grade 1 or 2 disease may benefit Hh pathway inhibition in combination with chemotherapy (Table 1). Previous studies have shown that the hedgehog pathway is dysregulated in chondrosarcoma and clinical trials evaluating Hh inhibition have shown that there could be a modest clinical benefit in chondrosarcoma (49–52).
PI3K-AKT- mTOR Pathway in Chondrosarcoma
The mTOR pathway is a crucial regulator of cell metabolism, survival and proliferation (54). The growth factor regulated phosphoinositol 3-kinase (PI3K)-AKT signaling network has many direct and indirect downstream effects on cellular metabolism (6). Physiologically, PI3K-AKT pathway is activated in response to metabolic factors such as insulin, growth factors and cytokines to regulate glucose metabolism, biosynthesis of macromolecules and redox balance (54). These processes support metabolic homeostasis and the growth and metabolism of individual cells (55–58). Pathologically, oncogenic activation of PI3K-AKT reprograms cellular metabolism to anabolism and to promote cancer growth (59). AKT promotes glycolysis and glucose dependency for survival. Protein synthesis is also induced downstream of AKT through mTOR signaling (59).
The mTOR/PI3K pathway as well as IGFR-1 have been studied as a therapeutic anticancer target and this work has served as a foundation for studies in chondrosarcoma (55, 56). Inhibitors of the mTOR pathway have been utilized as single agents or in combination with other agents in both solid and hematologic malignancies (54–58). Human chondrosarcoma-derived cell lines demonstrated phosphorylation of receptor tyrosine kinases (RTKs), particularly AKT, MEK and S6 kinase suggesting clinical relevance of the PI3K/ATK/mTOR pathway in chondrosarcoma (54–58). RTK inhibition in the mTOR/PI3K pathway was studied preclinically utilizing a dual inhibitor, BEZ235 (54). Dual PI3K/mTOR inhibition was studied in a chondrosarcoma xenograft model and demonstrated significant suppression of growth utilizing BEZ235 and GCD-041 with rapamycin (54). Baicalin, a flavonoid extract utilized in traditional Chinese medicine, was found to induce apoptosis in a human chondosarcoma cell line via targeting of PI3K, AKT and mTOR pathways (57). Similarly, the mTOR pathway was studied in a preclinical rat chondrosarcoma model by utilizing the mTOR inhibitor everolimus which blocked cell proliferation as well as GLUT1 and HIF1a expression in vivo (58). Activation of the HIF-1 pathway plays a central role in the adaptive response of tumor cells to hypoxia, and HIF-1 alpha expression is associated with high-grade chondrosarcomas and reduced disease free survival (60, 61). Everolimus reduced GLUT1 expression and HIF1a activity (60). However, single agent everolimus did not induce tumor regression in chondrosarcoma rat models (60). Additionally, doxorubicin was utilized in combination with everolimus and the combination had synergistic activity (61). One proposed mechanism for the synergy between everolimus and doxorubicin is by inhibiting both mTOR and AKT pathways simultaneously. The mTOR inhibitors sapanisertib and rapamycin have also been studied in chondrosarcoma cell lines and they induce decreased oxidative and glycolytic metabolism, independent of IDH status (7). Interestingly, chondrosarcoma cell lines treated with sapanisertib or rapamycin reduced cell viability with a decrease in glucose dependence and a shift towards glutaminolysis and fatty acid catabolism, and sapanisertib had the strongest response (57). Clinical studies of mTOR pathway inhibition in chondrosarcoma hold promise given the encouraging pre-clinical data.
Targeting Angiogenesis in Chondrosarcoma
Tumors and require high rates of metabolite uptake from the vasculature to support their growth and have high vascularization (6). PI3K activation promotes vascular endothelial growth factor-A (VEGF-A) expression leading to angiogenesis, a key component of tumor growth and metastases (44, 62). Catabolites implicated in metabolic coupling include the monocarboxylates lactate, pyruvate, and ketone bodies. Monocarboxylate transporters (MCT) are critically necessary for release and uptake of these catabolites (63). MCT4 is involved in the release of monocarboxylates from cells, is regulated by catabolic transcription factors such as hypoxia inducible factor 1 alpha (HIF1A) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and is highly expressed in cancer cells as well as cancer-associated fibroblasts and has been shown to be highly expressed in sarcomas (63).
In chondrosarcoma human cell lines, angiogenesis is promoted by the fatty acid metabolic hormone adiponectin on VEGF-A (44). VEGF-A expression was induced by adiponectin through the utilization of the adiponectin receptor, PI3K, AKT, mTOR and HIF-1a pathways 7,59. Pretreatment of chondrosarcoma cells with PI3K, AKT or mTOR inhibitors abolished the adiponectin effect on VEGF-A. As a previous study on chondrosarcoma cells demonstrated that adiponectin was linked to metastasis, this could be a possible treatment target in chondrosarcoma (62). The chemokine CCL5 has also been found to promote VEGF-A expression and angiogenesis in chondrosarcoma by activating the PI3K pathway (64). Inhibition of the PI3K pathway abolished the CCL5 effect on VEGF-A and angiogenesis (64). Targeting angiogenesis through multi-tyrosine kinase inhibitors was also studied in preclinical chondrosarcoma models (65). An inhibitor of VEFR2, PDGFR-B and FGFR1 through SU668 was used in a high-grade chondrosarcoma animal model (65).
Another mediator of angiogenesis, cyclooxygenase-2 (COX-2) and prostaglandin activity was studied in chondrosarcoma to determine if inhibiting its activity can improve chemotherapy and radiation sensitivity (42). Preclinical studies utilized agent celecoxib and NS-398 in four high-grade cell lines and although there was initial tumor regression, there was tumor relapse by 6 weeks (42).
A single-arm, multicenter phase 2 trial examining VEGF-inhibition by the oral tyrosine kinase inhibitor (TKI) pazopanib in metastatic or unresectable conventional chondrosarcoma was performed (66). Of the 47 enrolled patients, the primary endpoint of disease control rate (DCR) at 16 weeks was 43%, although this was not statistically significant (66). However, OS was 17.6 months and the median PFS was 7.9 months, which are promising time points in chondrosarcoma clinical trials. Grade 3 adverse events such as hypertension were observed, which are well-described with VEGF inhibitors (66).
As mentioned in Table 1, phase II studies investigating the safety and efficacy of multi-targeted tyrosine kinase inhibitors in various forms of chondrosarcomas are ongoing. These studies highlight the promise of targeting signaling pathways for the treatment of chondrosarcoma.
Targeting the SRC Pathway in Chondrosarcoma
SRC proteins are cellular tyrosine kinases that have been studied in multiple solid tumor types and SRC modulates cell signaling, survival, angiogenesis, proliferation and migration. Cytoplasmic-RSC (c-SRC) contributes to receptor signaling from a number of growth factor receptors (47). Redox regulation of platelet derived growth factor receptor (PDGFR) stimulation, protein kinase C, PI3K and NADPH oxidase activity contribute to complex SRC kinase activation (67). In chondrosarcoma, negative regulators of the intrinsic or mitochondrial apoptotic pathway such as BCL2, which has crosstalk with SRC, are upregulated and mediate chemo-resistance in cancer (67). The multifunctional tyrosine kinase inhibitor Dasatinib (BMS-354825), which inhibits SRC and down-regulates BCL2 was studied in chondrosarcoma cell lines (66, 67). In chondrosarcoma, it has been studied in combination with doxorubicin in TP53 mutant chondrosarcoma cell lines (68). In the 8 doxorubicin resistant chondrosarcoma cell lines utilized in this study, SRC inhibition was found to overcome doxorubicin resistance by inducing apoptosis and inhibiting migration (68).
Another receptor tyrosine kinase target in chondrosarcoma is the platelet derived growth factor receptor (PDGFR), as preclinical data suggests that it’s signaling is induced in chondrosarcoma (67). Preclinical data has demonstrated that PDGFRa can mediate rapamycin-induced AKT phosphorylation in synovial sarcoma cell lines (67). As such, the PDGFR pathway was further studied in clinical trials utilizing single agent imatinib in chondrosarcoma without strong activity signals (see Table 2). The PDGFR pathway remains a promising therapeutic target in chondrosarcoma.

Table 2 Clinical trials utilizing Tyrosine Kinase Inhibitors (TKIs) in Chondrosarcoma.
Histone Deactylase Inhibitors in Chondrosarcoma
Histone dysregulation is a common future across multiple cancer subtypes. Histone acetylation helps alter the regulatory switch to disrupt homeostasis and cause tumor growth. Histone deacetylation and acetylation play key roles in chondrocyte differentiation. As such, histone deactylase inhibitors (HDAC inhibitors) have been well studied as treatment for various cancers, particularly chondrosarcoma (69). In a preclinical model, HDAC inhibitors were found to have antitumor effects by causing differentiation, apoptosis and inducing cell arrest (43). Subsequently, IDH wild type and mutant chondrosarcoma cell lines were studied with the combination of the HDAC inhibitor romidepsin and the glutaminase inhibitor CB-839 (22, 66). Another HDAC inhibitor, depsipeptide demonstrated antitumor activity in both chondrosarcoma cell lines as well as rat models (65). The promising results of these preclinical studies spurred further interest in clinical trials utilizing HDAC inhibitors, and we are awaiting results.
Mitochondrial Metabolism in Chondrosarcoma
The mitochondria are the main source of ATP and the main site of generation of reactive oxygen species (ROS) and its effects have been studied in the progression of various malignancies. ROS is a second messenger notably used in tumor stemness, cell cycle progression, cell proliferation, cell survival, energy metabolism, cell motility, and angiogenesis (70). There is an interest in targeting ROS to induce cell death through induction of mitochondrial ROS production or alternatively using antioxidant strategies. Critical nodes in ROS production or antioxidant activity utilize cofactors such as NADPH, glutathione and FADH. 38-79% of chondrosarcomas have IDH1/IDH2 mutations with an enzymatic neomorphic activity and they use NADPH or NADH as a cofactor. The wild-type enzymes help with conversion of isocitrate dehydrogenase to α-ketoglutarate in cytoplasm and mitochondria respectively and when mutated they drive 2-hydroxyglutarate production, which is a metabolite that drives HIF-1α signaling (60). Hence, there is interest in evaluating the role of mitochondrial metabolism and the redox state in the treatment of chondrosarcoma (22).
The effects of targeting mitochondrial metabolism and apoptosis have been studied in human chondrosarcoma cell lines including studying effects of drugs on BCL2, BAX, Bid, cytochrome c and caspase-3 (71). The fungal metabolite Trichodermin from N psidii demonstrated anti-tumor activity by inducing mitochondrial apoptosis in chondrosarcoma cell lines (53). Other derivatives such as benzimadazole, benzofuran and honokiol have also been studied as pro-apoptotic agents (71–73).
Alterations in mitochondrial metabolism have also been implicated in resistance to chemotherapeutic effect. Translocase of the outer mitochondrial membrane complex subunit 20 (TOMM20) is an important subunit that is responsible for recognizing and translocating mitochondrial proteins into the mitochondria from the cytoplasm (62). Cells with greater mitochondrial mass and activity have greater TOMM20 expression. TOMM20 has been studied in human chondrosarcoma cell lines and found to have higher expression in higher grade chondrosarcomas, indicating a role of increased mitochondrial metabolism, glucose and lactose utilization and oxygen consumption (74). In vivo, TOMM20 induced larger tumor size. Additionally, TOMM20 overexpression was found to induce resistance to cell cycle specific chemotherapy agents (74).
Immunotherapy in Chondrosarcoma
The altered metabolic landscape of tumor microenvironment can influence the suppression of immune cells and anti-tumor immunity (63). Metabolic dysregulation in cancer cells can also effect cell surface expression of proteins related to immune surveillance (75, 76). Specifically, PI3K-AKT-mTOR, HIF-1α and other pathways are implicated in immunotherapy resistance (63). The TCGCA does not include chondrosarcoma but IDH1 mutations have been associated with suppressed immune response pathways based on bioinformatics analyses of TCGA transcriptomic datasets (77). Chondrosarcomas are known to escape immune surveillance through the expression of immune checkpoints and express PD-L1/PD-L2 (78). Dedifferentiated chondrosarcomas are known to have tumor lymphocyte infiltration that correlates with PD-L1 expression and HLA expression, which suggest that these tumors might be sensitive to anticancer immunotherapy (78). Clinical trials utilizing immune checkpoint inhibition are listed in Table 3, and trials are ongoing to investigate the addition of immune checkpoint inhibition in conjunction with other immunotherapy or other novel agents. Preclinical studies of chondrosarcomas with hypomethylating agents in combination with immunotherapy with NY-ESO-1 and PRAME are promising (79). There is an ongoing phase I clinical trial utilizing this agent (see Table 3). Given the current ongoing research in sarcomas and immunotherapy (80), future directions for research may include incorporating metabolic agents in the treatment of chondrosarcoma with novel agents such as immunotherapy, T cell therapy and combination treatments.
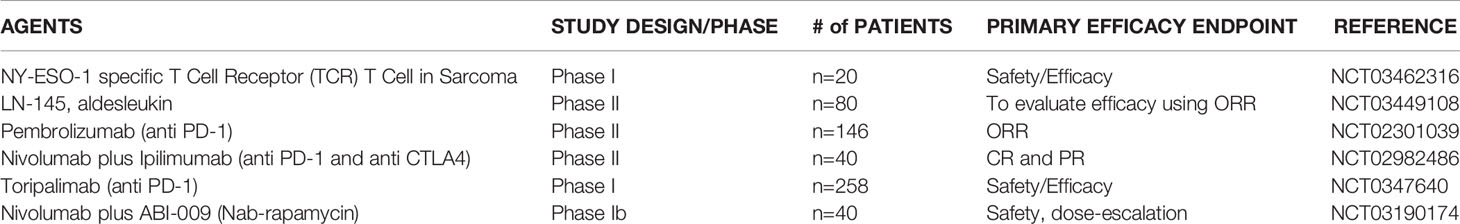
Table 3 Immunotherapy in Chondrosarcoma.
Targeting Extrinsic Apoptosis in Chondrosarcoma
Apoptosis is the process of death and elimination of unwanted cells in the body as a normal part of growth and development. Cancer cells are known to evade apoptosis (76). Apoptosis can be mediated through the intrinsic pathway via BCL 2 or extrinsic pathway via pro-apoptotic receptors like death receptors (DR), such as DR4/DR5. INBRX-109 (a DR5 agonist) is one such pro-apoptotic receptor agonist drug that has been tested in chondrosarcoma with promising results (76, 81). Interim analysis of 12 patients with chondrosarcoma treated in the phase 1 expansion cohort with INBRX-109 showed a decrease in tumor burden by RECIST in 67% (8/12) of patients and disease control rate of 92% (11/12), and a phase 2 study is planned. We await these results to elucidate the role of targeting apoptosis in chondrosarcoma therapy.
Conclusion
High grade, locally advanced and metastatic chondrosarcomas are frequently not amenable to surgery and are resistant to chemotherapy and radiation therapy. Preclinical studies focused on metabolism and/or oncogenic signaling pathways have led to clinical trials, but we still lack active anticancer agents in chondrosarcoma. Figure 1 summarizes potential metabolic and therapeutic targets in chondrosarcoma. Tables 1–3 list many relevant clinical therapeutic trials in chondrosarcoma. Targeting IDH mutations, lipid, glucose and mitochondrial metabolic targets, hedgehog, PI3K/Akt/MTOR, HIF and SRC pathways have yielded mixed results in clinical studies without clear therapeutic options in chondrosarcoma. While metabolic targets have been instrumental in understanding tumor development, the complex, heterogeneous roles of metabolism in chondrosarcoma likely renders mixed therapeutic results. As such, the therapy paradigm in chondrosarcoma aims to incorporate new approaches with anti-angiogenic agents and immunotherapy. Clinical trials will reveal the role of anti-angiogenic agents, immunotherapy and combination treatments and further research will be pivotal in illustrating their future therapeutic role with metabolic agents.
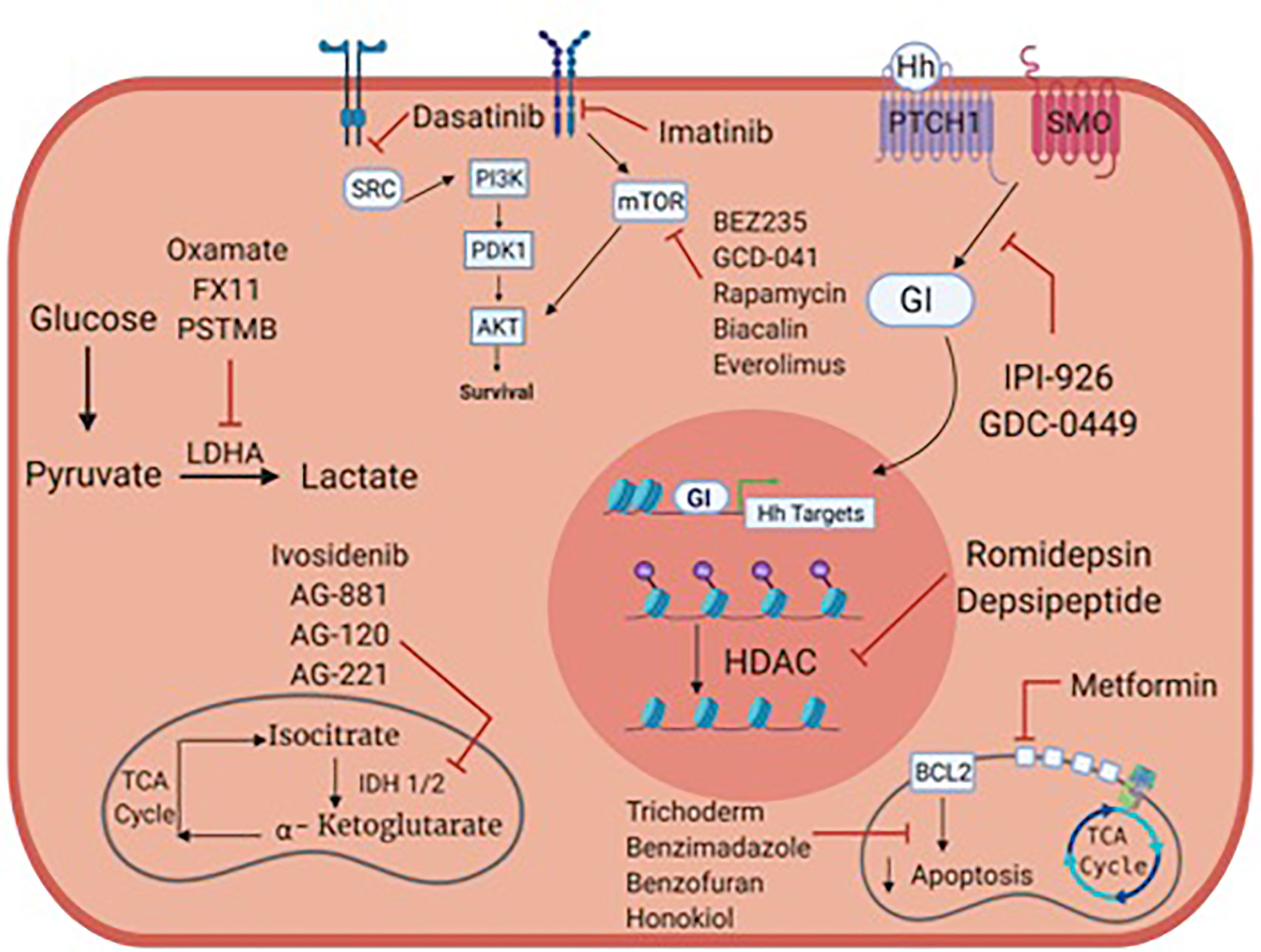
Figure 1 Metabolic targets and potential therapies in chondrosarcoma.
Author Contributions
IM wrote, read and approved the final manuscript. MR edited and aided with images. MY aided in editing the manuscript and citations. UM-O wrote and edited the manuscript. AB wrote and edited the final manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
IDH, Isocitrate dehydrogenase; LDH, Lactate dehydrogenase; PD-1, Programmed-Death Receptor-1; PDL-1, Programmed-Death Receptor Ligand 1.
References
1. Dorfman HD, Czerniak B. Bone Cancers. Cancer (1995) 75:203–10. doi: 10.1002/1097-0142(19950101)75:1+<203::AID-CNCR2820751308>3.0.CO;2-V
2. Bovee JV, Cleton-Jansen AM, Taminiau AH, Hogendoorn PC. Emerging Pathways in the Development of Chondrosarcoma of Bone and Implications for Targeted Treatment. Lancet Oncol (2005) 6:599–607. doi: 10.1016/S1470-2045(05)70282-5
3. van Maldegem AM, Gelderblom H, Palmerini E, Dijkstra SD, Gambarotti M, Ruggieri P, et al. Outcome of Advanced, Unresectable Conventional Central Chondrosarcoma. Cancer (2014) 120:3159–64. doi: 10.1002/cncr.28845
4. Italiano A, Mir O, Cioffi A, Palmerini E, Piperno-Neumann S, Perrin C, et al. Advanced Chondrosarcomas: Role of Chemotherapy and Survival. Ann Oncol (2013) 24:2916–22. doi: 10.1093/annonc/mdt374
5. Biermann JS, Chow W, Reed DR, Lucas D, Adkins DR, Agulnik M, et al. NCCN Guidelines Insights: Bone Cancer, Version 2.2017. J Natl Compr Canc Netw (2017) 15:155–67. doi: 10.6004/jnccn.2017.0017
6. Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP. Cancer Metabolism: A Therapeutic Perspective. Nat Rev Clin Oncol (2017) 14:11–31. doi: 10.1038/nrclinonc.2016.60
7. Addie RD, de Jong Y, Alberti G, Kruisselbrink AB, Que I, Baelde H, et al. Exploration of the Chondrosarcoma Metabolome; the mTOR Pathway as an Important Pro-Survival Pathway. J Bone Oncol (2019) 15:100222. doi: 10.1016/j.jbo.2019.100222
8. Pirozzi CJ, Yan H. The Implications of IDH Mutations for Cancer Development and Therapy. Nat Rev Clin Oncol (2021) 18:645–61. doi: 10.1038/s41571-021-00521-0
9. Zhu GG, Nafa K, Agaram N, Zehir A, Benayed R, Sadowska J, et al. Genomic Profiling Identifies Association of IDH1/IDH2 Mutation With Longer Relapse-Free and Metastasis-Free Survival in High-Grade Chondrosarcoma. Clin Cancer Res (2020) 26:419–27. doi: 10.1158/1078-0432.CCR-18-4212
10. Jin Y, Elalaf H, Watanabe M, Tamaki S, Hineno S, Matsunaga K, et al. Mutant IDH1 Dysregulates the Differentiation of Mesenchymal Stem Cells in Association With Gene-Specific Histone Modifications to Cartilage- and Bone-Related Genes. PloS One (2015) 10:e0131998. doi: 10.1371/journal.pone.0131998
11. Hirata M, Sasaki M, Cairns RA, Inoue S, Puviindran V, Li WY, et al. Mutant IDH Is Sufficient to Initiate Enchondromatosis in Mice. Proc Natl Acad Sci USA (2015) 112:2829–34. doi: 10.1073/pnas.1424400112
12. Li L, Hu X, Eid JE, Rosenberg AE, Wilky BA, Ban Y, et al. Mutant IDH1 Depletion Downregulates Integrins and Impairs Chondrosarcoma Growth. Cancers (Basel) (2020) 12:141. doi: 10.3390/cancers12010141
13. Vuong HG, Ngo TNM, Dunn IF. Prognostic Importance of IDH Mutations in Chondrosarcoma: An Individual Patient Data Meta-Analysis. Cancer Med (2021) 10:4415–23. doi: 10.1002/cam4.4019
14. Chen C, Zhou H, Wei F, Jiang L, Liu X, Liu Z, et al. Increased Levels of Hypoxia-Inducible Factor-1alpha Are Associated With Bcl-xL Expression, Tumor Apoptosis, and Clinical Outcome in Chondrosarcoma. J Orthop Res (2011) 29:143–51. doi: 10.1002/jor.21193
15. Kim H, Cho Y, Kim HS, Kang D, Cheon D, Kim YJ, et al. A System-Level Approach Identifies HIF-2alpha as a Critical Regulator of Chondrosarcoma Progression. Nat Commun (2020) 11:5023. doi: 10.1038/s41467-020-18817-7
16. Tarade D, Ohh M. The HIF and Other Quandaries in VHL Disease. Oncogene (2018) 37:139–47. doi: 10.1038/onc.2017.338
17. Marinozzi A, Papapietro N, Barnaba SA, Di Martino A, Tonini G, Denaro V. Chondrosarcoma of the Iliac Wing in Von Hippel-Lindau Disease. J Exp Clin Cancer Res (2007) 26:599–601.
18. Song MJ, Won KY, Park S. Clear Cell Chondrosarcoma of the Talus in Von Hippel-Lindau Disease: A Rare Tumor in an Unusual Location and Uncommon Co-Presentation. Int J Clin Exp Pathol (2020) 13:266–71.
19. Dreijerink KMA, van Leeuwaarde RS, Hackeng WM, Giles RH, de Leng WWJ, Jutte PC, et al. Clear Cell Chondrosarcoma in Von Hippel-Lindau Disease. Fam Cancer (2020) 19:41–5. doi: 10.1007/s10689-019-00149-1
20. Chen C, Zhou H, Liu X, Liu Z, Ma Q. Reduced Expression of Von Hippel-Lindau Protein Correlates With Decreased Apoptosis and High Chondrosarcoma Grade. J Bone Joint Surg Am (2011) 93:1833–40. doi: 10.2106/JBJS.I.01553
21. Pathmanapan S, Ilkayeva O, Martin JT, Loe AKH, Zhang H, Zhang GF, et al. Mutant IDH and non-Mutant Chondrosarcomas Display Distinct Cellular Metabolomes. Cancer Metab (2021) 9:13. doi: 10.1186/s40170-021-00247-8
22. Peterse EFP, Niessen B, Addie RD, de Jong Y, Cleven AHG, Kruisselbrink AB, et al. Targeting Glutaminolysis in Chondrosarcoma in Context of the IDH1/2 Mutation. Br J Cancer (2018) 118:1074–83. doi: 10.1038/s41416-018-0050-9
23. Zhang H, Wei Q, Tsushima H, Puviindran V, Tang YJ, Pathmanapan S, et al. Intracellular Cholesterol Biosynthesis in Enchondroma and Chondrosarcoma. JCI Insight (2019) 5:e127232. doi: 10.1172/jci.insight.127232
24. Li L, Paz AC, Wilky BA, Johnson B, Galoian K, Rosenberg A, et al. Treatment With a Small Molecule Mutant IDH1 Inhibitor Suppresses Tumorigenic Activity and Decreases Production of the Oncometabolite 2-Hydroxyglutarate in Human Chondrosarcoma Cells. PloS One (2015) 10:e0133813. doi: 10.1371/journal.pone.0133813
25. Suijker J, Oosting J, Koornneef A, Struys EA, Salomons GS, Schaap FG, et al. Inhibition of Mutant IDH1 Decreases D-2-HG Levels Without Affecting Tumorigenic Properties of Chondrosarcoma Cell Lines. Oncotarget (2015) 6:12505–19. doi: 10.18632/oncotarget.3723
26. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of Alpha-Ketoglutarate-Dependent Dioxygenases. Cancer Cell (2011) 19:17–30. doi: 10.1016/j.ccr.2010.12.014
27. Grassian AR, Parker SJ, Davidson SM, Divakaruni AS, Green CR, Zhang X, et al. IDH1 Mutations Alter Citric Acid Cycle Metabolism and Increase Dependence on Oxidative Mitochondrial Metabolism. Cancer Res (2014) 74:3317–31. doi: 10.1158/0008-5472.CAN-14-0772-T
28. Tap WD, Villalobos VM, Cote GM, Burris H, Janku F, Mir O, et al. Phase I Study of the Mutant IDH1 Inhibitor Ivosidenib: Safety and Clinical Activity in Patients With Advanced Chondrosarcoma. J Clin Oncol (2020) 38:1693–701. doi: 10.1200/JCO.19.02492
29. Khurshed M, Molenaar RJ, van Linde ME, Mathot RA, Struys EA, van Wezel T, et al. A Phase Ib Clinical Trial of Metformin and Chloroquine in Patients With IDH1-Mutated Solid Tumors. Cancers (Basel) (2021) 13:2474. doi: 10.3390/cancers13102474
30. Snaebjornsson MT, Janaki-Raman S, Schulze A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab (2020) 31:62–76. doi: 10.1016/j.cmet.2019.11.010
31. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol Induces CD8(+) T Cell Exhaustion in the Tumor Microenvironment. Cell Metab (2019) 30:143–156 e5. doi: 10.1016/j.cmet.2019.04.002
32. Kuzu OF, Noory MA, Robertson GP. The Role of Cholesterol in Cancer. Cancer Res (2016) 76:2063–70. doi: 10.1158/0008-5472.CAN-15-2613
33. Kopecka J, Trouillas P, Gasparovic AC, Gazzano E, Assaraf YG, Riganti C. Phospholipids and Cholesterol: Inducers of Cancer Multidrug Resistance and Therapeutic Targets. Drug Resist Updat (2020) 49:100670. doi: 10.1016/j.drup.2019.100670
34. Ding X, Zhang W, Li S, Yang H. The Role of Cholesterol Metabolism in Cancer. Am J Cancer Res (2019) 9:219–27.
35. Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, et al. SREBP Activity Is Regulated by Mtorc1 and Contributes to Akt-Dependent Cell Growth. Cell Metab (2008) 8:224–36. doi: 10.1016/j.cmet.2008.07.007
36. Tsushima H, Tang YJ, Puviindran V, Hsu SC, Nadesan P, Yu C, et al. Intracellular Biosynthesis of Lipids and Cholesterol by Scap and Insig in Mesenchymal Cells Regulates Long Bone Growth and Chondrocyte Homeostasis. Development (2018) 145:dev162396. doi: 10.1242/dev.162396
37. Yamashita A, Morioka M, Kishi H, Kimura T, Yahara Y, Okada M, et al. Statin Treatment Rescues FGFR3 Skeletal Dysplasia Phenotypes. Nature (2014) 513:507–11. doi: 10.1038/nature13775
38. Terabe K, Takahashi N, Cobb M, Askew EB, Knudson CB, Knudson W. Simvastatin Promotes Restoration of Chondrocyte Morphology and Phenotype. Arch Biochem Biophys (2019) 665:1–11. doi: 10.1016/j.abb.2019.01.038
39. Park JK, Coffey NJ, Limoges A, Le A. The Heterogeneity of Lipid Metabolism in Cancer. Adv Exp Med Biol (2021) 1311:39–56. doi: 10.1007/978-3-030-65768-0_3
40. Wilde L, Tanson K, Curry J, Martinez-Outschoorn U. Autophagy in Cancer: A Complex Relationship. Biochem J (2018) 475:1939–54. doi: 10.1042/BCJ20170847
41. Zeng W, Xiao T, Cai A, Cai W, Liu H, Liu J, et al. Inhibiting ROS-TFEB-Dependent Autophagy Enhances Salidroside-Induced Apoptosis in Human Chondrosarcoma Cells. Cell Physiol Biochem (2017) 43:1487–502. doi: 10.1159/000481971
42. Schrage YM, Machado I, Meijer D, Briaire-de Bruijn I, van den Akker BE, Taminiau AH, et al. COX-2 Expression in Chondrosarcoma: A Role for Celecoxib Treatment? Eur J Cancer (2010) 46:616–24. doi: 10.1016/j.ejca.2009.11.002
43. Sweeney C, Mackintosh D, Mason RM. UDP-Sugar Metabolism in Swarm Rat Chondrosarcoma Chondrocytes. Biochem J (1993) 290( Pt 2):563–70. doi: 10.1042/bj2900563
44. Lee HP, Lin CY, Shih JS, Fong YC, Wang SW, Li TM, et al. Adiponectin Promotes VEGF-A-Dependent Angiogenesis in Human Chondrosarcoma Through PI3K, Akt, mTOR, and HIF-Alpha Pathway. Oncotarget (2015) 6:36746–61. doi: 10.18632/oncotarget.5479
45. Zhang J, Zhou F, Wu X, Zhang X, Chen Y, Zha BS, et al. Cellular Pharmacokinetic Mechanisms of Adriamycin Resistance and its Modulation by 20(S)-Ginsenoside Rh2 in MCF-7/Adr Cells. Br J Pharmacol (2012) 165:120–34. doi: 10.1111/j.1476-5381.2011.01505.x
46. Hua G, Liu Y, Li X, Xu P, Luo Y. Targeting Glucose Metabolism in Chondrosarcoma Cells Enhances the Sensitivity to Doxorubicin Through the Inhibition of Lactate Dehydrogenase-A. Oncol Rep (2014) 31:2727–34. doi: 10.3892/or.2014.3156
47. Song YD, Zhang KF, Liu D, Guo YQ, Wang DY, Cui MY, et al. Inhibition of EGFR-Induced Glucose Metabolism Sensitizes Chondrosarcoma Cells to Cisplatin. Tumour Biol (2014) 35:7017–24. doi: 10.1007/s13277-014-1902-4
48. Kim EY, Chung TW, Han CW, Park SY, Park KH, Jang SB. And Ha KT. A Novel Lactate Dehydrogenase Inhibitor, 1-(Phenylseleno)-4-(Trifluoromethyl) Benzene, Suppresses Tumor Growth Through Apoptotic Cell Death. Sci Rep (2019) 9:3969. doi: 10.1038/s41598-019-40617-3
49. Tiet TD, Hopyan S, Nadesan P, Gokgoz N, Poon R, Lin AC, et al. Constitutive Hedgehog Signaling in Chondrosarcoma Up-Regulates Tumor Cell Proliferation. Am J Pathol (2006) 168:321–30. doi: 10.2353/ajpath.2006.050001
50. Chen YJ, Kuo CD, Chen SH, Chen WJ, Huang WC, Chao KS, et al. Small-Molecule Synthetic Compound Norcantharidin Reverses Multi-Drug Resistance by Regulating Sonic Hedgehog Signaling in Human Breast Cancer Cells. PloS One (2012) 7:e37006. doi: 10.1371/journal.pone.0037006
51. Campbell VT, Nadesan P, Ali SA, Wang CY, Whetstone H, Poon R, et al. Hedgehog Pathway Inhibition in Chondrosarcoma Using the Smoothened Inhibitor IPI-926 Directly Inhibits Sarcoma Cell Growth. Mol Cancer Ther (2014) 13:1259–69. doi: 10.1158/1535-7163.MCT-13-0731
52. Xiang W, Jiang T, Guo F, Gong C, Yang K, Wu Y, et al. Hedgehog Pathway Inhibitor-4 Suppresses Malignant Properties of Chondrosarcoma Cells by Disturbing Tumor Ciliogenesis. Oncol Rep (2014) 32:1622–30. doi: 10.3892/or.2014.3372
53. Su CM, Wang SW, Lee TH, Tzeng WP, Hsiao CJ, Liu SC, et al. Trichodermin Induces Cell Apoptosis Through Mitochondrial Dysfunction and Endoplasmic Reticulum Stress in Human Chondrosarcoma Cells. Toxicol Appl Pharmacol (2013) 272:335–44. doi: 10.1016/j.taap.2013.06.010
54. Zhang YX, van Oosterwijk JG, Sicinska E, Moss S, Remillard SP, van Wezel T, et al. Functional Profiling of Receptor Tyrosine Kinases and Downstream Signaling in Human Chondrosarcomas Identifies Pathways for Rational Targeted Therapy. Clin Cancer Res (2013) 19:3796–807. doi: 10.1158/1078-0432.CCR-12-3647
55. Mazzoletti M, Bortolin F, Brunelli L, Pastorelli R, Di Giandomenico S, Erba E, et al. Combination of PI3K/mTOR Inhibitors: Antitumor Activity and Molecular Correlates. Cancer Res (2011) 71:4573–84. doi: 10.1158/0008-5472.CAN-10-4322
56. Quek R, Wang Q, Morgan JA, Shapiro GI, Butrynski JE, Ramaiya N, et al. Combination mTOR and IGF-1R Inhibition: Phase I Trial of Everolimus and Figitumumab in Patients With Advanced Sarcomas and Other Solid Tumors. Clin Cancer Res (2011) 17:871–9. doi: 10.1158/1078-0432.CCR-10-2621
57. Zhu M, Ying J, Lin C, Wang Y, Huang K, Zhou Y, et al. Baicalin Induces Apoptotic Death of Human Chondrosarcoma Cells Through Mitochondrial Dysfunction and Downregulation of the PI3K/Akt/mTOR Pathway. Planta Med (2019) 85:360–9. doi: 10.1055/a-0791-1049
58. Perez J, Decouvelaere AV, Pointecouteau T, Pissaloux D, Michot JP, Besse A, et al. Inhibition of Chondrosarcoma Growth by mTOR Inhibitor in an In Vivo Syngeneic Rat Model. PloS One (2012) 7:e32458. doi: 10.1371/journal.pone.0032458
59. Hoxhaj G, Manning BD. The PI3K-AKT Network at the Interface of Oncogenic Signalling and Cancer Metabolism. Nat Rev Cancer (2020) 20:74–88. doi: 10.1038/s41568-019-0216-7
60. Kubo T, Sugita T, Shimose S, Matsuo T, Arihiro K, Ochi M. Expression of Hypoxia-Inducible Factor-1alpha and its Relationship to Tumour Angiogenesis and Cell Proliferation in Cartilage Tumours. J Bone Joint Surg Br (2008) 90:364–70. doi: 10.1302/0301-620X.90B3.19806
61. Boeuf S, Bovee JV, Lehner B, Hogendoorn PC, Richter W. Correlation of Hypoxic Signalling to Histological Grade and Outcome in Cartilage Tumours. Histopathology (2010) 56:641–51. doi: 10.1111/j.1365-2559.2010.03528.x
62. Chiu YC, Shieh DC, Tong KM, Chen CP, Huang KC, Chen PC, et al. Involvement of AdipoR Receptor in Adiponectin-Induced Motility and Alpha2beta1 Integrin Upregulation in Human Chondrosarcoma Cells. Carcinogenesis (2009) 30:1651–9. doi: 10.1093/carcin/bgp156
63. Wilde L, Roche M, Domingo-Vidal M, Tanson K, Philp N, Curry J, et al. Metabolic Coupling and the Reverse Warburg Effect in Cancer: Implications for Novel Biomarker and Anticancer Agent Development. Semin Oncol (2017) 44:198–203. doi: 10.1053/j.seminoncol.2017.10.004
64. Liu GT, Chen HT, Tsou HK, Tan TW, Fong YC, Chen PC, et al. CCL5 Promotes VEGF-Dependent Angiogenesis by Down-Regulating miR-200b Through PI3K/Akt Signaling Pathway in Human Chondrosarcoma Cells. Oncotarget (2014) 5:10718–31. doi: 10.18632/oncotarget.2532
65. Sakimura R, Tanaka K, Yamamoto S, Matsunobu T, Li X, Hanada M, et al. The Effects of Histone Deacetylase Inhibitors on the Induction of Differentiation in Chondrosarcoma Cells. Clin Cancer Res (2007) 13:275–82. doi: 10.1158/1078-0432.CCR-06-1696
66. Chow W, Frankel P, Ruel C, Araujo DM, Milhem M, Okuno S, et al. Results of a Prospective Phase 2 Study of Pazopanib in Patients With Surgically Unresectable or Metastatic Chondrosarcoma. Cancer (2020) 126:105–11. doi: 10.1002/cncr.32515
67. van Oosterwijk JG, van Ruler MA, Briaire-de Bruijn IH, Herpers B, Gelderblom H, van de Water B, et al. Src Kinases in Chondrosarcoma Chemoresistance and Migration: Dasatinib Sensitises to Doxorubicin in TP53 Mutant Cells. Br J Cancer (2013) 109:1214–22. doi: 10.1038/bjc.2013.451
68. Chow WA. Chondrosarcoma: Biology, Genetics, and Epigenetics. F1000Res (2018) 7(F1000 Faculty Rev):1826. doi: 10.12688/f1000research.15953.1
69. Sheikh TN, Patwardhan PP, Cremers S, Schwartz GK. Targeted Inhibition of Glutaminase as a Potential New Approach for the Treatment of NF1 Associated Soft Tissue Malignancies. Oncotarget (2017) 8:94054–68. doi: 10.18632/oncotarget.21573
70. Liou GY, Storz P. Reactive Oxygen Species in Cancer. Free Radic Res (2010) 44:479–96. doi: 10.3109/10715761003667554
71. Su CM, Chen CY, Lu T, Sun Y, Li W, Huang YL, et al. A Novel Benzofuran Derivative, ACDB, Induces Apoptosis of Human Chondrosarcoma Cells Through Mitochondrial Dysfunction and Endoplasmic Reticulum Stress. Oncotarget (2016) 7:83530–43. doi: 10.18632/oncotarget.13171
72. Chen YJ, Wu CL, Liu JF, Fong YC, Hsu SF, Li TM, et al. Honokiol Induces Cell Apoptosis in Human Chondrosarcoma Cells Through Mitochondrial Dysfunction and Endoplasmic Reticulum Stress. Cancer Lett (2010) 291:20–30. doi: 10.1016/j.canlet.2009.08.032
73. Li TM, Lin TY, Hsu SF, Wu CM, Su YC, Kao ST, et al. The Novel Benzimidazole Derivative, MPTB, Induces Cell Apoptosis in Human Chondrosarcoma Cells. Mol Carcinog (2011) 50:791–803. doi: 10.1002/mc.20749
74. Roche ME, Lin Z, Whitaker-Menezes D, Zhan T, Szuhai K, Bovee J, et al. Translocase of the Outer Mitochondrial Membrane Complex Subunit 20 (TOMM20) Facilitates Cancer Aggressiveness and Therapeutic Resistance in Chondrosarcoma. Biochim Biophys Acta Mol Basis Dis (2020) 1866:165962. doi: 10.1016/j.bbadis.2020.165962
75. Ramapriyan R, Caetano MS, Barsoumian HB, Mafra ACP, Zambalde EP, Menon H, et al. Altered Cancer Metabolism in Mechanisms of Immunotherapy Resistance. Pharmacol Ther (2019) 195:162–71. doi: 10.1016/j.pharmthera.2018.11.004
76. Ashkenazi A. Directing Cancer Cells to Self-Destruct With Pro-Apoptotic Receptor Agonists. Nat Rev Drug Discovery (2008) 7:1001–12. doi: 10.1038/nrd2637
77. Unruh D, Zewde M, Buss A, Drumm MR, Tran AN, Scholtens DM, et al. Methylation and Transcription Patterns Are Distinct in IDH Mutant Gliomas Compared to Other IDH Mutant Cancers. Sci Rep (2019) 9:8946. doi: 10.1038/s41598-019-45346-1
78. Kostine M, Cleven AH, de Miranda NF, Italiano A, Cleton-Jansen AM, Bovee JV. Analysis of PD-L1, T-Cell Infiltrate and HLA Expression in Chondrosarcoma Indicates Potential for Response to Immunotherapy Specifically in the Dedifferentiated Subtype. Mod Pathol (2016) 29:1028–37. doi: 10.1038/modpathol.2016.108
79. Pollack SM, Li Y, Blaisdell MJ, Farrar EA, Chou J, Hoch BL, et al. NYESO-1/LAGE-1s and PRAME Are Targets for Antigen Specific T Cells in Chondrosarcoma Following Treatment With 5-Aza-2-Deoxycitabine. PloS One (2012) 7:e32165. doi: 10.1371/journal.pone.0032165
80. Heymann MF, Schiavone K, Heymann D. Bone Sarcomas in the Immunotherapy Era. Br J Pharmacol (2021) 178:1955–72. doi: 10.1111/bph.14999
Keywords: chondrosarcoma, metabolism, IDH, LdhA, lactate dehydrogenase, mTOR, PI3K - AKT pathway
Citation: Micaily I, Roche M, Ibrahim MY, Martinez-Outschoorn U and Mallick AB (2021) Metabolic Pathways and Targets in Chondrosarcoma. Front. Oncol. 11:772263. doi: 10.3389/fonc.2021.772263
Received: 07 September 2021; Accepted: 18 November 2021;
Published: 06 December 2021.
Edited by:
Egidio Iorio, National Institute of Health (ISS), ItalyReviewed by:
Cinzia Domenicotti, Università di Genova, ItalyMaria Elena Pisanu, National Institute of Health (ISS), Italy
Copyright © 2021 Micaily, Roche, Ibrahim, Martinez-Outschoorn and Mallick. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Atrayee Basu Mallick, atrayee.basumallick@jefferson.edu