 Jiejun Hu
Jiejun Hu Dong Cai
Dong Cai Zhibo Zhao
Zhibo Zhao Guo-Chao Zhong
Guo-Chao Zhong- Department of Hepatobiliary Surgery, The Second Affiliated Hospital of Chongqing Medical University, Chongqing, China
Hepatocellular carcinoma (HCC), the most common malignant tumor, has high fatality and recurrence rates. Accumulating evidence shows that heterogeneous nuclear ribonucleoprotein C (HNRNPC), which is mainly involved in RNA splicing, export, and translation, promotes progression and metastasis of multiple tumor types; however, the effects of HNRNPC in HCC are unknown. In the present study, high levels of HNRNPC were detected in tumor tissues compared with para-tumor tissues by immunohistochemical and western blot assays. Furthermore, Cox proportional hazards regression models, the Kaplan–Meier method, and clinicopathologic features analysis showed that HNRNPC was not only an independent prognostic factor for both overall and disease-free survival in HCC but also a predictor of large tumor size and advanced tumor stage. Functional experiments revealed that silencing of HNRNPC not only led to arrest of more HCC cells at G0/G1 phase to inhibit their proliferation, but also suppressed EMT process to block their invasion, and migration in vitro; this was related to the Ras/MAPK signaling pathway. In addition, blocking of HCC cell proliferation regulated by HNRNPC silencing was observed in vivo. Finally, rescue tests showed that after recovery of Ras/MAPK signaling pathway activity by treatment with Ras agonists, the proliferation, migration, and invasion suppression of Huh-7 and Hep 3B cell lines caused by HNRNPC knockdown was partially reversed. Taken together, these results indicate that HNRNPC knockdown inhibits HCC cell proliferation, migration and invasion, in part via the Ras/MAPK signaling pathway. Thus, HNRNPC may have an important role in the progression of HCC and represents a promising biomarker for evaluation of prognosis and a potential therapeutic target in HCC patients.
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common cancer and the fourth leading cause of cancer-associated deaths worldwide, with a gradually increasing global burden (1, 2). For patients with early-stage tumors, liver resection and transplantation are the main treatment modes (3–5), whereas for patients with advanced-stage tumors, multimodal treatment is increasingly popular; this includes immunological therapy, molecular targeted treatments, microwave ablation, and transarterial chemoembolization (6–11). Although an increasing number of therapies are used to treat HCC, the prognosis of HCC patients remains poor (12–14), with a 5-year overall survival rate of less than 20% (15–17), primarily owing to the difficulty of early-stage diagnosis (18). For these reasons, there is a need to explore novel biomarkers that could be used as targets for HCC therapy and for predicting the prognosis of HCC patients.
Alternative splicing enables the generation of vast protein diversity by cutting out the introns and connecting the exons of heterogeneous nuclear RNAs (19). Increasing numbers of reports indicate that alternative splicing is intimately related to tumor occurrence, progression, and therapeutic resistance (20–23). A known RNA-binding protein, heterogeneous nuclear ribonucleoprotein C (HNRNPC) plays an important part in RNA splicing (24, 25), stability (26, 27), and expression (28). HNRNPC has been identified as an oncogene, enhancing deterioration in multiple tumor types, including gastric cancer (29), breast cancer (27), esophageal squamous cell carcinoma (26), and oral squamous cell carcinoma (30). According to previous reports, HNRNPC primarily regulates the biological activity of the IFNβ signaling pathway (27), p53 gene (31), and AKT signaling pathway (32), which are associated with tumor proliferation, invasion, and metastasis. Bioinformatic analysis has shown that HNRNPC, as a gene correlated with N6-methyladenosine RNA methylation, predicts poor prognosis of patients with glioblastoma multiforme (33), lung adenocarcinoma (34), and head and neck squamous cell carcinoma (35). Thus, HNRNPC is considered to be a tumor-related gene. Nevertheless, its impact on patient prognosis and the mechanism by which it regulates the biological characteristics of tumor cells in HCC remain unknown.
The mitogen-activated protein kinase (MAPK) cascade consists of serine/threonine kinases, which mainly participate in the transduction of cellular signals (36, 37). There are four important protein kinases, Ras, Raf, MEK, and Erk, in the MAPK signaling pathway (38). In response to signals from epidermal growth factor receptor, insulin like growth factor-1 receptor (IGF-1R), and calmodulin, the GDP combined with Ras is replaced by GTP. Then, the activated Ras cascade phosphorylates its downstream targets Raf, MEK, and Erk to regulate cell proliferation, differentiation, and cycling (39–41). Additionally, Ras/MAPK signaling pathway promoting epithelial-mesenchymal transition (EMT) to facilitate tumor migration and invasion have been reported in recent years (42–44). Abnormal activation of the MAPK pathway is involved in the progression of many malignancies, including gastric cancer (45), cervical carcinoma (46), and HCC (47). Hence, targeting Ras/Raf/MEk/Erk is regarded as a potential approach to treating HCC (36), esophageal carcinoma (48), acute lymphoblastic leukemia (49), and melanoma (50).
In the present study, we used western blotting, immunohistochemistry (IHC), and bioinformatics analysis to demonstrate that HNRNPC had significantly elevated expression in HCC tumor tissues compared with para-tumor tissues. In addition, high levels of HNRNPC protein were related to larger tumor size, advanced TNM stage, and poor prognosis. Subsequently, functional experiments revealed that silencing of HNRNPC expression could inhibit HCC cells’ growth by G0/G1 arrest, in part via the Ras/MAPK signaling pathway. Meanwhile, in vitro tests, we also demonstrated that the inhibition of Ras/MAPK signaling pathway caused by HNRNPC suppression could negatively regulate EMT progress to hinder migration and invasion of HCC cells. In vivo, xenograft assays showed that HNRNPC knockdown could block the proliferation of HCC cells, with a statistically significant effect. Therefore, HNRNPC may be a promising biomarker for evaluating prognosis of HCC patients, as well as a potential therapeutic target.
Materials and Methods
Sample Collection
In order to detect differences in HNRNPC protein levels between tumor tissues and para-tumor tissues, we collected 12 pairs of fresh tissues (tumor and matched para-tumor tissue) from patients diagnosed with HCC by a pathologist after undergoing hepatectomy between Dec 2018 and Jan 2019 in the Department of Hepatobiliary Surgery of the Second Affiliated Hospital of Chongqing Medical University. For analysis of prognosis and clinical information, our study enrolled 147 available cases from Jul 2014 to Nov 2016 from the same department who had not received chemotherapy, radiofrequency ablation, or molecular targeted therapy before liver resection. These 147 cases included 128 male and 19 female patients, with an average age of 53.67 years. Specific patient information is given in Table 1. The selection criteria were as follows. Inclusion criteria: HCC confirmed by biopsy after surgery; exclusion criteria: patients with other cancers, distant metastasis, or grade C Child–Pugh liver function. A total of 147 tumor tissues and 82 available corresponding para-tumor tissues were used to construct tissue microarrays (TMAs). This research was approved by the Ethics Committee at the Second Affiliated Hospital of Chongqing Medical University ((2017)36), and informed consent was obtained from all patients.
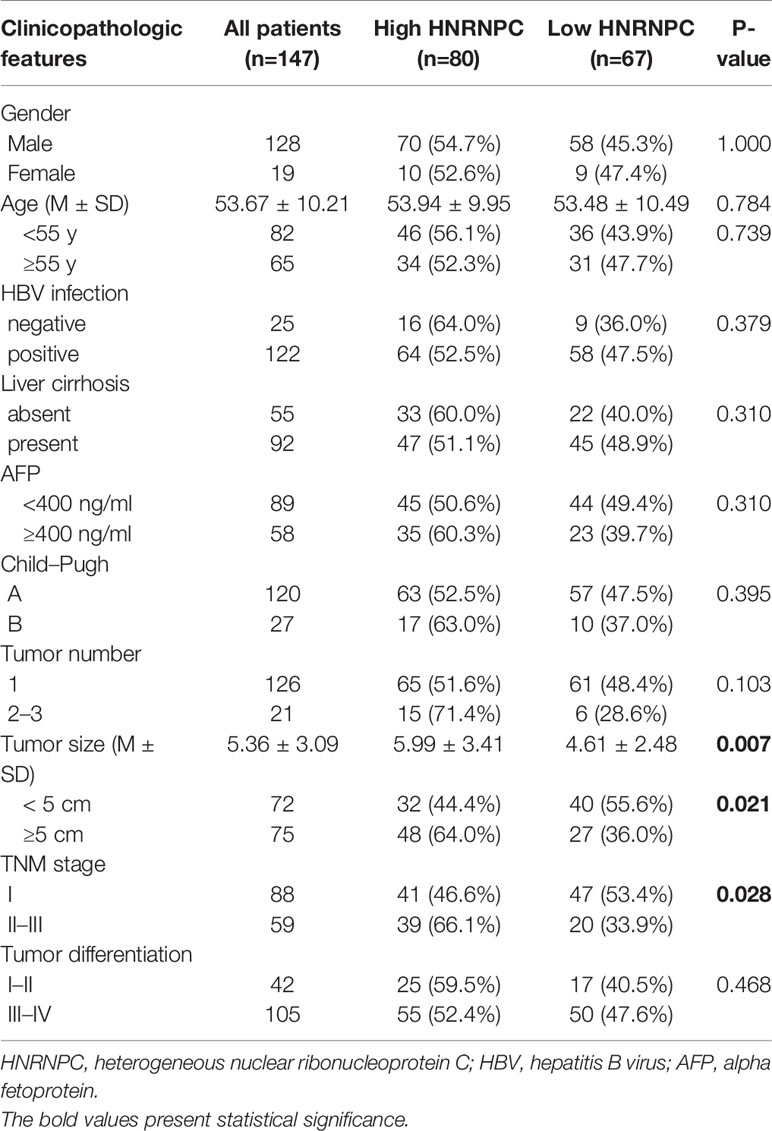
Table 1 Relationships of HNRNPC protein levels with clinicopathologic features in 147 HCC patients.
Follow-Up
Follow-up was carried out by phone, and alpha fetoprotein (AFP), liver function, and chest radiography were obtained at least every 2 months in the first 6 months after surgery, every 3 months for 6 months to 3 years following surgery, and every 6 months for 3–5 years after surgery. If necessary, magnetic resonance imaging or computed tomography was also used. Overall survival was defined as the time from liver resection to death or last follow-up; disease-free survival was defined as the time from liver resection to first recurrence or last follow-up.
IHC
Fresh tissues were immobilized with 4% formalin for 48 h at 37°C and subsequently washed with cool phosphate-buffered saline (PBS) three times for 5 min each time. Tissue samples were dehydrated using ethanol, embedded in paraffin, and then cut them into 4-μm-thick sections of 2 mm diameter to produce TMAs. The paraffin sections were dewaxed in an incubator at 65°C overnight, before being deparaffinized in xylene and rehydrated using a decreasing ethanol gradient. Sections were placed in ethylene diamine tetraacetic acid (cat.no. 0085; Beyotime) and heated in a microwave for 10 min for antigen repair. A 3% hydrogen peroxide was used to eliminate endogenous peroxidase at 37°C for 30 min. Sections were incubated with anti-HNRNPC (dilution 1:50, cat. no. ET1611-2; HUABIO), anti-ki67 (dilution 1:10000, cat. no. 27309; Proteintech), anti-CD34 (dilution 1:50, cat. no. ET1606-11; HUABIO), anti-Vimentin (dilution 1:100, cat. no. ET1610-39; HUABIO), and anti-E-Cadherin (dilution 1:50, cat. no. ET1607-75; HUABIO) antibodies at room temperature overnight, and then with secondary antibody (dilution: 1:1, cat.no. K5007; Dako) for 1 h at room temperature. Finally, sections were stained with an IHC kit (cat. no. K5007; Dako) according to the manufacturer’s protocol.
IHC Scoring
The proportion of positive cells and staining strength were used to evaluate the expression of HNRNPC. Color intensity was ranked using four grades: no staining (score = 0), weak staining (score = 1), moderate staining (score = 2), and intense staining (score = 3); and the proportions of positive cells were divided into five classifications: < 5% (score = 0), 5–25% (score =1), 26–50% (score = 2), 51–75% (score = 3), and 76–100% (score = 4). These two values were multiplied to acquire the final score. All patients were divided into high and low HNRNPC expression groups according to the median score of 7. The IHC results were analyzed by two pathologists; uncertain results were judged by a third pathologist.
Western Blot Analysis
We collected total protein from cells or tissues using RIPA buffer with protease and phosphatase inhibitors. After separation by sodium dodecyl polyacrylamide gel electrophoresis, proteins were transferred onto polyvinylidene fluoride membranes. Subsequently, the membranes were blocked with 5% skim milk at room temperature for 1 h, then incubated with primary antibody at 4°C overnight. On the second day, the membranes were incubated with secondary antibody for 1 h at 37°C. The antibodies used were as follows: HNRNPC (dilution 1: 2000, cat. no. ET1611-2; HUABIO), Ras (dilution 1:1000, cat. no. 3339T; CST), c-Raf (1:1000, cat. no. 9422T; CST), P-c-Raf (dilution 1:1000, cat. no. 9427T; CST), MEK1/2 (dilution 1:1000, cat. no. 8727T; CST), P-MEK1/2 (Ser217/221) (dilution 1:1000, cat. no. 9154T; CST), Erk1/2 (dilution 1:1000, cat. no. 4695T; CST), P-Erk1/2 (Thr202/Tyr204) (dilution 1:1000, cat. no. 4377T; CST), c-Myc (dilution 1:1000, cat. no. 5605T; CST), CDK4 (dilution 1:2000, cat. no. ET1612-1; HUABIO), cyclin E1 (dilution 1:1000, cat. no. ET1612-16; HUABIO), Vimentin (dilution 1:2000, cat. no. ET1610-39; HUABIO), E-Cadherin (dilution 1:1000, cat. no. ET1607-75; HUABIO), β-actin, (dilution 1:2000, cat. no. BM0627; Boster), goat anti-mouse (dilution 1:5000, cat. no. ZB-2305; ZSGB-BIO), and goat-anti-rabbit (dilution 1:5000, cat. no. ZB-2301; ZSGB-BIO). Finally, the membranes were imaged using a chemiluminescent horseradish peroxidase substrate (cat. no. WBKL S0100; Millipore). β-actin was used as the internal reference, and the western blot analysis was performed using Image Lab 6.0. The tests mentioned above were operated three times respectively.
Quantitative Real-Time PCR (q-RT-PCR)
Total RNA was extracted from cells using an Animal Total RNA Isolation Kit (cat. no. RE-03011; Foregene) according to the manufacturer’s instructions. Total RNA was reverse transcribed to cDNA with an iScript cDNA synthesis kit (cat. no. 1708891; Bio-Rad), following the manufacturer’s reaction protocol: 5 min at 25°C 20 min at 46°C, 1 min at 95°C, and holding at 4°C. Quantitation of cDNA was performed using a SYBR Green PCR kit (cat. no. 208054; QIAGEN). Relative HNRNPC expression was calculated by the 2-△△Cq method, with GAPDH as an internal reference. The primers were designed as follows: GAPDH forward, 5′-GGAGTCCACTGGCGTCTTCA-3′, reverse, 5′-GTCATGAGTCCTTC CACGATACC-3′; HNRNPC forward, 5′-GCAGAGCCAAAAGTGAACCG-3′, reverse, 5′-ACGTTTCGAGGGCACTACAG-3′. The reaction protocol was given by the manufacturer as follows: 2 min at 95°C, 5 sec at 95°C, and 10 sec at 60°C, repeated for 35 cycles. Each test was repeated dividedly in triplicate.
Cell Lines and Culture
Huh-7 and Hep 3B HCC cell lines were purchased from the Chinese Academy of Sciences Stem Cell Bank (Shanghai, China) and identified with STR Profile. Both cell lines were cultured in DMEM (cat.no. SH30022.01; HyClone) or MEM (cat.no. SH30024.01; Hyclone) supplied with 10% fetal bovine serum (cat.no. 04-001-1ACS; BI) in an incubator containing 5% CO2 at 37°C.
Cell Infection
Huh-7 and Hep 3B cells were cultured in six-well plates. When they reached 20–30% density, they were stably infected with HNRNPC lentivirus (Lv-HNRNPC) or a negative control lentivirus (Lv-NC) for 14 h (viral volume=MOI × cell number/viral titer; GeneChem, Shanghai, China). The lentiviral interference system was produced using a GV248 carrier loaded consecutively with the hU6 promoter, a multiple cloning site, the ubiquitin promoter, the EGFP gene, an IRES site, and the puromycin gene. A short hairpin RNA was designed to target the HNRNPC sequence (sh-HNRNPC): 5′-CTTCGTTCAGTATGTTAAT-3′; the negative sequence (sh-NC) was 5′-TTCTCCGAACGTGTCACGT-3′. After culture for 48 h, cells were filtrated with 2 µg/ml puromycin (cat. no. REVG1001; GeneChem, Shanghai, China) for 48 h; this was repeated twice. Infection and knockdown efficiency were detected by inverted fluorescence microscopy, and by western blotting and q-RT-PCR, respectively.
Colony Formation Assay
Approximately 500 cells were seeded in each well of the six-well plates and cultured in an incubator with 5% CO2 at 37°C for 2 weeks. After washing with PBS, the cells were immobilized with 4% paraformaldehyde for 15 min at room temperature and then stained with 0.1% crystal violet solution (cat. no. G1062; Solarbio) for 30 min. Finally, the colony numbers were counted under a microscope. All experiments were operated independently three times.
Cell Viability Analysis
Approximately 3000 (Huh-7) or 8000 (Hep 3B) cells were seeded in each well of a 96-well plate and cultured for 72 h. After being seeded in a 96-well plate, cell viability was measured using a CCK-8 kit (cat. no. ZP328-3; ZOMANBIO) according to the manufacturer’s instructions at 6, 24, 48 and 72h. Briefly, CCK-8 (10 µl) was added to each well, and the cells were incubated at 5% CO2 and 37°C. The absorbance (optical density) was detected by enzyme-linked immunosorbent array at a wavelength of 450 nm. Each test was performed dividedly in triplicate.
Scratch Wound for Cell Migration Assay
When the cells had grown to a density of 90% in the six-well plates, we used a sterile 200-µl pipette tip to scratch a wound at the middle line of each well bottom. Subsequently, the suspended cells were removed by PBS, and the culture medium was replaced with serum-free medium. Cells were sequentially cultured for 48 h in an incubator containing 5% CO2 at 37°C. The wound width was recorded under a light microscope at 0, 24 and 48h. The experiments mentioned above were operated three times respectively.
Cell Invasion Assay
To analyze the invasion ability of cells, a Corning BioCoat Matrigel Invasion Chamber (cat. no. 354480; Corning) was used according to the manufacturer’s instructions. Specifically, cells (5×104/well) were cultured with serum-free medium in the upper chambers, while the lower chambers were loaded with DMEM or MEM supplied with 10% fetal bovine serum. After culture for 48 h, the cells were washed with PBS and fixed with 4% paraformaldehyde for 15 min at room temperature. Invading cells were captured with 0.1% crystal violet solution under a light microscope, and non-invading cells were eliminated with cotton swabs. Specifically, five fields in each chamber were randomly recorded, and average values were calculated. Each test was repeated independently in triplicate.
Cell Cycle Analysis
Approximately 25×104/well cells were seeded in six-well plates. Subsequently, the cells were collected in 1.5-ml EP tubes and then fixed with 70% ethanol at 4°C overnight. The next day, the cells were stained with propidium iodide with RNase A at 37°C for 30 min in the dark. Finally, the results were analyzed by flow cytometry. A total of 2×104 cells were recorded for each sample. All experiments were performed three times respectively.
Groups and Treatments
To verify the upstream and downstream relationships between HNRNPC and the MAPK signaling pathway, we used an agonist for Ras (ML-098) (cat. no. HY-19800; MCE). The specific treatments were as follows. To measure the efficiency of the activation of the MAPK signaling pathway by ML-098, the Lv-NC group was treated with dimethyl sulfoxide (DMSO) for 48 h, and the Lv-HNRNPC group was treated with ML-098 (20 nmol/l) and DMSO, for 48 h. CCK-8 tests, cell cycle analysis, scratch wound assay, and cell invasion tests were performed using the method described above. To detect the rescue efficiency of ML-098 for colony formation, we used ML-098 (20 nmol/l) to treat the Lv-HNRNPC group twice sequentially for 5 days each time. The Lv-NC group was treated with the same amount of DMSO as a control.
In Vivo Assay for Tumor Growth
A total of 12 5-week-old male BALB/c nude mice were purchased from Beijing Huafukang Biotechnology and randomly divided into two groups (six per group). Lv-HNRNPC- and Lv-NC-group Huh-7 cells were suspended in cool PBS and subsequently injected into the backs of mice subcutaneously (2.5×106/mice). The mice were fed at 23–25°C and 60% humidity with a 12-h light/dark cycle. Tumor size was measured with a caliper every 3 days; the formula for the size calculation was as follows: volume = (length × width2)/2 cm3. Twenty-seven days after implantation, the mice were killed by cervical dislocation and the tumors were removed. Finally, the tumor samples were fixed with 4% formalin for IHC analysis. All animal experiments were approved by the Ethics Committee of Chongqing Medical University.
Bioinformatic Analysis
The expression of HNRNPC in HCC tissues was analyzed by UALCAN (ualcan.path.uab.edu), using RNA sequencing data downloaded from The Cancer Genome Atlas for 50 normal liver tissues and 371 primary HCC tissues. HNRNPC-correlated genes were also acquired from UALCAN; we selected 3517 significantly correlated genes (Pearson correlation coefficient ≥ 0.5) for Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis by WebGestalt 2013 (www.webgestalt.org/2013).
Statistical Analysis
IBM SPSS Statistics 23 was used for statistical analysis. Categorical data were analyzed by chi-square test, while continuous data between two groups were analyzed by t-test. Comparisons between multiple groups were performed by one-way analysis of variance (ANOVA), and the results were analyzed by LSD test and presented as mean ± standard deviation (SD). Univariate and multivariate Cox proportional hazards regression models were used to assess the risk factors associated with HCC. In addition, overall and disease-free survivals were computed by the Kaplan–Meier method and examined by log-rank test. Finally, a P-value less than 0.05 was considered to indicate a statistically significant difference.
Results
HNRNPC Is Upregulated in Tumor Tissues and Predicts Large Tumor Size, Advanced TNM Stage, and Poor Prognosis in HCC
Using the UALCAN database, we found that HNRNPC mRNA levels were higher in 371 primary HCC tissues than in 50 normal liver tissues (P < 0.001, Figure 1A); in addition, the expression of HNRNPC mRNA was elevated in stage 2 (P = 0.033, Figure 1B) and stage 3 (P < 0.001, Figure 1B) HCC patients compared with stage 1 HCC patients. To compare HNRNPC protein expression in tumor and para-tumor tissues, western blotting and IHC were performed. HNRNPC protein levels were obviously higher in tumor tissues than in para-tumor tissues, based on western blot analysis of 12 HCC tumor tissues and their matched para-tumor tissues (P< 0.001, Figures 1C, D). The IHC results showed high expression of HNRNPC protein, mainly located in nuclei, in 80 of 147 tumor tissues (54.40%) and in 13 (15.90%) of 82 paired para-tumor tissues (P<0.001, Figures 1E, F). Correlations of HNRNPC protein levels with clinicopathological features in HCC were analyzed by chi-square test; this demonstrated that HNRNPC protein levels were significantly associated with tumor size (P = 0.007) and tumor TNM stage (P=0.028) (Table 1). To explore the effects of HNRNPC protein levels on survival of HCC patients, Kaplan–Meier survival curves were constructed, showing that patients with high HNRNPC protein expression had poorer overall survival (P = 0.001, Figure 1G) and disease-free survival (P < 0.001, Figure 1H) than those with low HNRNPC protein expression. Univariate and multivariate analysis were used to evaluate the risk factors associated with HCC patients’ survival. According to our results, tumor size (P = 0.005, HR = 1.957), tumor TNM stage (P = 0.014, HR = 1.786), and high HNRNPC expression (P = 0.039, HR = 1.637) could be regarded as independent risk factors for overall survival of HCC patients (Table 2). Moreover, tumor TNM stage (P < 0.001, HR = 2.188) and high HNRNPC level (P = 0.002, HR = 1.883) could independently predict disease-free survival in HCC patients (Table 2).
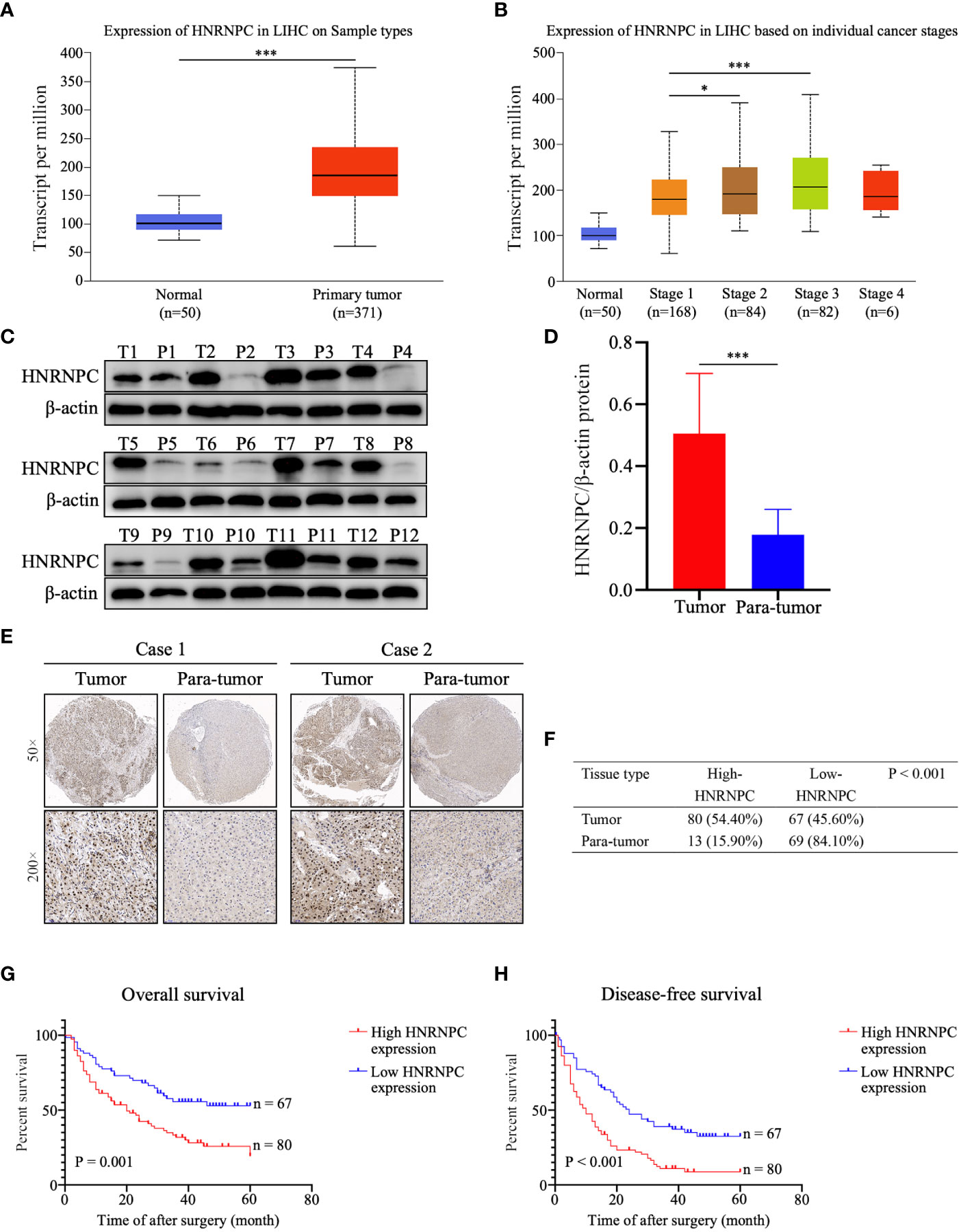
Figure 1 High HNRNPC expression was detected in HCC tumor tissues, and predicts a poor survival in HCC patients. (A) HNRNPC mRNA expression in HCC tissues and normal liver tissues acquired from UALCAN analysis. (B) UALCAN analysis showing HNRNPC mRNA expression at different cancer stages. (C, D) Western blot showing protein expression of HNRNPC in HCC tumor tissues and matched para-tumor tissues. (E) Classical cases showing HNRNPC protein expression in HCC tumor tissues and paired para-tumor tissues. (F) The χ2 test was used to assess HNRNPC protein expression in HCC tissues and adjacent tissues. (G) Kaplan–Meier analysis showing the correlation of HNRNPC level with overall survival of HCC patients. (H) Kaplan–Meier analysis showing the association between HNRNPC level and disease-free survival. The final results were presented as mean ± standard deviation (SD). *P < 0.05, ***P < 0.001.
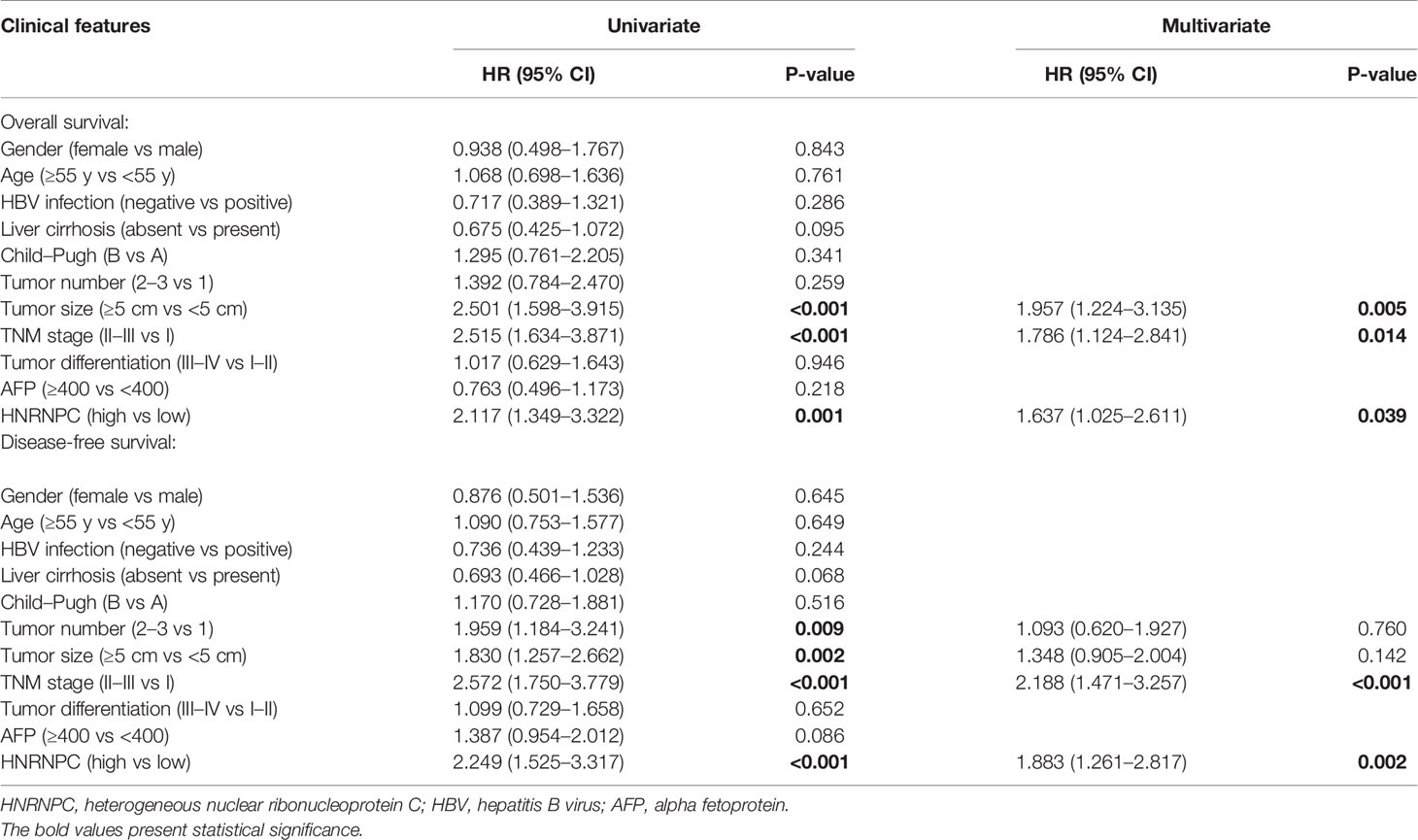
Table 2 Univariate and multivariate analysis for survival of 147 HCC patients.
Knockdown of HNRNPC Suppresses HCC Proliferation, Invasion, and Migration In Vitro
To observe the impact of HNRNPC on HCC proliferation, invasion, and migration, Lv-NC and Lv-HNRNPC lentiviruses were used to infect the Huh-7 and Hep 3B cell lines. After selection by puromycin, the infection efficiency of Huh-7 and Hep 3B cells was >90% (Supplementary Figure 1A), and lower HNRNPC expression was detected in Lv-HNRNPC Huh-7 and Hep 3B cells via western blotting and q-RT-PCR (Supplementary Figures 1B, C). CCK-8 and colony formation assays were used to analyze cell proliferation in vitro. The colony formation assay revealed that HNRNPC knockdown significantly inhibited proliferation of Huh-7 and Hep 3B cells (Figure 2A). In the CCK-8 test, the viability of Huh-7 and Hep 3B cells was also significantly suppressed by silencing of HNRNPC (Figure 2B). A transwell assay was performed to observe invasion; the results showed that inhibition of HNRNPC impaired the invasion activity of Huh-7 and Hep 3B cells (Figure 2C). The scratch wound assay showed decreased migration ability of Huh-7 and Hep 3B cells in the Lv-HNRNPC-group (Figure 2D).
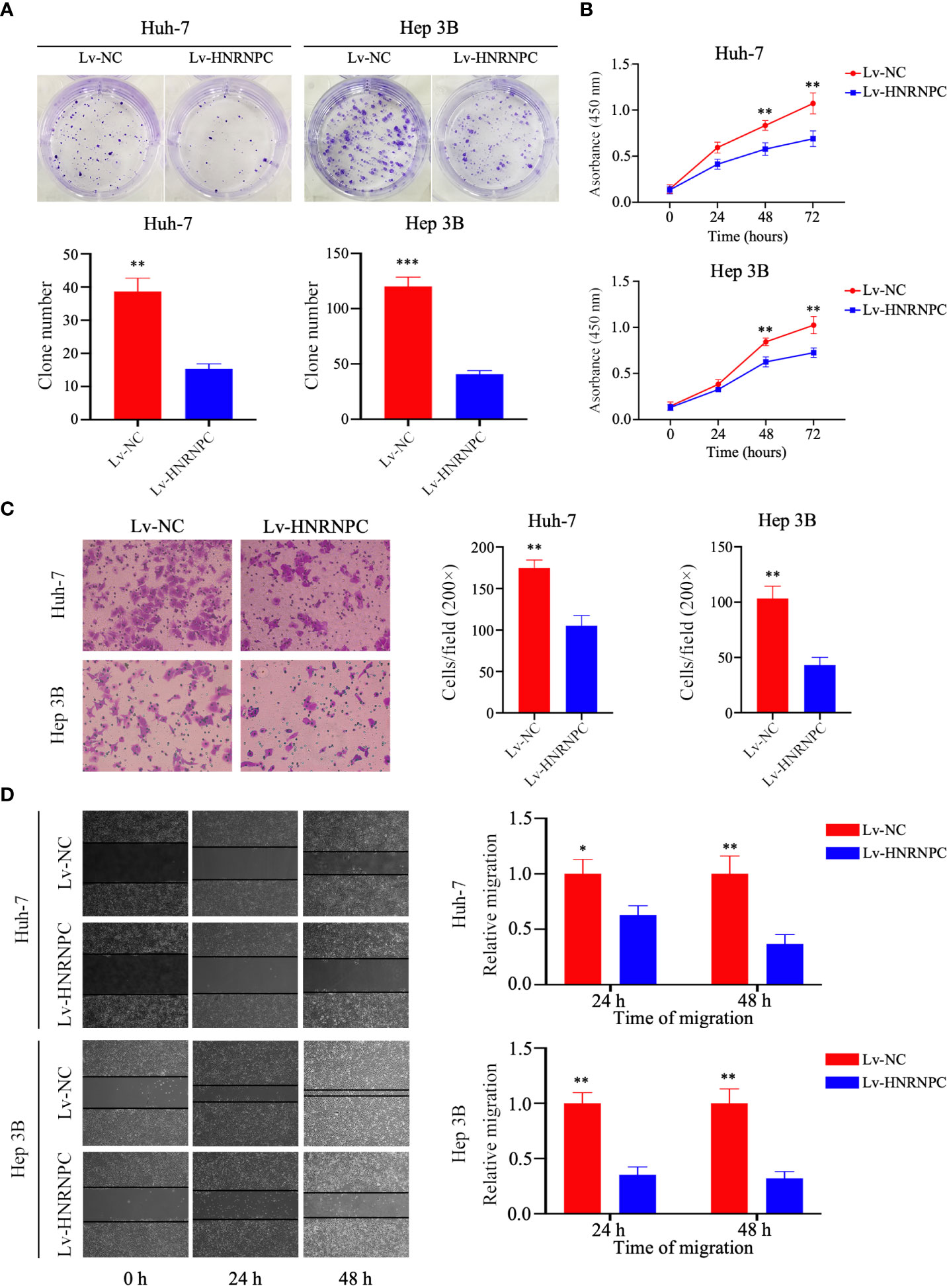
Figure 2 Knockdown of HNRNPC suppressed proliferation, invasion, and migration of HCC cells in vitro. (A) Colony formation assay showing the impact of HNRNPC knockdown on HCC cell proliferation. (B) CCK-8 test showing the effects of HNRNPC downregulation on HCC cell viability. (C) Transwell assay showing the influence of HNRNPC inhibition on HCC cell invasion. (D) Scratch wound test showing the influence of HNRNPC suppression on HCC cell migration. All experiments were independently repeated three times, and the results were showed as mean ± standard deviation (SD). *P < 0.05, **P < 0.01, ***P < 0.001.
Inhibition of HNRNPC Downregulates the Activity of the MAPK Signaling Pathway, Blocks Tumors at G0/G1 Phase, and Suppresses EMT Process In Vitro
To study the potential molecular mechanisms by which HNRNPC suppressed tumor growth, migration and invasion, we selected 3517 genes that were significantly correlated with HNRNPC (Pearson correlation coefficient ≥ 0.5) from the UALCAN database; these genes were subjected to KEGG analysis using WebGestalt (2013). Among the enriched KEGG pathways, “Cell cycle” and “Pathways in cancer” were highly related to tumor growth (Supplementary Figure 1D), and “Pathways in cancer” mainly involved the MAPK and AKT signaling pathways (Supplementary Figure 1E). Subsequently, the activity of the MAPK signaling pathway was detected by western blotting. The results showed that HNRNPC knockdown statistically decreased the expression of Ras and the relative levels of p-Raf, p-MEK1/2, and p-Erk1/2 in Huh-7 and Hep 3B cells (Figures 3A, B). As the MAPK pathway activates a series of downstream genes to regulate the cell cycle, thereby regulating cell proliferation, we used flow cytometry for cell cycle analysis. The results showed that HNRNPC downregulation blocked more cells at G0/G1 phase and reduced the proportion of S-phase cells among both Huh-7 and Hep 3B cells (Figures 3C, D). CDK4, cyclin E1, and c-myc are well known as important molecules that positively regulate G1-S transition. In the present study, silencing of HNRNPC markedly downregulated CDK4, cyclin E1, and c-myc protein levels, as confirmed by western blotting (Figures 3E, F). Ras/MAPK signaling pathway not only positively regulate G1-S transition to promote tumor proliferation, but also accelerate EMT process to facilitate tumor migration and invasion. We also detected the marker of EMT by western blotting, our results indicated that HNRNPC silencing decreased Vimentin level, and upregulated E-cadherin expression statistically (Figures 3G, H).
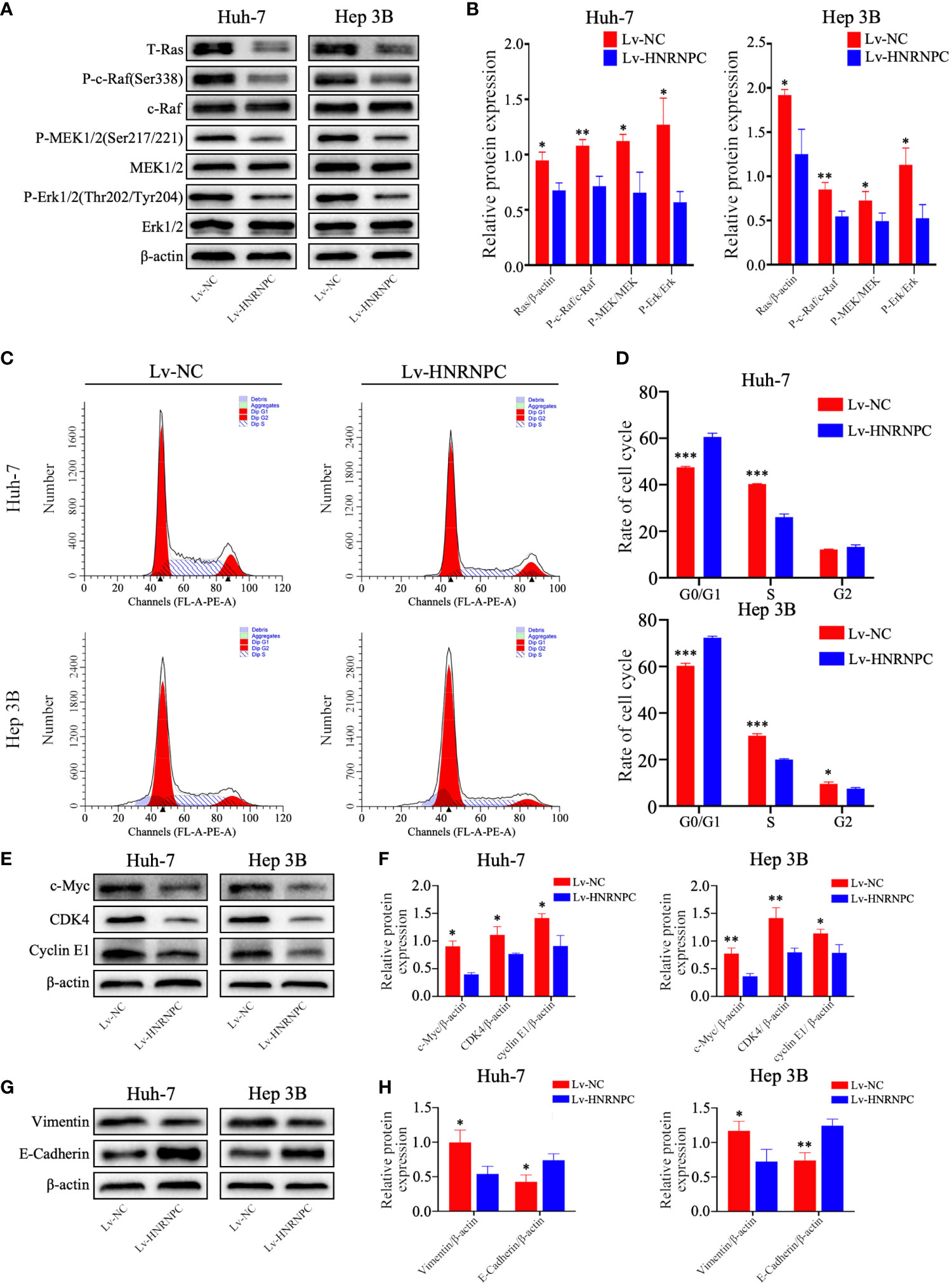
Figure 3 HNRNPC suppression inhibited the Ras/MAPK signaling pathway, leading to arrest of more cells at S phase and blocking of EMT in vitro. (A, B) Western blot to detect activation of the Ras/MAPK signaling pathway in Lv-NC Huh-7, Lv-HNRNPC Huh-7, Lv-NC Hep 3B, and Lv-HNRNPC Hep 3B cells. (C, D) Cell cycle analysis for the various cell groups mentioned above. (E, F) Western blot to detect c-Myc, CDK4, and cyclin E1 levels in the various cell groups. (G, H) Western blot to analyze Vimentin and E-Cadherin expression in various cell groups. Each test was performed independently in triplicate, and the results were presented as mean ± standard deviation (SD). *P < 0.05, **P < 0.01, ***P < 0.001.
HNRNPC Silencing Inhibits Tumor Growth In Vivo
To explore the impact of HNRNPC on tumor growth in vivo, Lv-NC and Lv-HNRNPC Huh-7 cells were implanted in nude mice subcutaneously. The tumors of the Lv-HNRNPC group showed obvious reductions in both size and weight compared with those of the Lv-NC group (Figures 4A–C). Subsequently, hematoxylin eosin (H&E) and IHC staining were performed to confirm the tumor tissues and HNRNPC knockdown efficiency, respectively (Figure 4D). In addition, Ki-67 was used to observe the proliferation of tumor cells via IHC; the Ki-67 staining intensity was stronger in the Lv-NC group than in the Lv-HNRNPC group (Figure 4D). Tumor migration and invasion are highly correlated with angiogenesis and EMT process, so Vimentin, E-cadherin, and CD34 were used to evaluate the ability of tumor invasion and migration by IHC. The results showed that Vimentin and CD34 staining intensity were weaker, and E-cadherin staining intensity was stronger in the Lv-HNRNPC group than in Lv-NC group (Figure 4E).
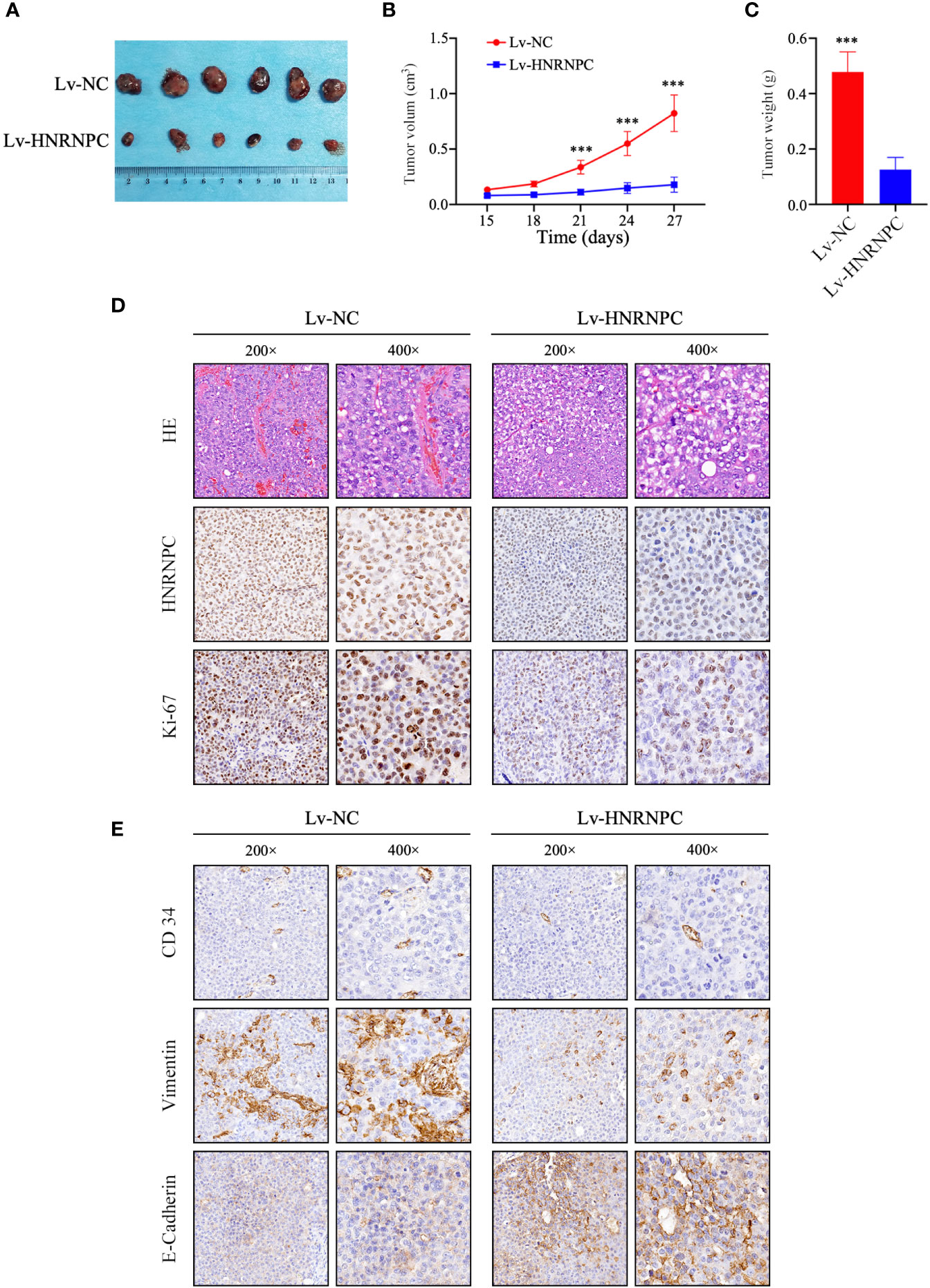
Figure 4 HNRNPC knockdown blocked HCC cell proliferation in vivo. (A) Huh-7-cell xenograft tumors in Lv-HNRNPC and Lv-NC groups. (B, C) t-test analysis showing tumor weights and sizes in Lv-HNRNPC and Lv-NC Huh-7 cell groups. (D) H&E staining to confirm xenograft tumors (upper panel); IHC to detect HNRNPC levels in xenograft tumors (middle panel); proliferation of xenograft tumors as determined by Ki-67 staining (nether panel). (E) IHC staining to analyze CD34 (upper panel), Vimentin (middle panel), and E-Cadherin (nether panel) expression in xenograft tumors. The tumor size and weight were demonstrated as mean ± standard deviation (SD). ***P < 0.001.
Ras Agonist Partly Recovers Growth, Migration, and Invasion Ability in Lv-HNRNPC Group
CDK4, cyclin E1, and c-myc are classical downstream genes of the MAPK signaling pathway. To confirm the upstream and downstream relationships between the MAPK signaling pathway and these cell cycle-related molecules, a Ras agonist (ML-098) was used to treat cells in the Lv-HNRNPC group. Details of this process are given in the Materials and Methods section. First, we performed western blotting to detect the recovery of MAPK pathway activity. After treatment with ML-098 for 48 h, the activity of the MAPK pathway was reversed completely in the Lv-HNRNPC group (Figures 5A, B). Moreover, colony formation and CCK-8 assays revealed that the proliferation ability of cells in the Lv-HNRNPC group was partly rescued by ML-098 (Figures 5C–E). Furthermore, the cell cycle assay showed that ML-098 treatment could partially reverse the G0/G1 arrest caused by HNRNPC knockdown in both Huh-7 and Hep 3B cells (Figures 6A, B). Finally, using western blotting, we showed that the effects of HNRNPC inhibition on CDK4, cyclin E1, and c-myc levels in HCC cells were reversed by ML-098 to some extent (Figures 6C, D). On the other hand, Ras/MAPK signaling pathway take part in EMT process. In present study, cell invasion and scratch wound assays showed that the invasion (Figures 7A, B) and migration (Figures 7C, D) abilities of the Lv-HNRNPC group cells were recovered by ML-098 partly. Furthermore, the western blotting results demonstrated that after treatment with ML-098, the EMT process inhibition of cells in the Lv-HNRNPC group was reversed in part too (Figures 7E, F). Taken together, these results indicate that HNRNPC inhibition not only arrests HCC cells in G0/G1 phase to inhibit tumor proliferation, but also suppresses EMT process to block invasion and migration of HCC cells in part via the Ras/Raf/MEk/Erk signaling pathway.
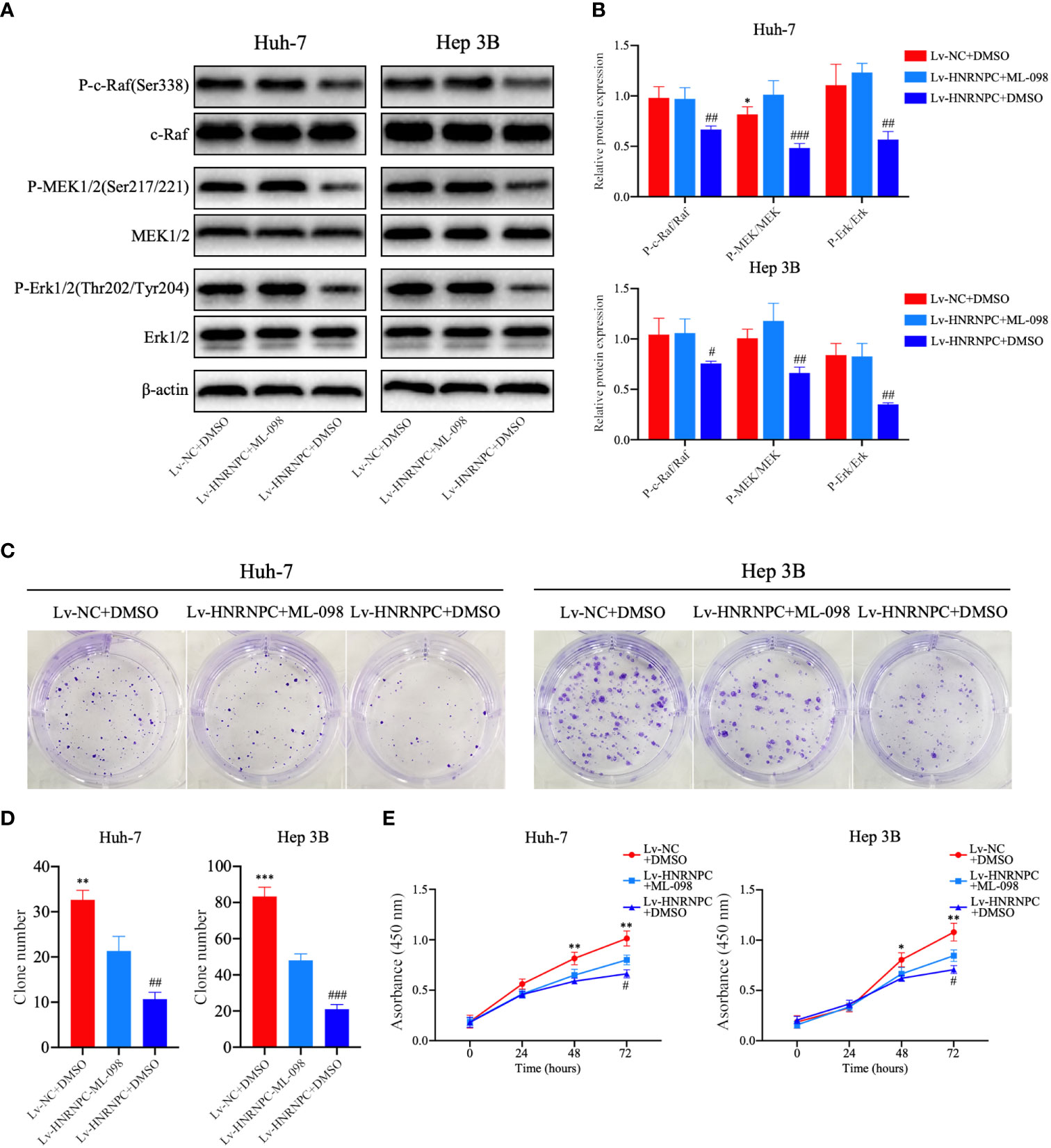
Figure 5 After treatment with ML-098 (20 nmol/l), MAPK signaling pathway activation was recovered completely in Lv-HNRNPC-group cells, and proliferation ability was partially reversed. (A, B) Western blot to detect recovery of activation of the MAPK signaling pathway in Lv-HNRNPC groups after treatment with ML-098. (C, D) Colony formation assay showing the reversed efficiency of ML-098 in Lv-HNRNPC group cell proliferation. (E) CCK-8 test showing the rescue by ML-098 of cell viability in the Lv-HNRNPC group. The tests mentioned above were operated three times respectively, and the results were reported as mean ± standard deviation (SD). *P < 0.05, **P < 0.01, ***P < 0.001, for Lv-NC+DMSO group compared with Lv-HNRNPC+ML-098 group; #P < 0.05, ##P < 0.01, ###P < 0.001, for Lv-HNRNPC+DMSO group compared with Lv-HNRNPC+ML-098 group.
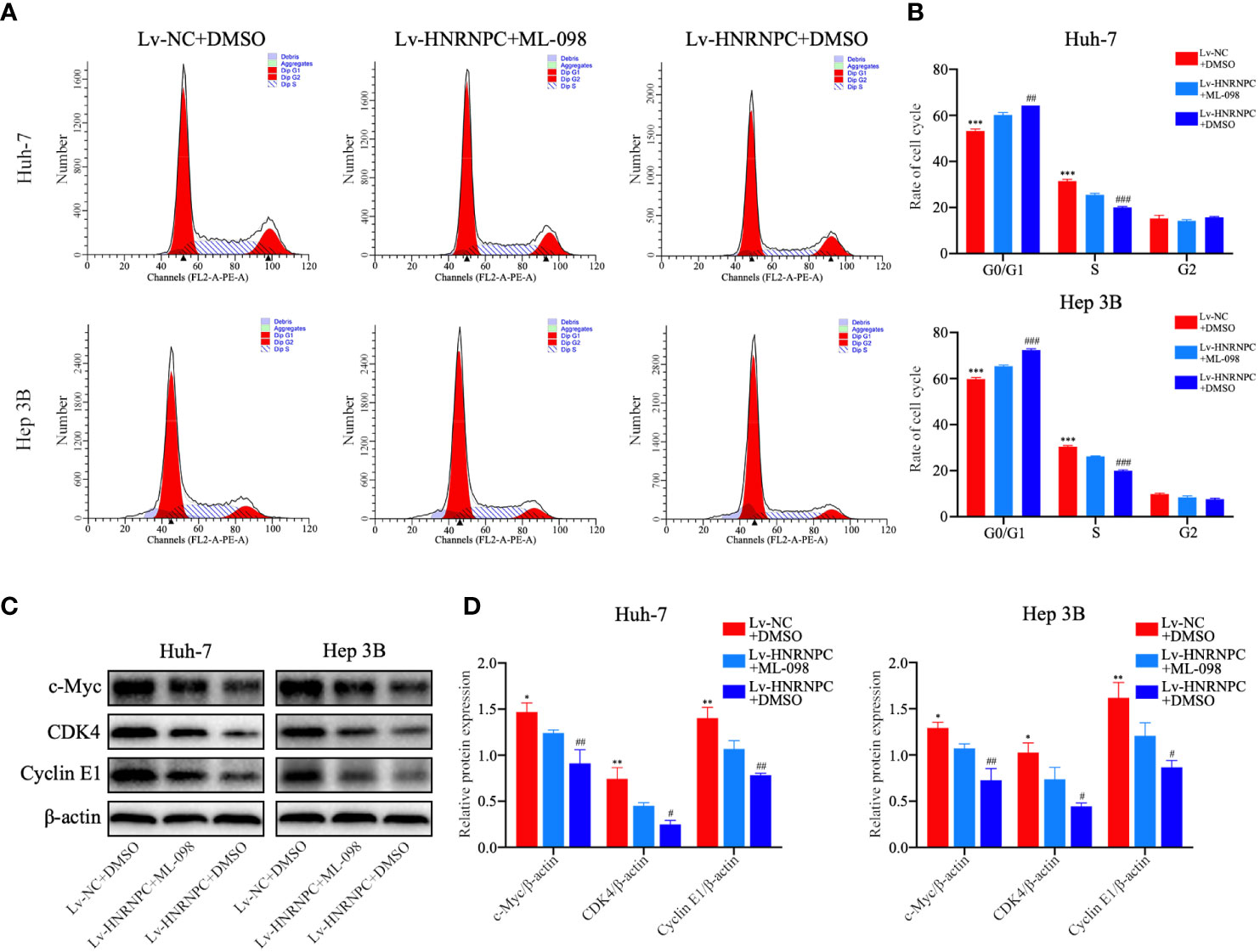
Figure 6 ML-098 partly reversed the reduction in numbers of S phase cells in the Lv-HNRNPC group. (A, B) Cell cycle analysis to detect the recovery of numbers of S phase cells in the Lv-HNRNPC groups after treatment with ML-098. (C, D) Western blot to detect the recovery of c-Myc, CDK4, and cyclin E1 in Lv-HNRNPC cell groups after treatment with ML-098. All tests were repeated independently in triplicate, and the results were showed as mean ± standard deviation (SD). *P < 0.05, **P < 0.01, ***P < 0.001, for Lv-NC+DMSO group compared with Lv-HNRNPC+ML-098 group; #P < 0.05, ##P < 0.01, ###P < 0.001, for Lv-HNRNPC+DMSO group compared with Lv-HNRNPC+ML-098 group.

Figure 7 ML-098 recovered the invasion and migration abilities of Lv-HNRNPC group cells in part. (A, B) Transwell assay demonstrating the recovered efficiency of ML-098 in Lv-HNRNPC group cell invasion. (C, D) Scratch wound test showing the rescue by ML-098 of cell migration in the Lv-HNRNPC group. (E, F) Western blot to detect the recovery of EMT process in Lv-HNRNPC cell groups after treatment with ML-098. Each test was repeated independently in triplicate, and the results were demonstrated as mean ± standard deviation (SD). *P < 0.05, **P < 0.01, ***P < 0.001, for Lv-NC+DMSO group compared with Lv-HNRNPC+ML-098 group; #P < 0.05, ##P < 0.01, for Lv-HNRNPC+DMSO group compared with Lv-HNRNPC+ML-098 group.
Discussion
With progress in medical treatment, molecular therapies are increasingly available for advanced HCC patients (51–53); however, the prognosis of these patients remains poor (1). This prompted us to explore novel pathogenetic mechanisms and therapeutic targets for HCC. According to present evidence, as a process fundamental to cancer (54), aberrant alternative splicing occurs more frequently in various malignant tumors, including renal cancer (55), lung cancer, and HCC (56); this makes it possible for cells with phenotypic variability to adapt to unfamiliar microenvironments, treatments, and immune responses (57). HNRNPs, which include HNRNP A2/B1, HNRNP L, HNRNP H, and HNRNPC, are primary factors for alternative splicing. HNRNPC is believed to be the founder of the HNRNP family (58), that is, it seems to be at the core of regulating alternative splicing. The relationships of HNRNP L (59), HNRNP A2/B1 (60), and HNRNP H (61) with HCC have been reported; however, any association of HNRNPC with HCC remained unknown.
Using two-dimensional difference electrophoresis, Sun et al. found that HNRNPC protein expression was higher in HCC tumor tissues than normal liver tissues (62). However, they did not further investigate any correlation of HNRNPC protein expression with prognosis or clinicopathologic characteristics in HCC. In the present study, we showed that HNRNPC protein levels were statistically higher in HCC tumor tissues than in para-tumor tissues by western blotting and IHC. Moreover, Kaplan–Meier analysis and Cox proportional hazards regression models were used to perform survival analysis; this demonstrated that a high HNRNPC protein level was an independent risk factor that could serve as a biomarker predicting poor overall survival and disease-free survival in HCC patients. By functional experiments, we showed that HNRNPC knockdown significantly inhibited proliferation, migration, and invasion of HCC cells in vitro. Finally, the xenograft assay showed that suppressing HNRNPC significantly restrained tumor growth in vivo.
HNRNPC has an important role in RNA combination and alternative splicing. Fischl et al. reported that HNRNPC regulates alternative cleavage and polyadenylation (APA) profiles in colon cancer, and by coding region APA, HNRNPC mainly affected MTHFD1L protein levels, which are strongly linked to tumor progression (24). In addition, Wu et al. extended the function of HNRNPC to alternative splicing in breast cancer: by controlling endogenous double-stranded RNA, HNRNPC regulated the activation of the IFNβ signaling pathway to affect the progression of breast cancer (27). In the present study, we performed bioinformatic analysis of HNRNPC-correlated genes, which brought the Ras/Raf/MEK/Erk and AKT signaling pathways to our attention. HNRNPC has previously been reported to cause AKT phosphorylation in ovarian cancer (32). We used western blotting to detect the activation of the Ras/Raf/MEK/Erk signaling pathway; the results indicated that HNRNPC knockdown significantly inhibited the activation of the MAPK signaling pathway. Ras mutation has a very important role in many cancers (63–65), and HCC is no exception (66, 67). Both H-ras and K-ras have respective splice variants (68, 69), which could be regarded as Ras mutations at the mRNA level. Barbier et al. reported that H-ras splice variants including premature translation termination codons were selectively degraded by the nonsense-mediated mRNA decay pathway to regulate Ras expression (69). However, the mechanism by which Ras downregulation was caused by HNRNPC inhibition was not investigated in our study; this will be explored in the future. This novel means of Ras mutation may provide a new approach to HCC therapy.
The MAPK signaling pathway regulates the majority of cell functions, in particular, cell proliferation, apoptosis, and metabolism (70–72). After being activated, the Ras cascade phosphorylates Raf, Mek, and Erk to regulate cell proliferation and apoptosis by controlling G1-S transition (73, 74). In the present study, we prove that inhibition of the MAPK signaling pathway by HNRNPC suppression causes the arrest of more HCC cells at G0/G1 phase. CDK4 (75), cyclin E1 (76), and c-myc (77) play important parts in regulating G1-S transition. As G1-S transition overactivation occurs in the majority of malignant tumors, inhibitors of CDK4 and cyclin E1 have emerged as candidate drugs for tumor treatment (78, 79). In our study, we showed that HNRNPC knockdown statistically inhibited CDK4, cyclin E1, and c-myc expression. Subsequently, rescue experiments revealed that activation of the MAPK signaling pathway could partly recover CDK4, cyclin E1, and c-myc expression, demonstrating that knockdown of HNRNPC induced G0/G1 arrest in part via the MAPK signaling pathway. Epithelial-mesenchymal transition is an essential factor to initiate tumor migration and invasion (80). In recent years, more and more research indicated that Ras/MAPK signaling pathway play an important role in EMT process (43, 81, 82). In the present study, we proved that HNRNPC inhibition suppressed the EMT process of HCC cells. Additionally, after treatment with ML-098, the EMT process of HCC cells in Lv-HNRNPC group was recovered in part, which support the above point again. Ras is the second mutated gene driver in many malignant tumors (83), and Ras/MAPK have been shown to be activated in 50–100% of HCC patients (84). MAPK-associated inhibitors have been used to treat HCC (85), but very few HCC patients benefited from this treatment (86), largely owing to the compensatory activation of other Ras-related pathways such as the AKT (87) and IGF/FGF (88) signaling pathways. Thus, exploration of the mechanisms of Ras mutation may provide a novel approach for HCC therapy. In our study, we reported that HNRNPC knockdown inhibited Ras/MAPK activation to block proliferation of HCC cells, which may represent a new mutation mechanism for Ras. However, the present study did not consider how HNRNPC regulates Ras; this will be explored in our future studies to determine the mechanisms of novel Ras mutations and offer a new approach for HCC therapy.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee at the Second Affiliated Hospital of Chongqing Medical University. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Chongqing Municipal Committee of Science Technology.
Author Contributions
The study was conceived and designed by JG. G-CZ, JH, and DC wrote the manuscript and performed the experiments. The statistical analysis was carried out by ZZ. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Laboratory of Pathology, West China Medical College, and Sichuan University for supplying the experimental platform, and Charlesworth Author Services (https://www.cwauthors.com/) for editing a draft of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.659676/full#supplementary-material
Supplementary Figure 1 | Knockdown efficiency of HNRNPC in Huh-7 and Hep 3B cells, and KEGG analysis. (A) EGFP expression showing the infective efficiency of the lentivirus. (B) Western blot to detect knockdown efficiency of HNRNPC in Huh-7 and Hep 3B cells. (C) q-RT-PCR to detect knockdown efficiency of HNRNPC in Huh-7 and Hep 3B cells. (D, E) KEGG analysis of 3517 HNRNPC-correlated genes by WebGestalt 2013. ***P<0.001.
Abbreviations
HCC, hepatocellular carcinoma; HNRNPC, heterogeneous nuclear ribonucleoprotein C; IHC, immunohistochemical; MAPK, mitogen-activated protein kinase; AFP, alpha fetoprotein; EMT, epithelial-mesenchymal transition; HBV, hepatitis B virus; Lv-NC, hepatocellular carcinoma cell lines infected with negative control lentiviruses; Lv-HNRNPC, hepatocellular carcinoma cell lines infected with targeted HNRNPC lentiviruses; EMT, epithelial-mesenchymal transition.
References
1. Akinyemiju T, Abera S, Ahmed M, Alam N, Alemayohu MA, Allen C, et al. The Burden of Primary Liver Cancer and Underlying Etiologies From 1990 to 2015 at the Global, Regional, and National Level: Results From the Global Burden of Disease Study 2015. JAMA Oncol (2017) 3(12):1683–91. doi: 10.1001/jamaoncol.2017.3055
2. Villanueva A. Hepatocellular Carcinoma. Reply. N Engl J Med (2019) 381(1):e2. doi: 10.1056/NEJMc1906565
3. Krenzien F, Schmelzle M, Struecker B, Raschzok N, Benzing C, Jara M, et al. Liver Transplantation and Liver Resection for Cirrhotic Patients with Hepatocellular Carcinoma: Comparison of Long-Term Survivals. J Gastrointest Surg (2018) 22(5):840–8. doi: 10.1007/s11605-018-3690-4
4. Lurje I, Czigany Z, Bednarsch J, Roderburg C, Isfort P, Neumann UP, et al. Treatment Strategies for Hepatocellular Carcinoma ⁻ a Multidisciplinary Approach. Int J Mol Sci (2019) 20(6):1465. doi: 10.3390/ijms20061465
5. Sapisochin G, Bruix J. Liver transplantation for hepatocellular carcinoma: outcomes and novel surgical approaches. Nat Rev Gastroenterol Hepatol (2017) 14(4):203–17. doi: 10.1038/nrgastro.2016.193
6. Boland P, Wu J. Systemic therapy for hepatocellular carcinoma: beyond sorafenib. Chin Clin Oncol (2018) 7(5):50. doi: 10.21037/cco.2018.10.10
7. Facciorusso A, Di Maso M, Muscatiello N. Microwave ablation versus radiofrequency ablation for the treatment of hepatocellular carcinoma: A systematic review and meta-analysis. Int J Hyperthermia (2016) 32(3):339–44. doi: 10.3109/02656736.2015.1127434
8. Galle PR, Tovoli F, Foerster F, Wörns MA, Cucchetti A, Bolondi L. The treatment of intermediate stage tumours beyond TACE: From surgery to systemic therapy. J Hepatol (2017) 67(1):173–83. doi: 10.1016/j.jhep.2017.03.007
9. Johnston MP, Khakoo SI. Immunotherapy for hepatocellular carcinoma: Current and future. World J Gastroenterol (2019) 25(24):2977–89. doi: 10.3748/wjg.v25.i24.2977
10. Kirstein MM, Wirth TC. [Multimodal treatment of hepatocellular carcinoma]. Internist (Berl) (2020) 61(2):164–9. doi: 10.1007/s00108-019-00722-x
11. Liu Z, Lin Y, Zhang J, Zhang Y, Li Y, Liu Z, et al. Molecular targeted and immune checkpoint therapy for advanced hepatocellular carcinoma. J Exp Clin Cancer Res (2019) 38(1):447. doi: 10.1186/s13046-019-1412-8
12. Lee KF, Chong CCN, Fong AKW, Fung AKY, Lok HT, Cheung YS, et al. Pattern of disease recurrence and its implications for postoperative surveillance after curative hepatectomy for hepatocellular carcinoma: experience from a single center. Hepatobiliary Surg Nutr (2018) 7(5):320–30. doi: 10.21037/hbsn.2018.03.17
13. Llovet JM, Montal R, Villanueva A. Randomized trials and endpoints in advanced HCC: Role of PFS as a surrogate of survival. J Hepatol (2019) 70(6):1262–77. doi: 10.1016/j.jhep.2019.01.028
14. Welling TH, Eddinger K, Carrier K, Zhu D, Kleaveland T, Moore DE, et al. Multicenter Study of Staging and Therapeutic Predictors of Hepatocellular Carcinoma Recurrence Following Transplantation. Liver Transpl (2018) 24(9):1233–42. doi: 10.1002/lt.25194
15. Javadian P, Nezhat F. Endometrial Carcinoma and its Precursors. Adv Exp Med Biol (2020) 1242:59–72. doi: 10.1007/978-3-030-38474-6_4
16. Sarveazad A, Agah S, Babahajian A, Amini N, Bahardoust M. Predictors of 5 year survival rate in hepatocellular carcinoma patients. J Res Med Sci (2019) 24:86. doi: 10.4103/jrms.JRMS_1017_18
17. Wu G, Wu J, Wang B, Zhu X, Shi X, Ding Y. Importance of tumor size at diagnosis as a prognostic factor for hepatocellular carcinoma survival: a population-based study. Cancer Manag Res (2018) 10:4401–10. doi: 10.2147/cmar.S177663
18. Dimitroulis D, Damaskos C, Valsami S, Davakis S, Garmpis N, Spartalis E, et al. From diagnosis to treatment of hepatocellular carcinoma: An epidemic problem for both developed and developing world. World J Gastroenterol (2017) 23(29):5282–94. doi: 10.3748/wjg.v23.i29.5282
19. Baralle FE, Giudice J. Alternative splicing as a regulator of development and tissue identity. Nat Rev Mol Cell Biol (2017) 18(7):437–51. doi: 10.1038/nrm.2017.27
20. Du JX, Zhu GQ, Cai JL, Wang B, Luo YH, Chen C, et al. Splicing factors: Insights into their regulatory network in alternative splicing in cancer. Cancer Lett (2020) 501:83–104. doi: 10.1016/j.canlet.2020.11.043
21. Wan L, Yu W, Shen E, Sun W, Liu Y, Kong J, et al. SRSF6-regulated alternative splicing that promotes tumour progression offers a therapy target for colorectal cancer. Gut (2019) 68(1):118–29. doi: 10.1136/gutjnl-2017-314983
22. Zhan YT, Li L, Zeng TT, Zhou NN, Guan XY, Li Y. SNRPB-mediated RNA splicing drives tumor cell proliferation and stemness in hepatocellular carcinoma. Aging (Albany NY) (2020) 12:537–54. doi: 10.18632/aging.202164
23. Zhang F, Deng CK, Wang M, Deng B, Barber R, Huang G. Identification of novel alternative splicing biomarkers for breast cancer with LC/MS/MS and RNA-Seq. BMC Bioinf (2020) 21(Suppl 9):541. doi: 10.1186/s12859-020-03824-8
24. Fischl H, Neve J, Wang Z, Patel R, Louey A, Tian B, et al. hnRNPC regulates cancer-specific alternative cleavage and polyadenylation profiles. Nucleic Acids Res (2019) 47(14):7580–91. doi: 10.1093/nar/gkz461
25. Zarnack K, König J, Tajnik M, Martincorena I, Eustermann S, Stévant I, et al. Direct competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell (2013) 152(3):453–66. doi: 10.1016/j.cell.2012.12.023
26. Zhang Y, Chen W, Pan T, Wang H, Zhang Y, Li C. LBX2-AS1 is activated by ZEB1 and promotes the development of esophageal squamous cell carcinoma by interacting with HNRNPC to enhance the stability of ZEB1 and ZEB2 mRNAs. Biochem Biophys Res Commun (2019) 511(3):566–72. doi: 10.1016/j.bbrc.2019.02.079
27. Wu Y, Zhao W, Liu Y, Tan X, Li X, Zou Q, et al. Function of HNRNPC in breast cancer cells by controlling the dsRNA-induced interferon response. EMBO J (2018) 37(23):e99017. doi: 10.15252/embj.201899017
28. Park YM, Hwang SJ, Masuda K, Choi KM, Jeong MR, Nam DH, et al. Heterogeneous nuclear ribonucleoprotein C1/C2 controls the metastatic potential of glioblastoma by regulating PDCD4. Mol Cell Biol (2012) 32(20):4237–44. doi: 10.1128/mcb.00443-12
29. Huang H, Han Y, Zhang C, Wu J, Feng J, Qu L, et al. HNRNPC as a candidate biomarker for chemoresistance in gastric cancer. Tumour Biol (2016) 37(3):3527–34. doi: 10.1007/s13277-015-4144-1
30. Chen Y, Bao C, Zhang X, Lin X, Fu Y. Knockdown of LINC00662 represses AK4 and attenuates radioresistance of oral squamous cell carcinoma. Cancer Cell Int (2020) 20:244. doi: 10.1186/s12935-020-01286-9
31. Shen Y, Liu S, Fan J, Jin Y, Tian B, Zheng X, et al. Nuclear retention of the lncRNA SNHG1 by doxorubicin attenuates hnRNPC-p53 protein interactions. EMBO Rep (2017) 18(4):536–48. doi: 10.15252/embr.201643139
32. Kleemann M, Schneider H, Unger K, Sander P, Schneider EM, Fischer-Posovszky P, et al. MiR-744-5p inducing cell death by directly targeting HNRNPC and NFIX in ovarian cancer cells. Sci Rep (2018) 8(1):9020. doi: 10.1038/s41598-018-27438-6
33. Wang LC, Chen SH, Shen XL, Li DC, Liu HY, Ji YL, et al. M6A RNA Methylation Regulator HNRNPC Contributes to Tumorigenesis and Predicts Prognosis in Glioblastoma Multiforme. Front Oncol (2020) 10:536875. doi: 10.3389/fonc.2020.536875
34. Zhuang Z, Chen L, Mao Y, Zheng Q, Li H, Huang Y, et al. Diagnostic, progressive and prognostic performance of m(6)A methylation RNA regulators in lung adenocarcinoma. Int J Biol Sci (2020) 16(11):1785–97. doi: 10.7150/ijbs.39046
35. Zhao X, Cui L. Development and validation of a m(6)A RNA methylation regulators-based signature for predicting the prognosis of head and neck squamous cell carcinoma. Am J Cancer Res (2019) 9(10):2156–69.
36. Yang S, Liu G. Targeting the Ras/Raf/MEK/ERK pathway in hepatocellular carcinoma. Oncol Lett (2017) 13(3):1041–7. doi: 10.3892/ol.2017.5557
37. Chang F, Steelman LS, Lee JT, Shelton JG, Navolanic PM, Blalock WL, et al. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia (2003) 17(7):1263–93. doi: 10.1038/sj.leu.2402945
38. Xu J, Pfarr N, Endris V, Mai EK, Md Hanafiah NH, Lehners N, et al. Molecular signaling in multiple myeloma: association of RAS/RAF mutations and MEK/ERK pathway activation. Oncogenesis (2017) 6(5):e337. doi: 10.1038/oncsis.2017.36
39. Wee P, Wang Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers (Basel) (2017) 9(5):52. doi: 10.3390/cancers9050052
40. Tang L, Yang J, Chen J, Yu J, Zhou Q, Lu X, et al. IGF-1R promotes the expression of cyclin D1 protein and accelerates the G1/S transition by activating Ras/Raf/MEK/ERK signaling pathway. Int J Clin Exp Pathol (2017) 10(12):11652–8.
41. Agell N, Bachs O, Rocamora N, Villalonga P. Modulation of the Ras/Raf/MEK/ERK pathway by Ca(2+), and calmodulin. Cell Signal (2002) 14(8):649–54. doi: 10.1016/s0898-6568(02)00007-4
42. Vuoriluoto K, Haugen H, Kiviluoto S, Mpindi JP, Nevo J, Gjerdrum C, et al. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene (2011) 30(12):1436–48. doi: 10.1038/onc.2010.509
43. Mulholland DJ, Kobayashi N, Ruscetti M, Zhi A, Tran LM, Huang J, et al. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res (2012) 72(7):1878–89. doi: 10.1158/0008-5472.Can-11-3132
44. David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, et al. TGF-β Tumor Suppression through a Lethal EMT. Cell (2016) 164(5):1015–30. doi: 10.1016/j.cell.2016.01.009
45. Gonzalez-Hormazabal P, Musleh M, Bustamante M, Stambuk J, Pisano R, Valladares H, et al. Polymorphisms in RAS/RAF/MEK/ERK Pathway Are Associated with Gastric Cancer. Genes (Basel) (2018) 10(1):20. doi: 10.3390/genes10010020
46. Zou Y, Liu FY, Wu J, Wan L, Fang SF, Zhang ZY, et al. Mutational analysis of the RAS/RAF/MEK/ERK signaling pathway in 260 Han Chinese patients with cervical carcinoma. Oncol Lett (2017) 14(2):2427–31. doi: 10.3892/ol.2017.6435
47. Li L, Zhao GD, Shi Z, Qi LL, Zhou LY, Fu ZX. The Ras/Raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC. Oncol Lett (2016) 12(5):3045–50. doi: 10.3892/ol.2016.5110
48. Chang YS, Liu JC, Fu HQ, Yu BT, Zou SB, Wu QC, et al. [Roles of targeting Ras/Raf/MEK/ERK signaling pathways in the treatment of esophageal carcinoma]. Yao Xue Xue Bao (2013) 48(5):635–41.
49. Knight T, Irving JA. Ras/Raf/MEK/ERK Pathway Activation in Childhood Acute Lymphoblastic Leukemia and Its Therapeutic Targeting. Front Oncol (2014) 4:160. doi: 10.3389/fonc.2014.00160
50. Wang AX, Qi XY. Targeting RAS/RAF/MEK/ERK signaling in metastatic melanoma. IUBMB Life (2013) 65(9):748–58. doi: 10.1002/iub.1193
51. Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol (2018) 15(10):599–616. doi: 10.1038/s41571-018-0073-4
52. Ikeda M, Morizane C, Ueno M, Okusaka T, Ishii H, Furuse J. Chemotherapy for hepatocellular carcinoma: current status and future perspectives. Jpn J Clin Oncol (2018) 48(2):103–14. doi: 10.1093/jjco/hyx180
53. Couri T, Pillai A. Goals and targets for personalized therapy for HCC. Hepatol Int (2019) 13(2):125–37. doi: 10.1007/s12072-018-9919-1
54. Urbanski LM, Leclair N, Anczuków O. Alternative-splicing defects in cancer: Splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip Rev RNA (2018) 9(4):e1476. doi: 10.1002/wrna.1476
55. Chen K, Xiao H, Zeng J, Yu G, Zhou H, Huang C, et al. Alternative Splicing of EZH2 pre-mRNA by SF3B3 Contributes to the Tumorigenic Potential of Renal Cancer. Clin Cancer Res (2017) 23(13):3428–41. doi: 10.1158/1078-0432.Ccr-16-2020
56. Li Y, Sun N, Lu Z, Sun S, Huang J, Chen Z, et al. Prognostic alternative mRNA splicing signature in non-small cell lung cancer. Cancer Lett (2017) 393:40–51. doi: 10.1016/j.canlet.2017.02.016
57. Marzese DM, Manughian-Peter AO, Orozco JIJ, Hoon DSB. Alternative splicing and cancer metastasis: prognostic and therapeutic applications. Clin Exp Metastasis (2018) 35(5-6):393–402. doi: 10.1007/s10585-018-9905-y
58. Geuens T, Bouhy D, Timmerman V. The hnRNP family: insights into their role in health and disease. Hum Genet (2016) 135(8):851–67. doi: 10.1007/s00439-016-1683-5
59. Klingenberg M, Groß M, Goyal A, Polycarpou-Schwarz M, Miersch T, Ernst AS, et al. The Long Noncoding RNA Cancer Susceptibility 9 and RNA Binding Protein Heterogeneous Nuclear Ribonucleoprotein L Form a Complex and Coregulate Genes Linked to AKT Signaling. Hepatology (2018) 68(5):1817–32. doi: 10.1002/hep.30102
60. Cui H, Wu F, Sun Y, Fan G, Wang Q. Up-regulation and subcellular localization of hnRNP A2/B1 in the development of hepatocellular carcinoma. BMC Cancer (2010) 10:356. doi: 10.1186/1471-2407-10-356
61. Xu H, Dong X, Chen Y, Wang X. Serum exosomal hnRNPH1 mRNA as a novel marker for hepatocellular carcinoma. Clin Chem Lab Med (2018) 56(3):479–84. doi: 10.1515/cclm-2017-0327
62. Sun W, Xing B, Sun Y, Du X, Lu M, Hao C, et al. Proteome analysis of hepatocellular carcinoma by two-dimensional difference gel electrophoresis: novel protein markers in hepatocellular carcinoma tissues. Mol Cell Proteomics (2007) 6(10):1798–808. doi: 10.1074/mcp.M600449-MCP200
63. Wiesweg M, Kasper S, Worm K, Herold T, Reis H, Sara L, et al. Impact of RAS mutation subtype on clinical outcome-a cross-entity comparison of patients with advanced non-small cell lung cancer and colorectal cancer. Oncogene (2019) 38(16):2953–66. doi: 10.1038/s41388-018-0634-0
64. Huang Y, Wei J, Liu BR. [Research advances of K-ras mutation in the prognosis and targeted therapy of gastric cancer]. Zhonghua Zhong Liu Za Zhi (2016) 38(2):81–5. doi: 10.3760/cma.j.issn.0253-3766.2016.02.001
65. Hacioglu BM, Kodaz H, Erdogan B, Cinkaya A, Tastekin E, Hacibekiroglu I, et al. K-RAS and N-RAS mutations in testicular germ cell tumors. Bosn J Basic Med Sci (2017) 17(2):159–63. doi: 10.17305/bjbms.2017.1764
66. Luo D, Liu QF, Gove C, Naomov N, Su JJ, Williams R. Analysis of N-ras gene mutation and p53 gene expression in human hepatocellular carcinomas. World J Gastroenterol (1998) 4(2):97–9. doi: 10.3748/wjg.v4.i2.97
67. Cullen JM, Williams C, Zadrozny L, Otstot JT, Solomon GG, Sills RC, et al. H-ras consensus sequence and mutations in primary hepatocellular carcinomas of lemurs and lorises. Vet Pathol (2011) 48(4):868–74. doi: 10.1177/0300985810388526
68. Zolfaghari N, Shahbazi S, Torfeh M, Khorasani M, Hashemi M, Mahdian R. Identification of Differentially Expressed K-Ras Transcript Variants in Patients With Leiomyoma. Reprod Sci (2017) 24(10):1438–43. doi: 10.1177/1933719116689596
69. Barbier J, Dutertre M, Bittencourt D, Sanchez G, Gratadou L, de la Grange P, et al. Regulation of H-ras splice variant expression by cross talk between the p53 and nonsense-mediated mRNA decay pathways. Mol Cell Biol (2007) 27(20):7315–33. doi: 10.1128/mcb.00272-07
70. Wang C, Li P, Xuan J, Zhu C, Liu J, Shan L, et al. Cholesterol Enhances Colorectal Cancer Progression via ROS Elevation and MAPK Signaling Pathway Activation. Cell Physiol Biochem (2017) 42(2):729–42. doi: 10.1159/000477890
71. Sui X, Kong N, Ye L, Han W, Zhou J, Zhang Q, et al. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett (2014) 344(2):174–9. doi: 10.1016/j.canlet.2013.11.019
72. Cui X, Qian DW, Jiang S, Shang EX, Zhu ZH, Duan JA. Scutellariae Radix and Coptidis Rhizoma Improve Glucose and Lipid Metabolism in T2DM Rats via Regulation of the Metabolic Profiling and MAPK/PI3K/Akt Signaling Pathway. Int J Mol Sci (2018) 19(11)3634. doi: 10.3390/ijms19113634
73. Rodenak-Kladniew B, Castro A, Stärkel P, De Saeger C, García de Bravo M, Crespo R. Linalool induces cell cycle arrest and apoptosis in HepG2 cells through oxidative stress generation and modulation of Ras/MAPK and Akt/mTOR pathways. Life Sci (2018) 199:48–59. doi: 10.1016/j.lfs.2018.03.006
74. Guo N, Xiao Y, Chen D, Wang J. Antiproliferative effects of Norartocarpetin isoflavone in human lung carcinoma cells are mediated via targeting Ras/Raf/MAPK signalling pathway, mitochondrial mediated apoptosis, S-phase cell cycle arrest and suppression of cell migration and invasion. J buon (2020) 25(2):855–61.
75. Chen L, Wang X, Cheng H, Zhang W, Liu Y, Zeng W, et al. Cyclin Y binds and activates CDK4 to promote the G1/S phase transition in hepatocellular carcinoma cells via Rb signaling. Biochem Biophys Res Commun (2020) 533(4):1162–9. doi: 10.1016/j.bbrc.2020.09.127
76. Liu SL, Liu Z, Zhang LD, Zhu HQ, Guo JH, Zhao M, et al. GSK3β-dependent cyclin D1 and cyclin E1 degradation is indispensable for NVP-BEZ235 induced G0/G1 arrest in neuroblastoma cells. Cell Cycle (2017) 16(24):2386–95. doi: 10.1080/15384101.2017.1383577
77. Wang C, Yang Y, Zhang G, Li J, Wu X, Ma X, et al. Long noncoding RNA EMS connects c-Myc to cell cycle control and tumorigenesis. Proc Natl Acad Sci USA (2019) 116(29):14620–9. doi: 10.1073/pnas.1903432116
78. O’Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol (2016) 13(7):417–30. doi: 10.1038/nrclinonc.2016.26
79. Milioli HH, Alexandrou S, Lim E, Caldon CE. Cyclin E1 and cyclin E2 in ER+ breast cancer: prospects as biomarkers and therapeutic targets. Endocr Relat Cancer (2020) 27(5):R93–r112. doi: 10.1530/erc-19-0501
80. De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer (2013) 13(2):97–110. doi: 10.1038/nrc3447
81. Okada T, Sinha S, Esposito I, Schiavon G, López-Lago MA, Su W, et al. Author Correction: The Rho GTPase Rnd1 suppresses mammary tumorigenesis and EMT by restraining Ras-MAPK signalling. Nat Cell Biol (2019) 21(4):534. doi: 10.1038/s41556-019-0288-3
82. Fattet L, Yang J. RREB1 Integrates TGF-β and RAS Signals to Drive EMT. Dev Cell (2020) 52(3):259–60. doi: 10.1016/j.devcel.2020.01.020
83. Chen S, Li F, Xu D, Hou K, Fang W, Li Y. The Function of RAS Mutation in Cancer and Advances in its Drug Research. Curr Pharm Des (2019) 25(10):1105–14. doi: 10.2174/1381612825666190506122228
84. Delire B, Stärkel P. The Ras/MAPK pathway and hepatocarcinoma: pathogenesis and therapeutic implications. Eur J Clin Invest (2015) 45(6):609–23. doi: 10.1111/eci.12441
85. Sun S, He Z, Huang M, Wang N, He Z, Kong X, et al. Design and discovery of thioether and nicotinamide containing sorafenib analogues as multikinase inhibitors targeting B-Raf, B-Raf(V600E) and VEGFR-2. Bioorg Med Chem (2018) 26(9):2381–91. doi: 10.1016/j.bmc.2018.03.039
86. Takaya H, Namisaki T, Shimozato N, Kaji K, Kitade M, Moriya K, et al. ADAMTS13 and von Willebrand factor are useful biomarkers for sorafenib treatment efficiency in patients with hepatocellular carcinoma. World J Gastrointest Oncol (2019) 11(5):424–35. doi: 10.4251/wjgo.v11.i5.424
87. Li W, Dong X, He C, Tan G, Li Z, Zhai B, et al. LncRNA SNHG1 contributes to sorafenib resistance by activating the Akt pathway and is positively regulated by miR-21 in hepatocellular carcinoma cells. J Exp Clin Cancer Res (2019) 38(1):183. doi: 10.1186/s13046-019-1177-0
Keywords: HCC, HNRNPC, alternative splicing, Ras, MAPK
Citation: Hu J, Cai D, Zhao Z, Zhong G-C and Gong J (2021) Suppression of Heterogeneous Nuclear Ribonucleoprotein C Inhibit Hepatocellular Carcinoma Proliferation, Migration, and Invasion via Ras/MAPK Signaling Pathway. Front. Oncol. 11:659676. doi: 10.3389/fonc.2021.659676
Received: 28 January 2021; Accepted: 30 March 2021;
Published: 16 April 2021.
Edited by:
Yujun Shi, Sichuan University, ChinaReviewed by:
Urbain Weyemi, University of Texas at Austin, United StatesArun Upadhyay, Northwestern University, United States
Copyright © 2021 Hu, Cai, Zhao, Zhong and Gong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianping Gong, gongjianping@cqmu.edu.cn; Guo-Chao Zhong, gczhong1991@stu.cqmu.edu.cn
†These authors share senior authorship