 Priya Bhardwaj1,2
Priya Bhardwaj1,2 Kristy A. Brown1,2,3*
Kristy A. Brown1,2,3*- 1Department of Medicine, Weill Cornell Medicine, New York, NY, United States
- 2Graduate School of Medical Sciences, Weill Cornell Medicine, New York, NY, United States
- 3Meyer Cancer Center, Weill Cornell Medicine, New York, NY, United States
Obesity is an established risk factor for breast cancer growth and progression. A number of advances have been made in recent years revealing new insights into this link. Early events in breast cancer development involve the neoplastic transformation of breast epithelial cells to cancer cells. In obesity, breast adipose tissue undergoes significant hormonal and inflammatory changes that create a mitogenic microenvironment. Many factors that are produced in obesity have also been shown to promote tumorigenesis. Given that breast epithelial cells are surrounded by adipose tissue, the crosstalk between the adipose compartment and breast epithelial cells is hypothesized to be a significant player in the initiation and progression of breast cancer in individuals with excess adiposity. The present review examines this crosstalk with a focus on obese breast adipose-derived estrogen, inflammatory mediators and adipokines, and how they are mechanistically linked to breast cancer risk and growth through stimulation of oxidative stress, DNA damage, and pro-oncogenic transcriptional programs. Pharmacological and lifestyle strategies targeting these factors and their downstream effects are evaluated for feasibility and efficacy in decreasing the risk of obesity-induced breast epithelial cell transformation and consequently, breast cancer development.
Introduction
Breast cancer is the second most common cancer diagnosed in women in the U.S. and the most common cancer among women worldwide, accounting for an estimated 627,000 deaths in 2018 (1, 2). In 2020, over a quarter million breast cancers were diagnosed in the U.S., representing a slow but steady increase in new diagnoses over the last 30 years (3). There are a number of well-established risk factors that impact the prevalence of breast cancer, including genetics, age, reproductive history, breast density, and exposure to hormones (4, 5). Additionally, lifestyle factors like alcohol consumption, physical inactivity, and elevated bodyweight have been causally linked with breast cancer development and worse prognosis (6, 7). The link between elevated bodyweight and breast cancer is of particular significance in light of the rapidly rising rates of obesity worldwide, nearly tripling since 1975 (8). By 2025 global obesity prevalence is expected to reach 18% in men and surpass 21% in women (8), with some estimates predicting significantly faster growth (9). Meanwhile, the U.S. has already seen obesity rates reaching 42.4% as of 2018, with the highest rates observed in women (10). Therefore, understanding the mechanistic basis of how obesity contributes to elevated risk of breast cancer and worse outcomes are critical from the standpoint of prevention and management.
The Relationship Between Obesity and Breast Cancer
Obesity, as defined by a body mass index (BMI, kg/m2) greater than or equal to 30, has consistently been associated with breast cancer in postmenopausal women. In premenopausal women, an inverse association has been observed and attributed in part to higher rates of amenorrhea in obese women and consequently, decreased circulating estrogen levels (11, 12). However, among postmenopausal women, there is a strong causal relationship between obesity and estrogen receptor positive (ER+) breast cancer (13, 14). In 2016, the International Agency for Research on Cancer (IARC) conducted a systematic review of over 1000 epidemiological studies on the risk of cancer due to excess body fat. This large-scale review confirmed the relationship between obesity and postmenopausal breast cancer while also concluding that there was substantial evidence establishing an association between increased BMI and reduced survival among breast cancer patients (15). The composition of the breast lends some insight into how obesity may be mechanistically linked with breast cancer. The breast is comprised of three major components: fibrous tissue, glandular tissue, and fat (adipose) tissue. On a cellular level, the adipose tissue compartment consists of adipocytes that store lipid, preadipocytes (adipose stromal cells), immune cells, and endothelial cells. The glandular tissue refers to the epithelial cells that form lobules and ducts, which produce and transport milk within the breast, respectively. The cells within these compartments are plastic as the breast undergoes significant changes during development, pregnancy, and in response to environmental factors. For example, during cold exposure white adipocytes can undergo “browning” and differentiate into beige adipocytes which have thermogenic properties when activated (16). Additionally, during pregnancy secretory alveolar mammary epithelial cells expand to support lactation and undergo involution via apoptosis during the post-lactation period (17). Studies in pregnant mice have suggested the ability of white adipocytes to transdifferentiate into milk producing mammary epithelial cells containing lipid droplets and termed “pink” adipocytes (18).
During breast carcinogenesis, it is the epithelial cells that undergo neoplastic transformation into cancer cells. Notably, breast epithelial cells are embedded in adipose tissue, allowing for paracrine interaction between epithelial cells and cells within the adipose compartment. In a recent study, microdissection was performed on epithelium, adipose tissue, and stroma in breast sections from women before and after breast cancer diagnosis. Co-expression network analyses comparing tissue associations between compartments showed increased interaction between the epithelial compartment and surrounding adipose tissue prior to breast cancer development, confirming the existence of a crosstalk between breast epithelial cells and adjacent adipose tissue (19). This is a significant finding since in the setting of obesity, breast adipose tissue undergoes considerable dysregulation in the production of estrogens, adipokines, inflammatory mediators, and reactive oxygen species (ROS), creating a microenvironment primed for initiating breast cancer development and promoting progression (Figure 1).
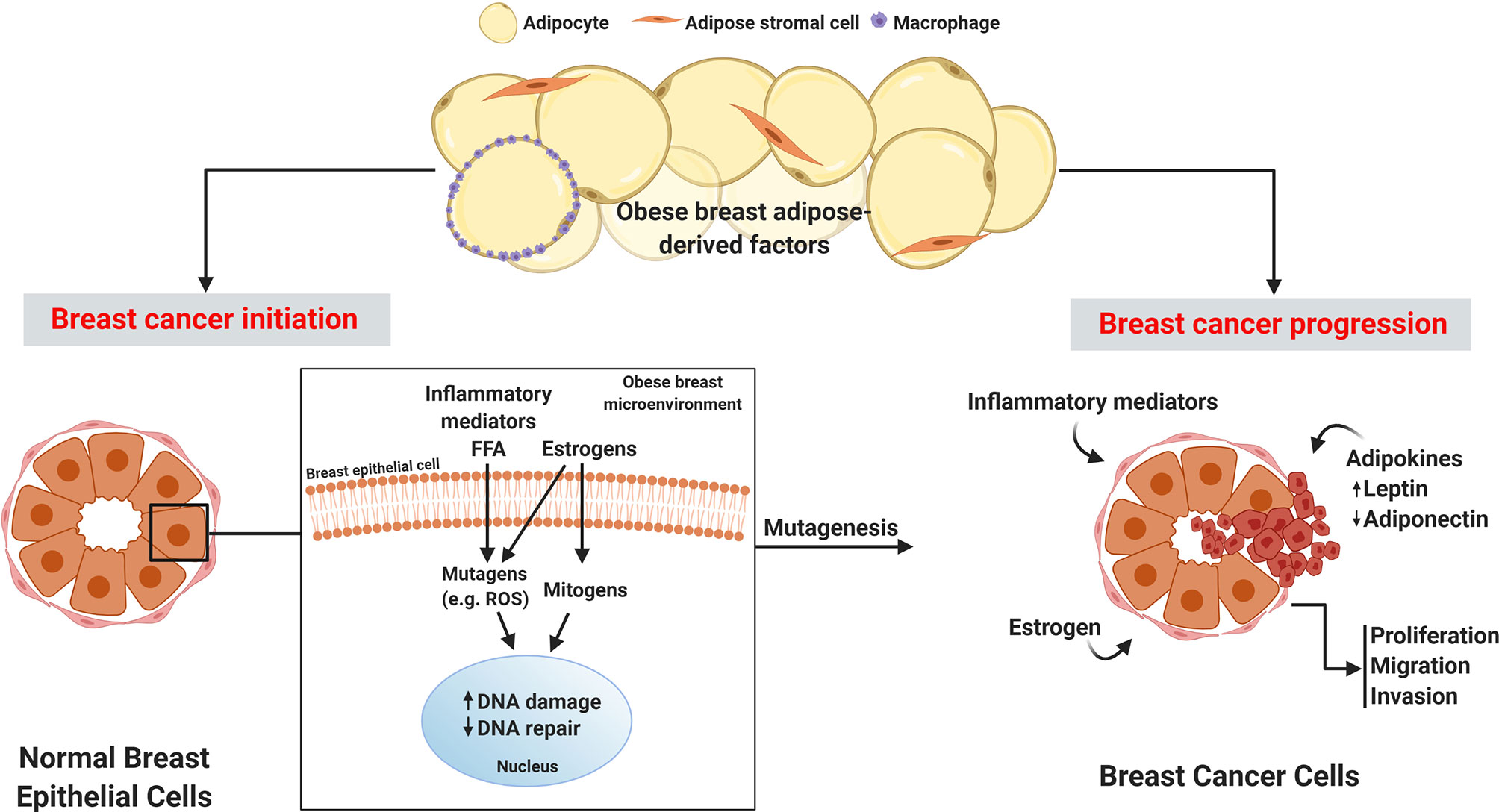
Figure 1 Obese breast adipose-derived factors contribute to the initiation and progression of breast cancer. Factors produced by obese breast adipose tissue including inflammatory mediators, free fatty acids (FFA), and estrogens can act as mutagens, for example by stimulating intracellular reactive oxygen species (ROS), which can lead to DNA damage in normal breast epithelial cells. Estrogen also has mitogenic effects that can lead to replication stress and consequently DNA damage. Elevation in DNA damage coupled with potential estrogen-induced dysfunctional DNA repair may lead to unresolved DNA damage, which is linked to mutagenesis and initiation of cancer. Breast cancer progression is fueled by inflammatory mediators, excess leptin, decreased adiponectin, and elevated estrogens in the obese breast adipose microenvironment which promote proliferation, migration, and invasion of breast cancer.
Obesity-Induced Dysregulation of the Breast Adipose Microenvironment
Estrogens
Estrogens, a class of steroid hormones, are major drivers of obesity-associated ER+ breast cancer. While they play an important role in normal mammary gland development and in metabolic processes, excessive or dysregulated estrogen signaling drives ER+ breast cancer through both genomic and non-genomic actions (20, 21). 17β-estradiol (E2), the most potent endogenously produced estrogen, binds to its receptor, ERα, causing dimerization, translocation to the nucleus and binding to estrogen response elements (EREs) on target genes to drive transcription of a variety of programs that promote cancer growth, including cell proliferation and decreased apoptosis (4, 22, 23). Estrogens can also have receptor-independent mutagenic effects, as reviewed in the next section. Lifetime exposure to estrogens, and especially high levels after menopause, has been consistently linked with elevated breast cancer risk (4, 24).
Before menopause, E2 is produced predominantly by the ovaries and is released into circulation. After menopause, ovarian estrogen production ceases and circulating levels diminish. However, it is still synthesized to a lesser extent in peripheral tissues including bone, brain, vascular tissue, and mainly, adipose tissue (25). In obesity, adipose tissue levels of E2 are elevated due to increased adipose stromal cell (ASC) expression of the enzyme aromatase, which catalyzes the conversion of androstenedione to estrone (E1), and testosterone to E2 (26–28). In addition to aromatase expression increasing in ASCs from obese individuals, it has also been suggested that the number of ASCs within the breast increases as adipose tissue expands in obesity, further compounding the elevation in aromatase levels and consequently, estrogen levels in postmenopausal breast tissue (29–32). Aromatase expression in ASCs is modulated by the adipose tissue microenvironment, including the presence of tumors (22) (Figure 2). For example, both tumors and macrophages in obese breast adipose tissue secrete the inflammatory mediator prostaglandin E2 (PGE2) which was shown to increase expression and nuclear localization of hypoxia inducible factor-1α (HIF-1α) which in turn, stimulates promoter II-driven aromatase expression in ASCs (33). PGE2 also stimulates aromatase expression through inhibition of 5’ AMP-activated protein kinase (AMPK), a negative regulator of aromatase, and by increasing expression and nuclear localization of transcription factors cAMP response element-binding protein (CREB), liver receptor homolog-1 (LRH-1), and transcription factor co-activator peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) which drive aromatase expression (23). The effects of PGE2 on aromatase expression in ASCs is further enhanced in the setting of obesity due to decreased expression of the gut-derived hormone ghrelin, which inhibits PGE2 signaling (34). Furthermore, obesity-induced modulation in the adipokines leptin and adiponectin results in a net inhibition of AMPK (35). Collectively, this increase in ASC aromatase expression leads to elevated estradiol production, which can fuel growth of nearby ER+ breast tumors. A recent study highlighted the importance of ASC-breast tumor crosstalk using an ex vivo organotypic breast model where mammary ASCs from lean and obese women were isolated and co-cultured with ER+ MCF7 breast cancer cells. It was demonstrated that ERE transactivation was heightened in MCF7 cells co-cultured with obese ASCs compared with lean ASCs due to increased expression of aromatase in obese ASCs (31).
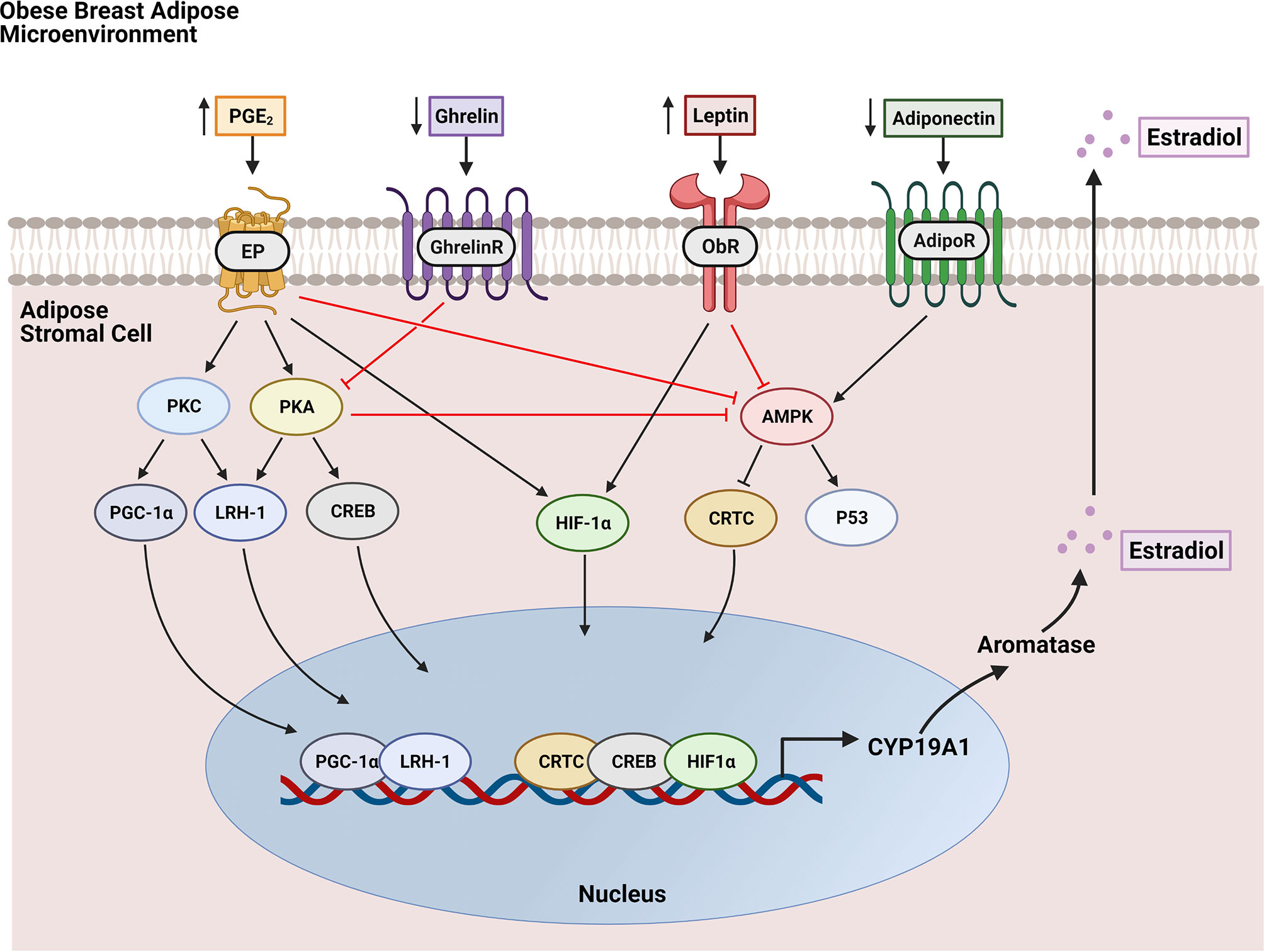
Figure 2 Regulation of aromatase expression in breast adipose stromal cells by the obese breast microenvironment. Hallmarks of the obese breast microenvironment include elevated PGE2 and leptin along with decreases in ghrelin and adiponectin. PGE2 binds EP receptors to activate signaling through protein kinase C (PKC) and protein kinase A (PKA) which results in increased expression and nuclear localization of transcription factor co-activator PGC-1α and transcription factors LRH-1 and CREB which both drive aromatase (CYP19A1) transcription. PGE2 also enhances aromatase expression through stimulation of HIF-1α-driven aromatase transcription and through inhibition of AMPK. AMPK activation negatively regulates aromatase by inhibiting entry into the nucleus of the aromatase transcription factor CRTC and by stimulating P53, which has been shown to translocate to the nucleus and inhibit aromatase transcription. Leptin signaling through its receptor ObR also increases HIF-1α expression and nuclear localization while inhibiting AMPK signaling. Both ghrelin and adiponectin negatively regulate aromatase expression through inhibition of PKA and through activation of AMPK, respectively. However, these factors are downregulated in the obese breast microenvironment, thereby decreasing their inhibitory effects on aromatase expression. The net effect of increased aromatase expression catalyzes the production of estradiol, which is released into the breast microenvironment.
Inflammatory Mediators
Adipose tissue expansion during weight gain and in obesity leads to enlarged, hypertrophic adipocytes as they fill with excess dietary lipids. This results in a hypoxic breast microenvironment as growing cells have higher oxygen demands and vascularization cannot keep up with the pace of adipose expansion (36, 37). Inadequate oxygen supply leads to adipocyte cell death which signals the infiltration macrophages to surround and engulf necrotic adipocytes, forming characteristic inflammatory foci termed “crown-like structures” (CLS) in white adipose tissue (38, 39). In a study of 107 breast cancer patients, there was a 3.2 and 6.9 times higher likelihood of CLS occurring in breast tissue from overweight and obese women, respectively, compared with healthy weight patients (40). Macrophages in CLS produce inflammatory mediators, including TNF, IL-1β, IL-6, and PGE2, which contributes to the characterization of obesity as a state of chronic low-grade inflammation (41–44). In addition to macrophages, adipocytes have also been shown to secrete inflammatory mediators (45). In an in vitro study, co-culture of adipocytes with breast cancer cells resulted in synergistically increased production of IL-6, IL-8, CXCL10, CCL2, and CCL5 in the culture media (46). This contributes to a microenvironment conducive to breast cancer growth as inflammatory cytokines have an established role in activating signaling pathways related to proliferation, invasion, and migration of breast cancer cells (47–51)(Figure 3).
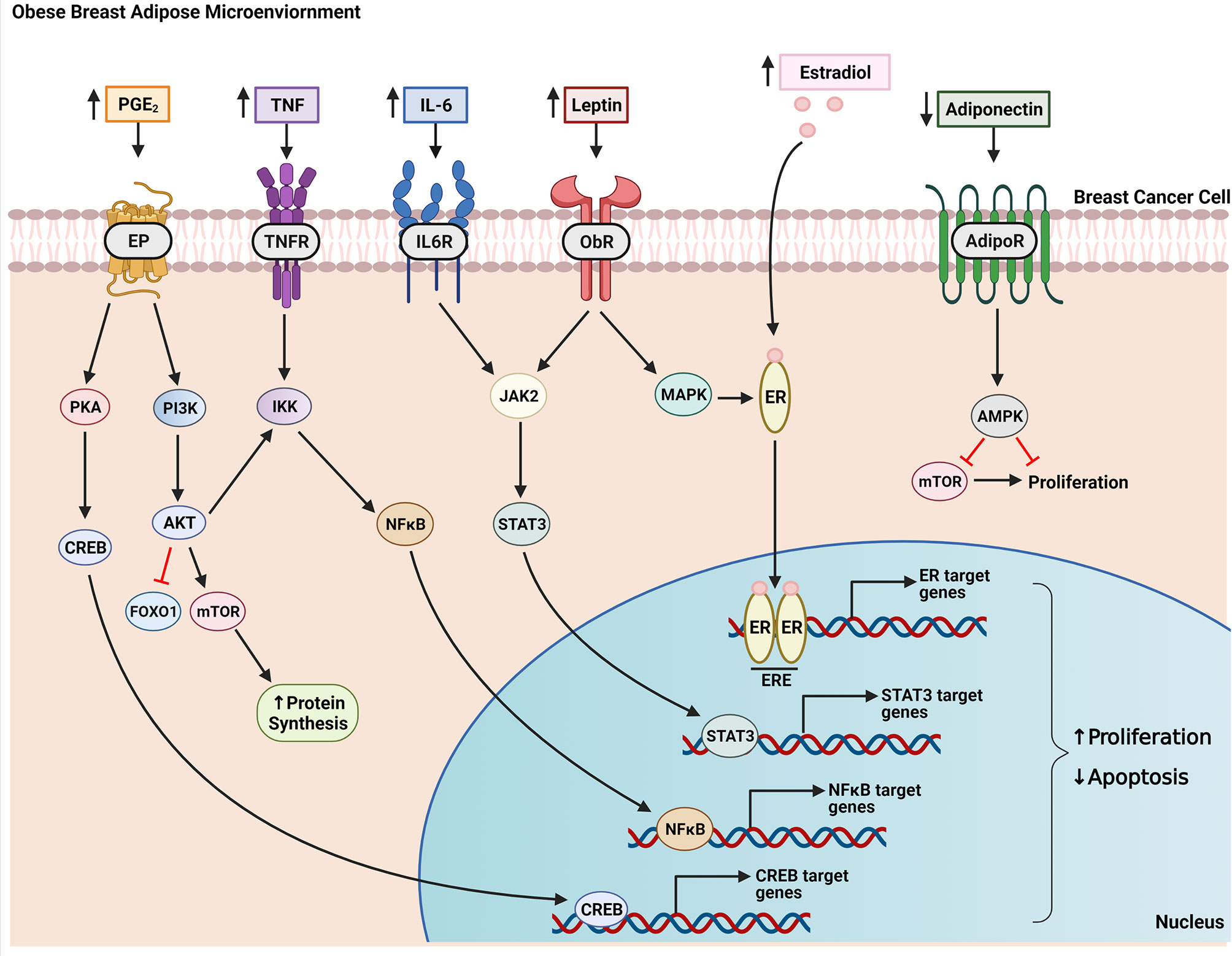
Figure 3 Factors produced in the obese breast microenvironment contribute to the growth of breast cancer. Obesity-induced changes to breast adipose tissue results in increased secretion of inflammatory mediators (e.g. PGE2, TNF, IL-6) and estradiol along with altered adipokine production. These factors act in a paracrine manner on breast tumor cells growing within this milieu to increase proliferation and decrease apoptosis. For example, PGE2 signals through EP receptors on tumor cells to activate PKA, which, in turn, stimulates CREB translocation to the nucleus and transcription of genes that promote cell growth. PGE2 also activates the PI3K pathway resulting in inhibition of FOXO1 and activation of mTOR via AKT, netting a decrease in apoptosis and increase in protein synthesis, a necessary precursor of cell growth. Both AKT and TNF signaling activate IKK leading to expression and translocation of transcription factor NFκB to the nucleus. IL-6 and leptin bind their respective receptors to activate JAK2/STAT3 signaling which promotes nuclear transcription of programs that enhance cell proliferation. Leptin also activates MAPK signaling which can lead to ligand-independent activation of the estrogen receptor (ER). Ligand-dependent (through binding of estradiol) or ligand-independent activation of ER causes receptor dimerization and binding to estrogen response elements (ERE) on target genes. Adiponectin signals through its receptor to activate AMPK signaling resulting in downstream inhibition of mTOR, which decreases proliferation. This inhibitory effect on cell proliferation is diminished in obesity due to decreased adiponectin production by obese breast adipose tissue.
Saturated fatty acids released by hypertrophic adipocytes undergoing lipolysis can also activate inflammatory cytokine production via ligand-mediated activation of toll-like receptors 2 and 4 (TLR2, TLR4) in both adipocytes and macrophages (52–54). In murine studies, high fat diet fed TLR2 and TLR4 deficient mice are protected from adipose inflammation, reinforcing the importance of these receptors as mediators of obesity-induced adipose inflammation (55–57). Recent work by Nishimoto et al. presented a novel mechanism for the development of adipose inflammation in obesity. Using in vitro and in vivo models, they showed that TLR9 activation facilitates the interaction between macrophages and adipocytes by increasing macrophage expression of monocyte chemoattractant protein-1 (MCP-1), which is a chemokine that signals macrophages to dead adipocytes (58). Furthermore, they found that obesity-related dying adipocytes shed cell-free DNA (cfDNA) that serve as an endogenous ligand for TLR9 in macrophages, thereby establishing an adipocyte cell death→cfDNA→TLR9 axis that leads to macrophage accumulation in obese adipose tissue. These studies were conducted exclusively in visceral fat, while breast tissue is considered subcutaneous fat. Therefore, further studies are warranted to determine whether this axis exists across fat depots. If so, it would represent an intriguing target for reducing breast adipose inflammation in obese postmenopausal women to protect against breast cancer development, or in a therapeutic setting to improve prognosis after diagnosis.
Adipokines
Beyond its role as a storage site for lipids, adipose tissue is recognized as an endocrine organ that secretes hormones called adipokines. Two major adipokines secreted by adipose tissue are leptin and adiponectin, which have widely been shown to promote or inhibit breast cancer, respectively, by directly interacting with breast cancer cells and by regulation of metabolic homeostasis in obesity.
Leptin is produced primarily by adipocytes and is secreted in abundance by obese adipose tissue as a function of adipocyte size, with levels correlating positively with BMI (59). While initially discovered in 1994 as a satiety hormone, it has since been shown to play a variety of roles in metabolic homeostasis, as an inflammatory mediator, and as a mitogen (60–62). Accordingly, elevated leptin levels are positively associated with breast cancer risk (63–65). Leptin acts by binding to its receptor, Ob-R, leading to signal transduction through phosphorylation of JAK2 and downstream activation of STAT3, MAPK/ERK1/2, AMPK and PI3K/AKT pathways. Activation of these pathways results in transcription of genes involved in cell growth, proliferation, angiogenesis and cell survival (62, 66–69) (Figure 3). In vitro work by Saxena et al. showed in a panel of breast cancer cell lines that leptin promotes proliferation through phosphorylation of JAK/STAT3 leading to translocation of phospho-STAT3 to the nucleus and transcription of cyclin D1, an important mediator of cell cycle (70). In addition to promoting breast cancer growth through stimulation of proliferation, leptin may also inhibit apoptosis of breast cancer cells by downregulating pro-apoptotic genes (71). However, this effect is not consistent across breast cancer cell lines.
Additionally, leptin promotes ER+ breast cancer cell proliferation through modulation of estrogen levels and signaling. For example, leptin was shown to transactivate ERα via signaling through ERK1/ERK2 in MCF7 breast cancer cells (72) and increase estrogen production in the local microenvironment by stimulating aromatase expression in ASCs (73–75) (Figure 2). Interestingly, another study also in MCF7 cells found that estrogen increases leptin and Ob-R mRNA (76), potentially representing a positive feedback loop between leptin and estrogen that amplifies expression of both in obesity. Strong et al. reported that leptin is not only secreted by adipocytes, but also by ASCs (77). Further, they showed that ASCs isolated from obese women had higher leptin expression than ASCs isolated from lean women. Co-culture of obese ASCs with several ER+ breast cancer cell lines increased proliferation, migration, and invasion, an effect that was lost after silencing leptin in the ASCs (77, 78).
Several studies have suggested a role for leptin in the early events that lead to cancer development. Using the non-neoplastic mammary epithelial cell line HMT-3522, Tenvooren et al. demonstrated that leptin disrupts apical-basal polarity of 3D cultured mammary acini via activation of PI3K/AKT signaling, an early hallmark of cellular transformation in breast cancer (79). Another study also implicated leptin in the early stages of breast cancer, showing that leptin treatment promotes epithelial-mesenchymal transition through activation of the kinases Src and FAK in MCF10A cells, a non-cancerous mammary epithelial cell line, which led to enhanced invasion through Matrigel (80). In a study of normal breast epithelium from women before and after breast cancer diagnosis compared with women who never developed breast cancer, leptin was upregulated 3.8 fold in the breast epithelium before cancer diagnosis, suggesting that increased leptin may be an early event associated with epithelial cell transformation (19).
Like leptin, adiponectin is an adipokine produced and secreted by adipocytes. However, unlike leptin, adiponectin is elevated in lean adipose tissue and decreases with obesity (23, 81). While leptin has pro-carcinogenic effects, adiponectin is inversely linked to breast cancer risk and aggressiveness (62, 82, 83). Adiponectin signals through ligand binding with its receptors AdipoR1 and AdipoR2 and recruitment of intracellular binding partner APPL1 which leads to downstream activation of the AMPK pathway and to a lesser extent p38 MAPK and PPARα pathways. It is both directly and indirectly protective against obesity-related breast cancer. Direct anti-cancer effects of adiponectin have been illustrated in several breast cancer cell lines where adiponectin inhibited proliferation, increased apoptotic response, and in one study, induced autophagic cell death (84–86). Indirect effects involve regulation of anti-inflammatory processes through stimulation of anti-inflammatory cytokine IL-10 and inhibition of inflammatory cytokine production from macrophages (87, 88). Additionally, adiponectin plays an important role as an insulin sensitizer through AMPK-mediated elevation in glucose utilization and increased fatty acid oxidation (89, 90). Therefore, obesity-induced elevation in breast adipose leptin and reduction in adiponectin play a significant role in establishing a breast microenvironment conducive to cellular transformation and tumor growth.
Obese Breast Adipose-Derived Sources of DNA Damage
In addition to fueling the growth of breast cancer, obesity has also been hypothesized to drive breast cancer initiation (Figure 1). A growing number of studies have highlighted an apparent genomic instability associated with obesity (91–94). This is significant since genomic instability can lead to mutations that lead to tumorigenesis (95). Genomic instability in the form of DNA damage has been demonstrated in various contexts in association with obesity in both preclinical and clinical studies. For example, in one study DNA damage in peripheral blood lymphocytes was measured utilizing the comet assay that quantitates breaks in DNA. Both BMI and waist circumference were positively associated with DNA damage (94). Similar findings were made in an aging study, where obesity was a stronger predictor of skeletal muscle DNA damage compared to aging (96). In preclinical work, obese zucker rats were found to have increased levels of DNA damage in several organ sites as measured by frequency of cells staining positive for phosphorylated histone variant H2AX (γH2AX), a marker of double strand breaks in DNA (97). While the mechanisms of obesity-driven DNA damage are incompletely understood, potential sources of DNA damage in obesity-related breast cancer include the elevation in breast adipose oxidative stress, which is a known inducer of DNA damage (98–100), and genotoxic effects of estrogen, produced in abundance in obese breast adipose tissue, as described in earlier sections.
Oxidative Stress
Reactive oxygen species (ROS) is a byproduct of cellular metabolism with both endogenous and exogenous sources. A major intracellular source of ROS is the mitochondria as a result of increased oxidative phosphorylation (101, 102). Mitochondrial ROS production occurs by the leakage of electrons from complexes I and III of the electron transport chain (ETC) during respiration, resulting in generation of the ROS species superoxide, a precursor for other ROS species, including hydrogen peroxide and hydroxyl. Other endogenous sources of ROS include the NOX family of NADPH oxidases and nitric acid synthases. If left uncontrolled and out of balance with antioxidants, excessive ROS production leads to oxidative stress and DNA damage which has been extensively linked to the initiation of cancer (103–105).
A number of factors secreted by obese breast adipose tissue have the ability to stimulate ROS production. Inflammatory cytokines increase ROS production in both cancer and non-cancerous cells (48, 106, 107). For example, Kastl et al. found that stimulation of murine hepatocytes with TNF-α resulted in up to a 150% increase in ROS production from mitochondria complexes I and III without causing cell death (108). TNF-α administration can also increase intracellular ROS by depleting cytosolic and mitochondrial antioxidants (109). In chondrocytes, Ansari et al. showed that stimulation with IL-1β leads to mitochondrial dysfunction and elevated levels of ROS (110). IL-6 stimulates intracellular ROS production by decreasing membrane potential leading to more electron leakage from the electron transport chain (ETC) (111).
Additionally, adipocytes store fatty acids as triacylglycerol, which is hydrolyzed and released as free fatty acids (FFA) when needed as source of fuel, such as during fasting. In obesity, FFA levels are often elevated due to enhanced lipolysis of expanding adipocytes and reduced clearance. Cells utilize fatty acids as a fuel source via the process of β-oxidation in which fatty acids are broken down to produce energy (ATP). Fatty acid β-oxidation produces substrates that are used by the tricarboxylic acid (TCA) cycle and the mitochondrial ETC resulting in ATP production. Importantly, this process results in the release of ROS. Increased mitochondrial ROS production in several disease contexts has been demonstrated to be due to oxidation of fatty acids (112, 113). Interestingly, Yamagishi et al. demonstrated that leptin also induces mitochondrial ROS production by increasing fatty acid oxidation in endothelial cells (114). Since obesity is characterized by an increase in both leptin and FFAs, β-oxidation-induced mitochondrial ROS production may be enhanced even further with excess bodyweight. The ability of leptin to drive ROS formation has been validated in other studies as well. Non-cancerous primary human mammary epithelial cells (HMEC) upregulated ROS production when stimulated with leptin in a NOX5 dependent manner, even at relatively low doses (115). One study also proposed that leptin-mediated elevation in oxidative stress is due to reduction in the activity the antioxidant enzyme PON1 (116).
Abundant evidence demonstrates the ability of obese breast adipose-derived factors, including inflammatory mediators, fatty acids, and leptin, to elevate ROS and consequently oxidative stress. While it is conceivable that this elevation in breast microenvironment oxidative stress leads to DNA damage in pre-neoplastic breast epithelial cells, studies directly demonstrating this link are lacking. Further studies are warranted to determine whether an increase in ROS in obese breast adipose tissue represents a direct mechanistic link between obesity and breast cancer initiation.
Estrogens
Estrogens stimulate DNA damage in three ways: 1) ligand binding to ERα stimulates proliferation which can lead to replication stress and accumulation of errors when the volume of replication errors overwhelms DNA damage repair capacity. Stimulation of proliferation also increases cellular respiration, which produces ROS as a byproduct of oxidative phosphorylation (4, 117, 118). 2) The metabolism of estrogen involves the redox cycling of catechol estrogen metabolites between semi-quinone and quinone states, a process that produces ROS (119–121). 3) Catechol estrogen metabolites can also directly interact with DNA to form mutagenic depurinating adducts, a form of DNA damage (122). For example, in vitro studies have demonstrated that the estrogen catechol metabolites 4-hydroxy-estradiol (4-OH-E2), 2-hydroxy-estradiol (2-OH-E2), and 2-methoxy-estradiol (2-MeO-E2) increase ROS and induce DNA damage in normal human mammary epithelial cells (123–125). Given the multiple avenues of estrogen-induced DNA damage, proficient DNA repair is crucial to limiting the propagation of damaged DNA. However, in addition to stimulating DNA damage, estrogens have also been shown to disrupt DNA repair machinery and consequently, response to damage (126, 127), further compounding the risk of DNA damage-induced mutagenesis due to excessive estrogen exposure, as in the setting of obesity.
Prevention or Reversal of Obesity-Induced Adipose Dysfunction and DNA Damage as a Modifier of Breast Cancer Risk
Targeting obesity-induced changes in breast adipose tissue has long been a proposed strategy to decrease the risk of breast cancer development. Given the genomic instability associated with obesity, it is also possible that interventions focused on decreasing obesity-induced DNA damage in breast epithelial cells could prevent chromosomal defects, including mutations, that leads to tumor formation. Interventions that target body weight and pharmacological approaches that target obesity-induced changes in hormones and signaling pathways have shown promise in decreasing the risk of breast cancer (Figure 4).
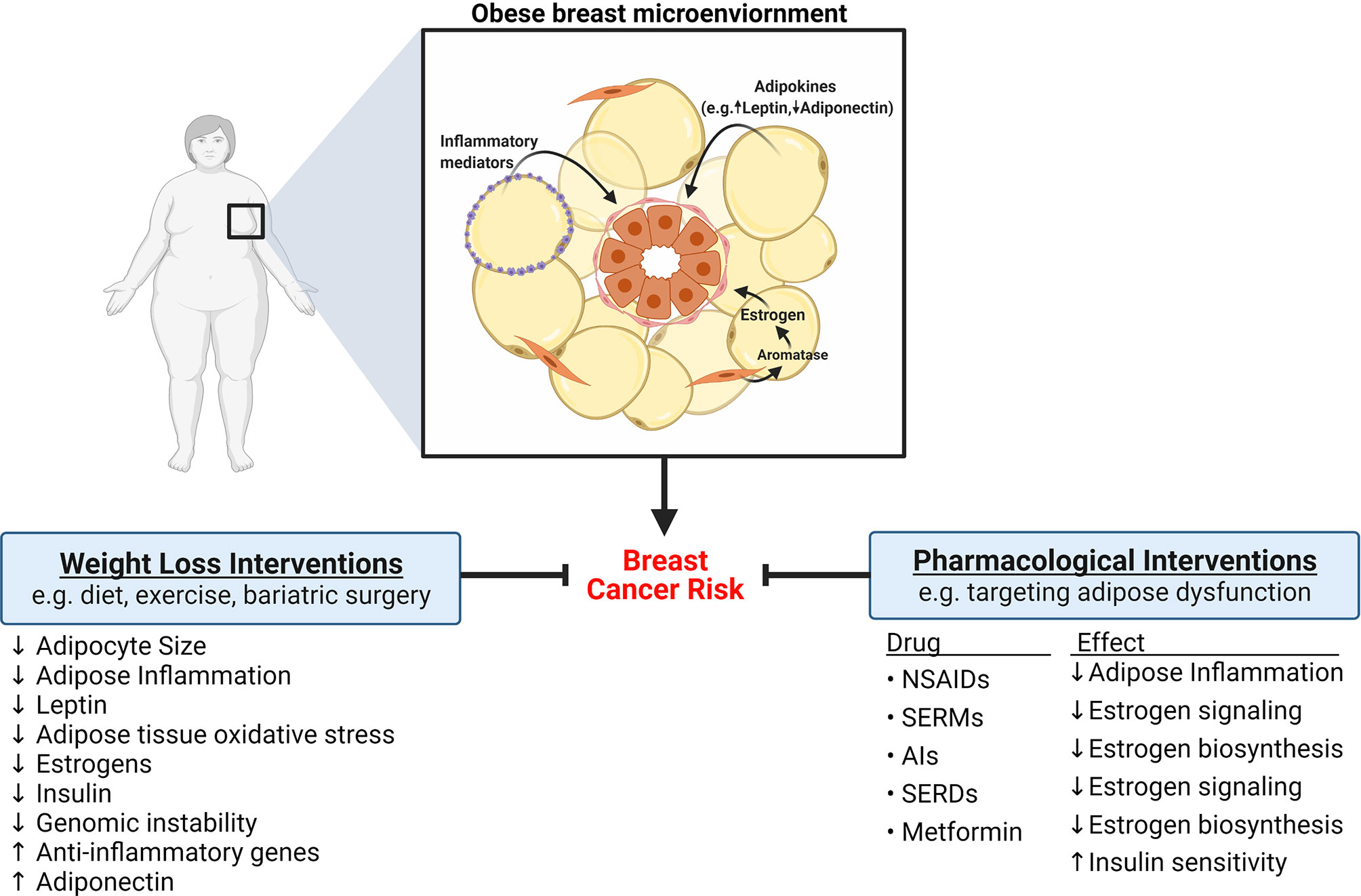
Figure 4 Strategies to reduce the risk of breast cancer development in obese women. Obesity is associated with significant changes to breast adipose tissue that increase the risk of developing breast cancer, including elevation in adipose inflammation, altered adipokine secretion, and elevation in estrogens. Ample evidence indicates that weight loss, achieved through diet, exercise, or bariatric surgery, can reverse or diminish some obesity-induced changes to adipose tissue and consequently reduce the risk of developing breast cancer. Pharmacological interventions that target adipose dysfunction have also been shown to be efficacious at reducing breast cancer risk. For example, nonsteroidal anti-inflammatory drugs (NSAIDs) decrease adipose inflammation. Drugs like selective estrogen receptor modulators (SERMS), aromatase inhibitors (AIs), and selective estrogen receptor degraders (SERDS), exert protective effects by inhibiting estrogen signaling. Metformin may also reduce risk of breast cancer through regulation of estrogen biosynthesis and improvement of insulin sensitivity.
Weight Loss Interventions
Weight loss, achieved through reduction of caloric intake, physical activity, or surgery, has been examined as a risk reduction strategy in both preclinical models and in human studies. The 2016 IARC working group’s review of body weight and cancer risk literature determined that weight loss, either through changes in lifestyle or by bariatric surgery, may reduce breast cancer risk, although the number and quality of the studies were deemed “insufficient for formal evaluation” (15). The mixed outcomes of weight loss studies can be explained in part by the wide variety of methods used to achieve weight loss, variability in degree of weight reduction, whether reduction in weight was sustained, and characteristics of study participants. Since the 2016 IARC report, a large-scale study analyzing data from more than 180,000 women across 10 prospective studies found that sustained weight loss in women over 50 was significantly associated with lower breast cancer risk compared with women who did not lose weight (128). Similar findings have been made in other studies where weight loss after menopause (129) or weight loss due to bariatric surgery (130) were associated with reduced breast cancer risk. Considerable observational data shows that exercise may also reduce breast cancer risk (131–134).
Mechanistically, beyond simply reducing the volume of dysfunctional adipose tissue, weight loss also mitigates many of the obesity-induced changes in adipose tissue that create a pro-carcinogenic microenvironment. In pre-clinical studies, weight loss after diet-induced obesity reduced mammary adipocyte size in association with decreases in adipose inflammation, aromatase expression and oxidative stress-related genes (135–137). Similarly, a number of human studies examining the impact of weight loss on inflammation have found reductions in inflammatory biomarkers including CRP, TNF, and IL-6 (138–140). In one study, weight loss induced by consumption of very low calorie diet significantly decreased inflammatory gene expression in subcutaneous adipose tissue while increasing anti-inflammatory gene expression (141). In a randomized controlled trial, Magkos et al. recently highlighted the dynamic nature of weight loss, showing that 5% weight loss was sufficient to illicit many metabolically beneficial effects, including downregulation of adipose tissue oxidative stress, yet was not sufficient to reduce adipose inflammation. Greater weight loss (16%), however, was associated with decreased adipose tissue inflammation and additional downregulation of transcriptional changes associated with oxidative stress (142). Furthermore, weight reduction has been associated with decreased levels of estradiol, leptin, and insulin along with an increase in adiponectin and steroid hormone binding globulin (SHBG), which binds free estradiol to reduce its availability in target tissues (138, 143, 144).
Pharmacological Interventions
While lifestyle interventions that reduce weight have been reported to be effective at ameliorating the adipose tissue dysfunction associated with obesity and reducing breast cancer risk, major limitations of lifestyle approaches include the difficulty in adherence to diet and/or exercise regimens and the lack of sustainability of long-term weight loss (145, 146). Alternative risk reduction strategies involve pharmacological approaches that target inflammatory and hormonal changes in the obese breast microenvironment. For example, in a study of 5,078 women in the Nashville Breast Health Study (NBHS), regular use of nonsteroidal anti-inflammatory drugs (NSAIDs) was inversely associated with breast cancer risk in overweight women (147). Preclinical studies have shown NSAIDs to be effective at reducing or reversing obesity-induced adipose inflammation as well as decreasing adipose tissue leptin levels (148, 149). Leptin has also been targeted directly by Catalano et al. who reported on a novel leptin antagonist peptide which was found to inhibit breast cancer growth in vitro and in vivo (150).
Additionally, several drugs already used in the treatment of breast cancer inhibit estrogen signaling. Tamoxifen, a selective estrogen receptor modulator (SERM), competitively binds the estrogen receptor causing a conformational change that prevents binding by estrogen in the breast (151). However, while acting as an estrogen antagonist in the breast, tamoxifen acts as an estrogen agonist in other tissues, such as the endometrium and bone (151). These agonistic effects limit the feasibility of broadly employing tamoxifen in the preventative setting in obese women to inhibit estrogen signaling due to the potential of endometrial hyperplasia and other sides effects that may outweigh the preventative benefit. Nevertheless, several randomized control trials have found that tamoxifen and raloxifene, another SERM with fewer side effects, significantly reduce the risk of invasive breast cancer development in women who are at high risk (152–154). This led to the FDA approving both drugs for use in the preventative setting, making tamoxifen and raloxifene the only two breast cancer drugs approved for chemoprevention (155). Whether the elevated risk for breast cancer development associated with obesity in postmenopausal women represents a substantial enough risk to warrant the prophylactic use of these SERMs in this population remains to be studied. Other drugs that inhibit estrogen action include fulvestrant, an estrogen receptor degrader, which is a second line therapy approved for advanced ER+ breast cancer, and aromatase inhibitors (AIs). AIs are a commonly prescribed first line treatment of postmenopausal ER+ breast cancer, used with the goal of lowering levels of estrogens by preventing their synthesis from androgen precursors. Although it has been reported that AIs may lower the risk of breast cancer development in high-risk women, they are currently not approved for use in the prevention setting, due in part to significant side effects like osteoporosis and arthralgia, although additional prevention trials with AIs are ongoing (155, 156).
A more tenable strategy to reduce estrogen in the obese breast adipose microenvironment may be through use of metformin, a low cost, well-tolerated antidiabetic drug that has emerged as an intriguing drug candidate for breast cancer prevention. Although primarily used to reduce gluconeogenesis and increase insulin sensitivity in patients with type 2 diabetes, some, but not all, observational studies have reported lower incidence of breast cancer among diabetic patients taking metformin (157–159). While improving insulin sensitivity is likely to contribute to the decreased breast cancer risk, another important function of metformin in the context of obesity-associated breast cancer is its ability to inhibit aromatase expression in breast ASCs (160, 161). A recent preclinical study utilizing a model of postmenopausal carcinogen-induced breast cancer in obese rats found that metformin inhibited ASC aromatase expression in association with a reduction in size of existing mammary tumors and prevention of new tumor formation (162). Other studies have also supported an anti-tumor function of metformin, showing that treatment of several breast cancer cell lines inhibits growth, decreases colony formation, and induces cell cycle arrest (163–165). These actions are dependent on metformin-induced activation of AMP-activated protein kinase (AMPK), an important regulator of energy homeostasis (160, 164). Metformin may be especially beneficial in obese patients where AMPK activation also improves various aspects of obesity-associated adipose dysfunction, including decreasing adipose inflammation (166).
Targeting Obesity-Induced DNA Damage
As reviewed above, many of the factors found to be reduced after weight loss or by pharmacological intervention, including estrogen, inflammatory mediators, and leptin have roles in promoting DNA damage and inhibiting repair processes. Therefore, it is possible that reduced DNA damage and improved DNA repair capacity represents an additional mechanistic link between the therapeutics that reduces these factors and lower risk of breast cancer. In a randomized controlled trial where participants were placed on a 6-month calorie restriction regimen, a significant decrease in DNA damage measured in blood by the comet assay was reported (167). Similar reductions in DNA damage have also been reported in blood and in lymphocytes of morbidly obese patients after weight loss surgery (168–170). Setayesh et al. showed in a mouse model of diet-induced obesity that weight loss significantly decreased DNA damage in the colon, liver, and testes in association with reductions in circulating levels of IL-6, MCP-1, leptin, and TNF and increased adiponectin (171). Interestingly, in a study of 220 healthy volunteers engaging in varying degrees of physical activity, physical activity at any intensity was associated with increased leukocyte repair capacity (172). To date, no studies have looked at the effects of weight loss on DNA damage or DNA repair capacity in normal breast epithelial cells. However, it stands to reason that reducing factors that have known genotoxic effects in the obese breast adipose microenvironment may be an effective strategy to prevent or reduce DNA damage in pre-neoplastic breast epithelial cells and consequently, mitigate the risk of breast tumorigenesis.
Discussion
Abundant evidence demonstrates the importance of the crosstalk between obese breast adipose tissue and neighboring normal and neoplastic breast epithelial cells. Excess caloric intake and subsequent weight gain initiates a cascade of effects in breast adipose tissue beginning with adipocyte hypertrophy and progressing to oxidative stress, chronic inflammation, altered adipokine secretion, and elevated estrogen production by ASCs. While these are major, well-established changes in the breast microenvironment, a wide variety of additional changes occur in the setting of obesity, which can also promote breast cancer and have been reviewed by others, for example extracellular matrix stiffening, immune cell dysfunction, and dysregulation of insulin. The current review highlights the effects of breast adipose-derived estrogen, inflammatory mediators, leptin and decreased adiponectin on two aspects of breast cancer: initial events leading to carcinogenesis and progression. These factors have the ability to initiate breast cancer through regulation of oxidative stress, DNA damage, and DNA repair response. Further large-scale studies are warranted to elucidate whether there is a direct mechanistic link between adipose derived factors and breast tumorigenesis through stimulation of genomic instability. Promotion of breast cancer growth occurs through regulation of pro-proliferative and anti-apoptotic transcriptional programs in cancer cells and through effects on microenvironment oxidative stress. Targeting obese breast adipose dysfunction with weight reduction or pharmacological approaches has so far shown promise at decreasing breast cancer risk. With obesity rates expected to reach over 50% in the next 10 years in the U.S., additional studies aimed at disrupting the crosstalk between obese breast adipose tissue and neighboring breast epithelial cells may elucidate novel, effective prevention strategies for obese women at elevated risk for breast cancer development.
Author Contributions
PB prepared first draft, KB reviewed and edited. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH R01 CA215797, Anne Moore Breast Cancer Research Fund, and NIH 5 F31 CA236306-02.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
All graphics were created with BioRender.com.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J Clin (2018) 68(6):394–424. doi: 10.3322/caac.21492
2. Centers for Disease Control and Prevention. Breast Cancer Statistics. U.S. Cancer Statistics Data Visualizations Tool. U.S. Department of Health and Human Services (2020).
4. Yager JD, Davidson NE. Estrogen Carcinogenesis in Breast Cancer. New Engl J Med (2006) 354(3):270–82. doi: 10.1056/NEJMra050776
5. Samavat H, Kurzer MS. Estrogen metabolism and breast cancer. Cancer Lett (2015) 356(2):231–43. doi: 10.1016/j.canlet.2014.04.018
6. Tamimi RM, Spiegelman D, Smith-Warner SA, Wang M, Pazaris M, Willett WC, et al. Population Attributable Risk of Modifiable and Nonmodifiable Breast Cancer Risk Factors in Postmenopausal Breast Cancer. Am J Epidemiol (2016) 184(12):884–93. doi: 10.1093/aje/kww145
7. Zhang SM, Lee I-M, Manson JE, Cook NR, Willett WC, Buring JE. Alcohol Consumption and Breast Cancer Risk in the Women’s Health Study. Am J Epidemiol (2007) 165(6):667–76. doi: 10.1093/aje/kwk054
8. NCD-RisC. Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19·2 million participants. Lancet (2016) 387(10026):1377–96. doi: 10.1016/S0140-6736(16)30054-X
9. Finkelstein EA, Khavjou OA, Thompson H, Trogdon JG, Pan L, Sherry B, et al. Obesity and Severe Obesity Forecasts Through 2030. Am J Prev Med (2012) 42(6):563–70. doi: 10.1016/j.amepre.2011.10.026
10. Hales CM, Carroll MD, Fryar CD, Ogden CL. Prevalence of Obesity and Severe Obesity Among Adults: United States, 2017–2018. In: . Services USDoHaH. Hyattsville, MD: National Center for Health Statistics (2020).
11. Schoemaker MJ, Nichols HB, Wright LB, Brook MN, Jones ME, O’Brien KM, et al. Association of Body Mass Index and Age With Subsequent Breast Cancer Risk in Premenopausal Women. JAMA Oncol (2018) 4(11):e181771. doi: 10.1001/jamaoncol.2018.1771
12. Weiderpass E, Braaten T, Magnusson C, Kumle M, Vainio H, Lund E, et al. A prospective study of body size in different periods of life and risk of premenopausal breast cancer. Cancer Epidemiol Biomarkers Prev (2004) 13(7):1121–7.
13. Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet (2008) 371(9612):569–78. doi: 10.1016/S0140-6736(08)60269-X
14. Arnold M, Pandeya N, Byrnes G, Renehan AG, Stevens GA, Ezzati M, et al. Global burden of cancer attributable to high body-mass index in 2012: a population-based study. Lancet Oncol (2015) 16(1):36–46. doi: 10.1016/S1470-2045(14)71123-4
15. Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, Straif K. Body Fatness and Cancer — Viewpoint of the IARC Working Group. New Engl J Med (2016) 375(8):794–8. doi: 10.1056/NEJMsr1606602
16. Wu J, Boström P, Sparks M, Lauren, Ye L, Choi H, et al. Beige Adipocytes Are a Distinct Type of Thermogenic Fat Cell in Mouse and Human. Cell (2012) 150(2):366–76. doi: 10.1016/j.cell.2012.05.016
17. Watson CJ, Khaled WT. Mammary development in the embryo and adult: a journey of morphogenesis and commitment. Development (2008) 135(6):995–1003. doi: 10.1242/dev.005439
18. Cinti S. Pink Adipocytes. Trends Endocrinol Metab (2018) 29(9):651–66. doi: 10.1016/j.tem.2018.05.007
19. Marino N, German R, Rao X, Simpson E, Liu S, Wan J, et al. Upregulation of lipid metabolism genes in the breast prior to cancer diagnosis. NPJ Breast Cancer (2020) 6(1):50. doi: 10.1038/s41523-020-00191-8
20. Brisken C, O’Malley B. Hormone Action in the Mammary Gland. Cold Spring Harbor Perspect Biol (2010) 2(12):a003178–a. doi: 10.1101/cshperspect.a003178
21. Monteiro R, Teixeira D, Calhau C. Estrogen signaling in metabolic inflammation. Mediators Inflamm (2014) 2014:615917. doi: 10.1155/2014/615917
22. Bhardwaj P, Au CC, Benito-Martin A, Ladumor H, Oshchepkova S, Moges R, et al. Estrogens and breast cancer: Mechanisms involved in obesity-related development, growth and progression. J Steroid Biochem Mol Biol (2019) 189:161–70. doi: 10.1016/j.jsbmb.2019.03.002
23. Gérard C, Brown KA. Obesity and breast cancer – Role of estrogens and the molecular underpinnings of aromatase regulation in breast adipose tissue. Mol Cell Endocrinol (2018) 466:15–30. doi: 10.1016/j.mce.2017.09.014
24. Group TEHABCC. Endogenous Sex Hormones and Breast Cancer in Postmenopausal Women: Reanalysis of Nine Prospective Studies. Cancer Spectr Knowl Environ (2002) 94(8):606–16. doi: 10.1093/jnci/94.8.606
25. Simpson ER. Sources of estrogen and their importance. J Steroid Biochem Mol Biol (2003) 86(3-5):225–30. doi: 10.1016/S0960-0760(03)00360-1
26. Bulun SE, Chen D, Moy I, Brooks DC, Zhao H. Aromatase, breast cancer and obesity: a complex interaction. Trends Endocrinol Metab (2012) 23(2):83–9. doi: 10.1016/j.tem.2011.10.003
27. Price T, Aitken J, Head J, Mahendroo M, Means G, Simpson E. Determination of aromatase cytochrome P450 messenger ribonucleic acid in human breast tissue by competitive polymerase chain reaction amplification. J Clin Endocrinol Metab (1992) 74(6):1247–52. doi: 10.1210/jcem.74.6.1592866
28. Wang X, Simpson ER, Brown KA. Aromatase overexpression in dysfunctional adipose tissue links obesity to postmenopausal breast cancer. J Steroid Biochem Mol Biol (2015) 153:35–44. doi: 10.1016/j.jsbmb.2015.07.008
29. Hillers LE, D’Amato JV, Chamberlin T, Paderta G, Arendt LM. Obesity-Activated Adipose-Derived Stromal Cells Promote Breast Cancer Growth and Invasion. Neoplasia (2018) 20(11):1161–74. doi: 10.1016/j.neo.2018.09.004
30. Zhang Y, Daquinag AC, Amaya-Manzanares F, Sirin O, Tseng C, Kolonin MG. Stromal Progenitor Cells from Endogenous Adipose Tissue Contribute to Pericytes and Adipocytes That Populate the Tumor Microenvironment. Cancer Res (2012) 72(20):5198–208. doi: 10.1158/0008-5472.CAN-12-0294
31. Morgan MM, Arendt LM, Alarid ET, Beebe DJ, Johnson BP. Mammary adipose stromal cells derived from obese women reduce sensitivity to the aromatase inhibitor anastrazole in an organotypic breast model. FASEB J (2019) 33(7):8623–33. doi: 10.1096/fj.201802347RRR
32. Bulun SE, Lin Z, Imir G, Amin S, Demura M, Yilmaz B, et al. Regulation of Aromatase Expression in Estrogen-Responsive Breast and Uterine Disease: From Bench to Treatment. Pharmacol Rev (2005) 57(3):359–83. doi: 10.1124/pr.57.3.6
33. Samarajeewa NU, Yang F, Docanto MM, Sakurai M, Mcnamara KM, Sasano H, et al. HIF-1α stimulates aromatase expression driven by prostaglandin E2 in breast adipose stroma. Breast Cancer Res (2013) 15(2):R30. doi: 10.1186/bcr3410
34. Docanto MM, Yang F, Callaghan B, Au CC, Ragavan R, Wang X, et al. Ghrelin and des-acyl ghrelin inhibit aromatase expression and activity in human adipose stromal cells: suppression of cAMP as a possible mechanism. Breast Cancer Res Treat (2014) 147(1):193–201. doi: 10.1007/s10549-014-3060-1
35. Jeon S-M. Regulation and function of AMPK in physiology and diseases. Exp Mol Med (2016) 48(7):e245–e. doi: 10.1038/emm.2016.81
36. Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, et al. Hypoxia-Inducible Factor 1α Induces Fibrosis and Insulin Resistance in White Adipose Tissue. Mol Cell Biol (2009) 29(16):4467–83. doi: 10.1128/MCB.00192-09
37. Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, et al. Adipose Tissue Hypoxia in Obesity and Its Impact on Adipocytokine Dysregulation. Diabetes (2007) 56(4):901–11. doi: 10.2337/db06-0911
38. Weisberg SP, Mccann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest (2003) 112(12):1796–808. doi: 10.1172/JCI200319246
39. Wang L, Zhao R-P, Song X-Y, Wu W-F. Targeting ERβ in Macrophage Reduces Crown-like Structures in Adipose Tissue by Inhibiting Osteopontin and HIF-1α. Sci Rep (2019) 9(1):15762. doi: 10.1038/s41598-019-52265-8
40. Vaysse C, Lømo J, Garred Ø, Fjeldheim F, Lofteroed T, Schlichting E, et al. Inflammation of mammary adipose tissue occurs in overweight and obese patients exhibiting early-stage breast cancer. NPJ Breast Cancer (2017) 3(1):19. doi: 10.1038/s41523-017-0015-9
41. Olefsky JM, Glass CK. Macrophages, Inflammation, and Insulin Resistance. Annu Rev Physiol (2010) 72(1):219–46. doi: 10.1146/annurev-physiol-021909-135846
42. Sun X, Casbas-Hernandez P, Bigelow C, Makowski L, Joseph Jerry D, Smith Schneider S, et al. Normal breast tissue of obese women is enriched for macrophage markers and macrophage-associated gene expression. Breast Cancer Res Treat (2012) 131(3):1003–12. doi: 10.1007/s10549-011-1789-3
43. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest (2007) 117(1):175–84. doi: 10.1172/JCI29881
44. Hotamisligil GS. Inflammation and metabolic disorders. Nature (2006) 444(7121):860–7. doi: 10.1038/nature05485
45. Chu DT, Phuong TNT, Tien NLB, Tran DK, Nguyen TT, Thanh VV, et al. The Effects of Adipocytes on the Regulation of Breast Cancer in the Tumor Microenvironment: An Update. Cells (2019) 8(8):857. doi: 10.3390/cells8080857
46. Picon-Ruiz M, Pan C, Drews-Elger K, Jang K, Besser AH, Zhao D, et al. Interactions between Adipocytes and Breast Cancer Cells Stimulate Cytokine Production and Drive Src/Sox2/miR-302b–Mediated Malignant Progression. Cancer Res (2016) 76(2):491–504. doi: 10.1158/0008-5472.CAN-15-0927
47. Gilbert CA, Slingerland JM. Cytokines, Obesity, and Cancer: New Insights on Mechanisms Linking Obesity to Cancer Risk and Progression. Annu Rev Med (2013) 64(1):45–57. doi: 10.1146/annurev-med-121211-091527
48. Grivennikov SI, Karin M. Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Ann Rheum Dis (2011) 70(Suppl 1):i104–i8. doi: 10.1136/ard.2010.140145
49. Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res (2011) 71(7):2455–65. doi: 10.1158/0008-5472.CAN-10-3323
50. Esquivel-Velázquez M, Ostoa-Saloma P, Palacios-Arreola MI, Nava-Castro KE, Castro JI, Morales-Montor J. The Role of Cytokines in Breast Cancer Development and Progression. J Interferon Cytokine Res (2015) 35(1):1–16. doi: 10.1089/jir.2014.0026
51. Walter M, Liang S, Ghosh S, Hornsby PJ, Li R. Interleukin 6 secreted from adipose stromal cells promotes migration and invasion of breast cancer cells. Oncogene (2009) 28(30):2745–55. doi: 10.1038/onc.2009.130
52. Hwang DH, Kim J-A, Lee JY. Mechanisms for the activation of Toll-like receptor 2/4 by saturated fatty acids and inhibition by docosahexaenoic acid. Eur J Pharmacol (2016) 785:24–35. doi: 10.1016/j.ejphar.2016.04.024
53. Rogero M, Calder P. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients (2018) 10(4):432. doi: 10.3390/nu10040432
54. Mckernan K, Varghese M, Patel R, Singer K. Role of TLR4 in the induction of inflammatory changes in adipocytes and macrophages. Adipocyte (2020) 9(1):212–22. doi: 10.1080/21623945.2020.1760674
55. Suganami T, Mieda T, Itoh M, Shimoda Y, Kamei Y, Ogawa Y. Attenuation of obesity-induced adipose tissue inflammation in C3H/HeJ mice carrying a Toll-like receptor 4 mutation. Biochem Biophys Res Commun (2007) 354(1):45–9. doi: 10.1016/j.bbrc.2006.12.190
56. Kim K-A, Gu W, Lee I-A, Joh E-H, Kim D-H. High Fat Diet-Induced Gut Microbiota Exacerbates Inflammation and Obesity in Mice via the TLR4 Signaling Pathway. PloS One (2012) 7(10):e47713. doi: 10.1371/journal.pone.0047713
57. Davis JE, Braucher DR, Walker-Daniels J, Spurlock ME. Absence of Tlr2 protects against high-fat diet-induced inflammation and results in greater insulin-stimulated glucose transport in cultured adipocytes. J Nutr Biochem (2011) 22(2):136–41. doi: 10.1016/j.jnutbio.2009.12.008
58. Nishimoto S, Fukuda D, Higashikuni Y, Tanaka K, Hirata Y, Murata C, et al. Obesity-induced DNA released from adipocytes stimulates chronic adipose tissue inflammation and insulin resistance. Sci Adv (2016) 2(3):e1501332. doi: 10.1126/sciadv.1501332
59. Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, et al. Leptin levels in human and rodent: Measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med (1995) 1(11):1155–61. doi: 10.1038/nm1195-1155
60. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature (1994) 372(6505):425–32. doi: 10.1038/372425a0
61. Stern H,J, Rutkowski M,J, Scherer E,P. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis through Adipose Tissue Crosstalk. Cell Metab (2016) 23(5):770–84. doi: 10.1016/j.cmet.2016.04.011
62. Jardé T, Perrier S, Vasson M-P, Caldefie-Chézet F. Molecular mechanisms of leptin and adiponectin in breast cancer. Eur J Cancer (2011) 47(1):33–43. doi: 10.1016/j.ejca.2010.09.005
63. Niu J, Jiang L, Guo W, Shao L, Liu Y, Wang L. The Association between Leptin Level and Breast Cancer: A Meta-Analysis. PloS One (2013) 8(6):e67349. doi: 10.1371/journal.pone.0067349
64. Harris HR, Tworoger SS, Hankinson SE, Rosner BA, Michels KB. Plasma Leptin Levels and Risk of Breast Cancer in Premenopausal Women. Cancer Prev Res (2011) 4(9):1449–56. doi: 10.1158/1940-6207.CAPR-11-0125
65. Wu M-H, Chou Y-C, Chou W-Y, Hsu G-C, Chu C-H, Yu C-P, et al. Circulating levels of leptin, adiposity and breast cancer risk. Br J Cancer (2009) 100(4):578–82. doi: 10.1038/sj.bjc.6604913
66. Gonzalez RR, Cherfils S, Escobar M, Yoo JH, Carino C, Styer AK, et al. Leptin Signaling Promotes the Growth of Mammary Tumors and Increases the Expression of Vascular Endothelial Growth Factor (VEGF) and Its Receptor Type Two (VEGF-R2). J Biol Chem (2006) 281(36):26320–8. doi: 10.1074/jbc.M601991200
67. Sánchez-Jiménez F, Pérez-Pérez A, De La Cruz-Merino L, Sánchez-Margalet V. Obesity and Breast Cancer: Role of Leptin. Front Oncol (2019) 9:596. doi: 10.3389/fonc.2019.00596
68. Cava AL, Matarese G. The weight of leptin in immunity. Nat Rev Immunol (2004) 4(5):371–9. doi: 10.1038/nri1350
69. Haque I, Ghosh A, Acup S, Banerjee S, Dhar K, Ray A, et al. Leptin-induced ER-α-positive breast cancer cell viability and migration is mediated by suppressing CCN5-signaling via activating JAK/AKT/STAT-pathway. BMC Cancer (2018) 18(1):99. doi: 10.1186/s12885-018-3993-6
70. Saxena NK, Vertino PM, Anania FA, Sharma D. Leptin-induced Growth Stimulation of Breast Cancer Cells Involves Recruitment of Histone Acetyltransferases and Mediator Complex toCYCLIN D1Promoter via Activation of Stat3. J Biol Chem (2007) 282(18):13316–25. doi: 10.1074/jbc.M609798200
71. Perera CN, Chin HG, Duru N, Camarillo IG. Leptin-regulated gene expression in MCF-7 breast cancer cells: mechanistic insights into leptin-regulated mammary tumor growth and progression. J Endocrinol (2008) 199(2):221–33. doi: 10.1677/JOE-08-0215
72. Catalano S, Mauro L, Marsico S, Giordano C, Rizza P, Rago V, et al. Leptin Induces, via ERK1/ERK2 Signal, Functional Activation of Estrogen Receptor α in MCF-7 Cells. J Biol Chem (2004) 279(19):19908–15. doi: 10.1074/jbc.M313191200
73. Brown KA, Mcinnes KJ, Hunger NI, Oakhill JS, Steinberg GR, Simpson ER. Subcellular Localization of Cyclic AMP-Responsive Element Binding Protein-Regulated Transcription Coactivator 2 Provides a Link between Obesity and Breast Cancer in Postmenopausal Women. Cancer Res (2009) 69(13):5392–9. doi: 10.1158/0008-5472.CAN-09-0108
74. Zahid H, Subbaramaiah K, Iyengar NM, Zhou XK, Chen I-C, Bhardwaj P, et al. Leptin regulation of the p53-HIF1α/PKM2-aromatase axis in breast adipose stromal cells: a novel mechanism for the obesity–breast cancer link. Int J Obes (2018) 42(4):711–20. doi: 10.1038/ijo.2017.273
75. Catalano S, Marsico S, Giordano C, Mauro L, Rizza P, Panno ML, et al. Leptin Enhances, via AP-1, Expression of Aromatase in the MCF-7 Cell Line. J Biol Chem (2003) 278(31):28668–76. doi: 10.1074/jbc.M301695200
76. Garofalo C. Increased Expression of Leptin and the Leptin Receptor as a Marker of Breast Cancer Progression: Possible Role of Obesity-Related Stimuli. Clin Cancer Res (2006) 12(5):1447–53. doi: 10.1158/1078-0432.CCR-05-1913
77. Strong AL, Strong TA, Rhodes LV, Semon JA, Zhang X, Shi Z, et al. Obesity associated alterations in the biology of adipose stem cells mediate enhanced tumorigenesis by estrogen dependent pathways. Breast Cancer Res (2013) 15(5):R102. doi: 10.1186/bcr3569
78. Strong AL, Ohlstein JF, Biagas BA, Rhodes LV, Pei DT, Tucker HA, et al. Leptin produced by obese adipose stromal/stem cells enhances proliferation and metastasis of estrogen receptor positive breast cancers. Breast Cancer Res (2015) 17(1):112. doi: 10.1186/s13058-015-0622-z
79. Tenvooren I, Jenks MZ, Rashid H, Cook KL, Muhlemann JK, Sistrunk C, et al. Elevated leptin disrupts epithelial polarity and promotes premalignant alterations in the mammary gland. Oncogene (2019) 38(20):3855–70. doi: 10.1038/s41388-019-0687-8
80. Olea-Flores M, Zuñiga-Eulogio M, Tacuba-Saavedra A, Bueno-Salgado M, Sánchez-Carvajal A, Vargas-Santiago Y, et al. Leptin Promotes Expression of EMT-Related Transcription Factors and Invasion in a Src and FAK-Dependent Pathway in MCF10A Mammary Epithelial Cells. Cells (2019) 8(10):1133. doi: 10.3390/cells8101133
81. Hung J, Mcquillan BM, Thompson PL, Beilby JP. Circulating adiponectin levels associate with inflammatory markers, insulin resistance and metabolic syndrome independent of obesity. Int J Obes (2008) 32(5):772–9. doi: 10.1038/sj.ijo.0803793
82. Miyoshi Y, Funahashi T, Kihara S, Taguchi T, Tamaki Y, Matsuzawa Y, et al. Association of serum adiponectin levels with breast cancer risk. Clin Cancer Res (2003) 9(15):5699–704.
83. Ye J, Jia J, Dong S, Zhang C, Yu S, Li L, et al. Circulating adiponectin levels and the risk of breast cancer. Eur J Cancer Prev (2014) 23(3):158–65. doi: 10.1097/CEJ.0b013e328364f293
84. Kang JH, Lee YY, Yu BY, Yang B-S, Cho K-H, Yoon DK, et al. Adiponectin induces growth arrest and apoptosis of MDA-MB-231 breast cancer cell. Arch Pharmacal Res (2005) 28(11):1263–9. doi: 10.1007/BF02978210
85. Dieudonne M-N, Bussiere M, Dos Santos E, Leneveu M-C, Giudicelli Y, Pecquery R. Adiponectin mediates antiproliferative and apoptotic responses in human MCF7 breast cancer cells. Biochem Biophys Res Commun (2006) 345(1):271–9. doi: 10.1016/j.bbrc.2006.04.076
86. Chung SJ, Nagaraju GP, Nagalingam A, Muniraj N, Kuppusamy P, Walker A, et al. ADIPOQ/adiponectin induces cytotoxic autophagy in breast cancer cells through STK11/LKB1-mediated activation of the AMPK-ULK1 axis. Autophagy (2017) 13(8):1386–403. doi: 10.1080/15548627.2017.1332565
87. Kumada M, Kihara S, Ouchi N, Kobayashi H, Okamoto Y, Ohashi K, et al. Adiponectin Specifically Increased Tissue Inhibitor of Metalloproteinase-1 Through Interleukin-10 Expression in Human Macrophages. Circulation (2004) 109(17):2046–9. doi: 10.1161/01.CIR.0000127953.98131.ED
88. Ouchi N, Walsh K. Adiponectin as an anti-inflammatory factor. Clin Chim Acta (2007) 380(1-2):24–30. doi: 10.1016/j.cca.2007.01.026
89. Lihn AS, Pedersen SB, Richelsen B. Adiponectin: action, regulation and association to insulin sensitivity. Obes Rev (2005) 6(1):13–21. doi: 10.1111/j.1467-789X.2005.00159.x
90. Ruan H, Dong LQ. Adiponectin signaling and function in insulin target tissues. J Mol Cell Biol (2016) 8(2):101–9. doi: 10.1093/jmcb/mjw014
91. Setayesh T, Nersesyan A, Mišík M, Ferk F, Langie S, Andrade VM, et al. Impact of obesity and overweight on DNA stability: Few facts and many hypotheses. Mutat Res/Rev Mutat Res (2018) 777:64–91. doi: 10.1016/j.mrrev.2018.07.001
92. Włodarczyk M, Nowicka G. Obesity DNA. Damage, and Development of Obesity-Related Diseases. Int J Mol Sci (2019) 20(5):1146. doi: 10.3390/ijms20051146
93. Lee SC, Chan JC. Evidence for DNA Damage as a Biological Link Between Diabetes and Cancer. Chin Med J (2015) 128(11):1543–8. doi: 10.4103/0366-6999.157693
94. Talon LDC, Ferraz APCR, Pierine DT, Minatel IO, Garcia JL, Nogueira V, et al. Increased BMI and Waist Circumference are Related to Increased DNA Damage in Women with Overweight and Metabolic Syndrome. Asian J Adv Res Rep (2020) 13(2):43–52. doi: 10.9734/ajarr/2020/v13i230306
95. Sieber O, Heinimann K, Tomlinson I. Genomic stability and tumorigenesis. Semin Cancer Biol (2005) 15(1):61–6. doi: 10.1016/j.semcancer.2004.09.005
96. Dungan CM, Peck BD, Walton RG, Huang Z, Bamman MM, Kern PA, et al. In vivo analysis of γH2AX+ cells in skeletal muscle from aged and obese humans. FASEB J (2020) 34(5):7018–35. doi: 10.1096/fj.202000111RR
97. Azzarà A, Chiaramonte A, Filomeni E, Pinto B, Mazzoni S, Piaggi S, et al. Increased level of DNA damage in some organs of obese Zucker rats by γ-H2AX analysis. Environ Mol Mutagen (2017) 58(7):477–84. doi: 10.1002/em.22115
98. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest (2004) 114(12):1752–61. doi: 10.1172/JCI21625
99. Crujeiras AB, Díaz-Lagares A, Carreira MC, Amil M, Casanueva FF. Oxidative stress associated to dysfunctional adipose tissue: a potential link between obesity, type 2 diabetes mellitus and breast cancer. Free Radical Res (2013) 47(4):243–56. doi: 10.3109/10715762.2013.772604
100. Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J (2003) 17(10):1195–214. doi: 10.1096/fj.02-0752rev
101. Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochem (Moscow) (2005) 70(2):200–14. doi: 10.1007/s10541-005-0102-7
102. Adam-Vizi V, Chinopoulos C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol Sci (2006) 27(12):639–45. doi: 10.1016/j.tips.2006.10.005
103. Liou G-Y, Storz P. Reactive oxygen species in cancer. Free Radical Res (2010) 44(5):479–96. doi: 10.3109/10715761003667554
104. Mahalingaiah PKS, Singh KP. Chronic Oxidative Stress Increases Growth and Tumorigenic Potential of MCF-7 Breast Cancer Cells. PloS One (2014) 9(1):e87371. doi: 10.1371/journal.pone.0087371
105. Kryston TB, Georgiev AB, Pissis P, Georgakilas AG. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat Res/Fundam Mol Mech Mutagen (2011) 711(1-2):193–201. doi: 10.1016/j.mrfmmm.2010.12.016
106. Blaser H, Dostert C, Mak TW, Brenner D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol (2016) 26(4):249–61. doi: 10.1016/j.tcb.2015.12.002
107. Aggarwal V, Tuli H, Varol A, Thakral F, Yerer M, Sak K, et al. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules (2019) 9(11):735. doi: 10.3390/biom9110735
108. Kastl L, Sauer SW, Ruppert T, Beissbarth T, Becker MS, Süss D, et al. TNF-α mediates mitochondrial uncoupling and enhances ROS-dependent cell migrationviaNF-κB activation in liver cells. FEBS Lett (2014) 588(1):175–83. doi: 10.1016/j.febslet.2013.11.033
109. Mariappan N, Soorappan RN, Haque M, Sriramula S, Francis J. TNF-α-induced mitochondrial oxidative stress and cardiac dysfunction: restoration by superoxide dismutase mimetic Tempol. Am J Physiol-Heart Circulatory Physiol (2007) 293(5):H2726–H37. doi: 10.1152/ajpheart.00376.2007
110. Ansari MY, Khan NM, Ahmad I, Haqqi TM. Parkin clearance of dysfunctional mitochondria regulates ROS levels and increases survival of human chondrocytes. Osteoarthr Cartil (2018) 26(8):1087–97. doi: 10.1016/j.joca.2017.07.020
111. Ji C, Chen X, Gao C, Jiao L, Wang J, Xu G, et al. IL-6 induces lipolysis and mitochondrial dysfunction, but does not affect insulin-mediated glucose transport in 3T3-L1 adipocytes. J Bioenerg Biomembr (2011) 43(4):367–75. doi: 10.1007/s10863-011-9361-8
112. Wang C, Shao L, Pan C, Ye J, Ding Z, Wu J, et al. Elevated level of mitochondrial reactive oxygen species via fatty acid β-oxidation in cancer stem cells promotes cancer metastasis by inducing epithelial–mesenchymal transition. Stem Cell Res Ther (2019) 10(1):175. doi: 10.1186/s13287-019-1265-2
113. Tumova J, Andel M, Trnka J. Excess of free fatty acids as a cause of metabolic dysfunction in skeletal muscle. Physiol Res (2016) 65(2):193–207. doi: 10.33549/physiolres.932993
114. Yamagishi S-I, Edelstein D, Du X-L, Kaneda Y, Guzmán M, Brownlee M. Leptin Induces Mitochondrial Superoxide Production and Monocyte Chemoattractant Protein-1 Expression in Aortic Endothelial Cells by Increasing Fatty Acid Oxidation via Protein Kinase A. J Biol Chem (2001) 276(27):25096–100. doi: 10.1074/jbc.M007383200
115. Mahbouli S, Der Vartanian A, Ortega S, Rougé S, Vasson M-P, Rossary A. Leptin induces ROS via NOX5 in healthy and neoplastic mammary epithelial cells. Oncol Rep (2017) 38(5):3254–64. doi: 10.3892/or.2017.6009
116. Beltowski J. Leptin decreases plasma paraoxonase 1 (PON1) activity and induces oxidative stress: the possible novel mechanism for proatherogenic effect of chronic hyperleptinemia. Atherosclerosis (2003) 170(1):21–9. doi: 10.1016/S0021-9150(03)00236-3
117. Tubbs A, Nussenzweig A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell (2017) 168(4):644–56. doi: 10.1016/j.cell.2017.01.002
118. Williamson LM, Lees-Miller SP. Estrogen receptor α-mediated transcription induces cell cycle-dependent DNA double-strand breaks. Carcinogenesis (2011) 32(3):279–85. doi: 10.1093/carcin/bgq255
119. Okoh V, Deoraj A, Roy D. Estrogen-induced reactive oxygen species-mediated signalings contribute to breast cancer. Biochim Biophys Acta (BBA) - Rev Cancer (2011) 1815(1):115–33. doi: 10.1016/j.bbcan.2010.10.005
120. Felty Q, Xiong W-C, Sun D, Sarkar S, Singh KP, Parkash J, et al. Estrogen-Induced Mitochondrial Reactive Oxygen Species as Signal-Transducing Messengers†. Biochemistry (2005) 44(18):6900–9. doi: 10.1021/bi047629p
121. Roy D, Cai Q, Felty Q, Narayan S. Estrogen-Induced Generation of Reactive Oxygen and Nitrogen Species, Gene Damage, and Estrogen-Dependent Cancers. J Toxicol Environ Health Part B (2007) 10(4):235–57. doi: 10.1080/15287390600974924
122. Santen RJ, Yue W, Wang J-P. Estrogen metabolites and breast cancer. Steroids (2015) 99:61–6. doi: 10.1016/j.steroids.2014.08.003
123. Savage KI, Matchett KB, Barros EM, Cooper KM, Irwin GW, Gorski JJ, et al. BRCA1 Deficiency Exacerbates Estrogen-Induced DNA Damage and Genomic Instability. Cancer Res (2014) 74(10):2773–84. doi: 10.1158/0008-5472.CAN-13-2611
124. Okoh VO, Felty Q, Parkash J, Poppiti R, Roy D. Reactive Oxygen Species via Redox Signaling to PI3K/AKT Pathway Contribute to the Malignant Growth of 4-Hydroxy Estradiol-Transformed Mammary Epithelial Cells. PloS One (2013) 8(2):e54206. doi: 10.1371/journal.pone.0054206
125. Fussell KC, Udasin RG, Smith PJS, Gallo MA, Laskin JD. Catechol metabolites of endogenous estrogens induce redox cycling and generate reactive oxygen species in breast epithelial cells. Carcinogenesis (2011) 32(8):1285–93. doi: 10.1093/carcin/bgr109
126. Caldon CE. Estrogen Signaling and the DNA Damage Response in Hormone Dependent Breast Cancers. Front Oncol (2014) 4:106. doi: 10.3389/fonc.2014.00106
127. Pedram A, Razandi M, Evinger AJ, Lee E, Levin ER. Estrogen Inhibits ATR Signaling to Cell Cycle Checkpoints and DNA Repair. Mol Biol Cell (2009) 20(14):3374–89. doi: 10.1091/mbc.e09-01-0085
128. Teras LR, Patel AV, Wang M, Yaun S-S, Anderson K, Brathwaite R, et al. Sustained Weight Loss and Risk of Breast Cancer in Women 50 Years and Older: A Pooled Analysis of Prospective Data. JNCI: J Natl Cancer Inst (2020) 112(9):929–37. doi: 10.1093/jnci/djaa026
129. Eliassen AH, Colditz GA, Rosner B, Willett WC, Hankinson SE. Adult Weight Change and Risk of Postmenopausal Breast Cancer. JAMA (2006) 296(2):193. doi: 10.1001/jama.296.2.193
130. Feigelson HS, Caan B, Weinmann S, Leonard AC, Powers JD, Yenumula PR, et al. Bariatric Surgery is Associated With Reduced Risk of Breast Cancer in Both Premenopausal and Postmenopausal Women. Ann Surg (2020) 272(6):1053–9. doi: 10.1097/SLA.0000000000003331
131. Phipps AI, Chlebowski RT, Prentice R, Mctiernan A, Stefanick ML, Wactawski-Wende J, et al. Body Size, Physical Activity, and Risk of Triple-Negative and Estrogen Receptor-Positive Breast Cancer. Cancer Epidemiol Biomarkers Prev (2011) 20(3):454–63. doi: 10.1158/1055-9965.EPI-10-0974
132. Thune I, Brenn T, Lund E, Gaard M. Physical Activity and the Risk of Breast Cancer. New Engl J Med (1997) 336(18):1269–75. doi: 10.1056/NEJM199705013361801
133. Steindorf K, Ritte R, Eomois P-P, Lukanova A, Tjonneland A, Johnsen NF, et al. Physical activity and risk of breast cancer overall and by hormone receptor status: The European prospective investigation into cancer and nutrition. Int J Cancer (2013) 132(7):1667–78. doi: 10.1002/ijc.27778
134. Eliassen AH, Hankinson SE, Rosner B, Holmes MD, Willett WC. Physical Activity and Risk of Breast Cancer Among Postmenopausal Women. Arch Internal Med (2010) 170(19):1758–64. doi: 10.1001/archinternmed.2010.363
135. Bhardwaj P, Du B, Zhou XK, Sue E, Harbus MD, Falcone DJ, et al. Caloric restriction reverses obesity-induced mammary gland inflammation in mice. Cancer Prev Res (Phila) (2013) 6(4):282–9. doi: 10.1158/1940-6207.CAPR-12-0467
136. Kosteli A, Sugaru E, Haemmerle G, Martin JF, Lei J, Zechner R, et al. Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J Clin Invest (2010) 120(10):3466–79. doi: 10.1172/JCI42845
137. Park S, Park N-Y, Valacchi G, Lim Y. Calorie Restriction with a High-Fat Diet Effectively Attenuated Inflammatory Response and Oxidative Stress-Related Markers in Obese Tissues of the High Diet Fed Rats. Mediators Inflamm (2012) 2012:1–11. doi: 10.1155/2012/984643
138. Byers T, Sedjo RL. Does intentional weight loss reduce cancer risk? Diab Obes Metab (2011) 13(12):1063–72. doi: 10.1111/j.1463-1326.2011.01464.x
139. Cancello R, Henegar C, Viguerie N, Taleb S, Poitou C, Rouault C, et al. Reduction of Macrophage Infiltration and Chemoattractant Gene Expression Changes in White Adipose Tissue of Morbidly Obese Subjects After Surgery-Induced Weight Loss. Diabetes (2005) 54(8):2277–86. doi: 10.2337/diabetes.54.8.2277
140. Forsythe LK, Wallace JMW, Livingstone MBE. Obesity and inflammation: the effects of weight loss. Nutr Res Rev (2008) 21(2):117–33. doi: 10.1017/S0954422408138732
141. Clément K, Viguerie N, Poitou C, Carette C, Pelloux V, Curat CA, et al. Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. FASEB J (2004) 18(14):1657–69. doi: 10.1096/fj.04-2204com
142. Magkos F, Fraterrigo G, Yoshino J, Luecking C, Kirbach K, Kelly C,S, et al. Effects of Moderate and Subsequent Progressive Weight Loss on Metabolic Function and Adipose Tissue Biology in Humans with Obesity. Cell Metab (2016) 23(4):591–601. doi: 10.1016/j.cmet.2016.02.005
143. Rock CL, Pande C, Flatt SW, Ying C, Pakiz B, Parker BA, et al. Favorable Changes in Serum Estrogens and Other Biologic Factors After Weight Loss in Breast Cancer Survivors Who are Overweight or Obese. Clin Breast Cancer (2013) 13(3):188–95. doi: 10.1016/j.clbc.2012.12.002
144. Mctiernan A, Wu L, Chen C, Chlebowski R, Mossavar-Rahmani Y, Modugno F, et al. Relation of BMI and Physical Activity to Sex Hormones in Postmenopausal Women*. Obesity (2006) 14(9):1662–77. doi: 10.1038/oby.2006.191
145. Dombrowski SU, Knittle K, Avenell A, Araujo-Soares V, Sniehotta FF. Long term maintenance of weight loss with non-surgical interventions in obese adults: systematic review and meta-analyses of randomised controlled trials. BMJ (2014) 348(may14 6):g2646–g. doi: 10.1136/bmj.g2646
146. Barte JCM, Ter Bogt NCW, Bogers RP, Teixeira PJ, Blissmer B, Mori TA, et al. Maintenance of weight loss after lifestyle interventions for overweight and obesity, a systematic review. Obes Rev (2010) 11(12):899–906. doi: 10.1111/j.1467-789X.2010.00740.x
147. Cui Y, Deming-Halverson SL, Shrubsole MJ, Beeghly-Fadiel A, Cai H, Fair AM, et al. Use of nonsteroidal anti-inflammatory drugs and reduced breast cancer risk among overweight women. Breast Cancer Res Treat (2014) 146(2):439–46. doi: 10.1007/s10549-014-3030-7
148. Hsieh P-S, Lu K-C, Chiang C-F, Chen C-H. Suppressive effect of COX2 inhibitor on the progression of adipose inflammation in high-fat-induced obese rats. Eur J Clin Invest (2010) 40(2):164–71. doi: 10.1111/j.1365-2362.2009.02239.x
149. Lu C-H, Hung Y-J, Hsieh P-S. Additional effect of metformin and celecoxib against lipid dysregulation and adipose tissue inflammation in high-fat fed rats with insulin resistance and fatty liver. Eur J Pharmacol (2016) 789:60–7. doi: 10.1016/j.ejphar.2016.07.012
150. Catalano S, Leggio A, Barone I, De Marco R, Gelsomino L, Campana A, et al. A novel leptin antagonist peptide inhibits breast cancer growthin vitroandin vivo. J Cell Mol Med (2015) 19(5):1122–32. doi: 10.1111/jcmm.12517
151. O’Regan RM, Jordan VC. The evolution of tamoxifen therapy in breast cancer: selective oestrogen-receptor modulators and downregulators. Lancet Oncol (2002) 3(4):207–14. doi: 10.1016/S1470-2045(02)00711-8
152. Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, et al. Tamoxifen for Prevention of Breast Cancer: Report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. JNCI: J Natl Cancer Inst (1998) 90(18):1371–88. doi: 10.1093/jnci/90.18.1371
153. Barrett-Connor E, Mosca L, Collins P, Geiger MJ, Grady D, Kornitzer M, et al. Effects of Raloxifene on Cardiovascular Events and Breast Cancer in Postmenopausal Women. New Engl J Med (2006) 355(2):125–37. doi: 10.1056/NEJMoa062462
154. Vogel VG, Constantino JP, Wickerham DL, Cronin WM, Cecchini RS, Atkins JN, et al. Effects of Tamoxifen vs Raloxifene on the Risk of Developing Invasive Breast Cancer and Other Disease Outcomes: The NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 Trial. JAMA (2006) 295(23):2727–41. doi: 10.1001/jama.295.23.joc60074
155. Waters EA, Mcneel TS, Stevens WM, Freedman AN. Use of tamoxifen and raloxifene for breast cancer chemoprevention in 2010. Breast Cancer Res Treat (2012) 134(2):875–80. doi: 10.1007/s10549-012-2089-2
156. Goss PE, Ingle JN, Alés-Martínez JE, Cheung AM, Chlebowski RT, Wactawski-Wende J, et al. Exemestane for Breast-Cancer Prevention in Postmenopausal Women. New Engl J Med (2011) 364(25):2381–91. doi: 10.1056/NEJMoa1103507
157. Evans JMM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ (2005) 330(7503):1304–5. doi: 10.1136/bmj.38415.708634.F7
158. Currie CJ, Poole CD, Gale EAM. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia (2009) 52(9):1766–77. doi: 10.1007/s00125-009-1440-6
159. Bodmer M, Meier C, Krahenbuhl S, Jick SS, Meier CR. Long-Term Metformin Use Is Associated With Decreased Risk of Breast Cancer. Diabetes Care (2010) 33(6):1304–8. doi: 10.2337/dc09-1791
160. Brown KA, Hunger NI, Docanto M, Simpson ER. Metformin inhibits aromatase expression in human breast adipose stromal cells via stimulation of AMP-activated protein kinase. Breast Cancer Res Treat (2010) 123(2):591–6. doi: 10.1007/s10549-010-0834-y
161. Samarajeewa NU, Ham S, Yang F, Simpson ER, Brown KA. Promoter-specific effects of metformin on aromatase transcript expression. Steroids (2011) 76(8):768–71. doi: 10.1016/j.steroids.2011.02.041
162. Giles ED, Jindal S, Wellberg EA, Schedin T, Anderson SM, Thor AD, et al. Metformin inhibits stromal aromatase expression and tumor progression in a rodent model of postmenopausal breast cancer. Breast Cancer Res (2018) 20(1):15. doi: 10.1186/s13058-018-0974-2
163. Alimova IN, Liu B, Fan Z, Edgerton SM, Dillon T, Lind SE, et al. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle (2009) 8(6):909–15. doi: 10.4161/cc.8.6.7933
164. Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res (2006) 66(21):10269–73. doi: 10.1158/0008-5472.CAN-06-1500
165. Queiroz EAIF, Puukila S, Eichler R, Sampaio SC, Forsyth HL, Lees SJ, et al. Metformin Induces Apoptosis and Cell Cycle Arrest Mediated by Oxidative Stress, AMPK and FOXO3a in MCF-7 Breast Cancer Cells. PloS One (2014) 9(5):e98207. doi: 10.1371/journal.pone.0098207
166. Bijland S, Mancini J,S, Salt P,I. Role of AMP-activated protein kinase in adipose tissue metabolism and inflammation. Clin Sci (2013) 124(8):491–507. doi: 10.1042/CS20120536
167. Heilbronn LK, de Jonge L, Frisard MI, DeLany JP, Larson-Meyer DE, Rood J, et al. Effect of 6-month calorie restriction on biomarkers of longevity, metabolic adaptation, and oxidative stress in overweight individuals: a randomized controlled trial. JAMA (2006) 295(13):1539–48. doi: 10.1001/jama.295.13.1539
168. Bankoglu EE, Gerber J, Kodandaraman G, Seyfried F, Stopper H. Influence of bariatric surgery induced weight loss on oxidative DNA damage. Mutat Res/Gen Toxicol Environ Mutagen (2020) 853:503194. doi: 10.1016/j.mrgentox.2020.503194
169. Bankoglu EE, Seyfried F, Arnold C, Soliman A, Jurowich C, Germer CT, et al. Reduction of DNA damage in peripheral lymphocytes of obese patients after bariatric surgery-mediated weight loss. Mutagenesis (2018) 33(1):61–7. doi: 10.1093/mutage/gex040
170. Monzo-Beltran L, Vazquez-Tarragón A, Cerdà C, Garcia-Perez P, Iradi A, Sánchez C, et al. One-year follow-up of clinical, metabolic and oxidative stress profile of morbid obese patients after laparoscopic sleeve gastrectomy. 8-oxo-dG as a clinical marker. Redox Biol (2017) 12:389–402. doi: 10.1016/j.redox.2017.02.003
171. Setayesh T, Mišík M, Langie SAS, Godschalk R, Waldherr M, Bauer T, et al. Impact of Weight Loss Strategies on Obesity-Induced DNA Damage. Mol Nutr Food Res (2019) 63(17):1900045. doi: 10.1002/mnfr.201900045
Keywords: obesity, breast cancer, development, growth, estrogen
Citation: Bhardwaj P and Brown KA (2021) Obese Adipose Tissue as a Driver of Breast Cancer Growth and Development: Update and Emerging Evidence. Front. Oncol. 11:638918. doi: 10.3389/fonc.2021.638918
Received: 07 December 2020; Accepted: 12 March 2021;
Published: 30 March 2021.
Edited by:
Manuel D. Gahete, Maimonides Biomedical Research Institute of Cordoba (IMIBIC), SpainReviewed by:
Amaia Rodríguez, University of Navarra, SpainDavid Rincón Fernández Pacheco, Cedars Sinai Medical Center, United States
Copyright © 2021 Bhardwaj and Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kristy A. Brown, kab2060@med.cornell.edu