BDNF signaling and survival of striatal neurons
 Maryna Baydyuk
Maryna Baydyuk Baoji Xu
Baoji Xu- 1National Institute of Neurological Diseases and Stroke, National Institutes of Health, Bethesda, MD, USA
- 2Department of Neuroscience, The Scripps Research Institute Florida, Jupiter, FL, USA
The striatum, a major component of the basal ganglia, performs multiple functions including control of movement, reward, and addiction. Dysfunction and death of striatal neurons are the main causes for the motor disorders associated with Huntington’s disease (HD). Brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family, is among factors that promote survival and proper function of this neuronal population. Here, we review recent studies showing that BDNF determines the size of the striatum by supporting survival of the immature striatal neurons at their origin, promotes maturation of striatal neurons, and facilitates establishment of striatal connections during brain development. We also examine the role of BDNF in maintaining proper function of the striatum during adulthood, summarize the mechanisms that lead to a deficiency in BDNF signaling and subsequently striatal degeneration in HD, and highlight a potential role of BDNF as a therapeutic target for HD treatment.
Introduction
The striatum is the largest component of the basal ganglia. It is responsible for movement control and is associated with addictive behaviors. The striatum receives projections from the entire cortical mantle; processes motor, sensory, and associational cortical information; and passes it to the output nuclei of the basal ganglia (Gerfen et al., 1990, 2002; Bolam et al., 2000). Most striatal functions are mediated by the medium-sized spiny neurons (MSNs), which comprise 95% of the striatum with the rest being interneurons (Parent and Hazrati, 1995; Kawaguchi, 1997). MSNs, which use γ-amino butyric acid (GABA) as a transmitter, are born in the ventricular/subventricular zones (VZ/SVZ) of the lateral ganglionic eminence (LGE) and migrate to the striatum during embryogenesis (Hamasaki et al., 2003). They are divided into two equal size populations based on their projection sites and protein expression pattern. The MSNs that form the direct pathway send projections directly to the output nuclei of the basal ganglia, such as the internal segment of the globus pallidus, the substantia nigra pars reticulata, and the ventral pallidum. The other population of the MSNs sits at the origin of the indirect pathway and projects to the output nuclei of the basal ganglia via the external segment of the globus pallidus and the subthalamic nucleus (Bolam et al., 2000). Under normal conditions, activation of the direct pathway leads to initiation of movement. On the other hand, activation of the indirect pathway leads to the opposite physiological effects such as termination of movement or suppression of unwanted movements (Mink and Thach, 1993). In addition to their distinct functions, the MSNs of the two pathways express different sets of neuropeptides and dopamine receptors: neurons of the direct pathway express substance P (SP) and the dopamine receptor D1a (DRD1a), while neurons of the indirect pathway produce enkephalin (Enk) and the dopamine receptor D2 (DRD2; Kawaguchi, 1997). These distinct expression patterns in the MSNs of the two pathways are further confirmed in bacterial artificial chromosome (BAC) transgenic mice expressing fluorescent proteins under the control of the promoter for either DRD1a or DRD2 (Day et al., 2006; Shuen et al., 2008).
Recent studies have shown that striatal neurons are dependent on neurotrophins for their development, survival, and proper function. Increased attention has been given to brain-derived neurotrophic factor (BDNF) and its function in normal and pathological conditions. BDNF is a member of the neurotrophin family, which also includes nerve growth factor (NGF), neurotrophin-3 (NT-3), and neurotrophin-4/5 (NT-4/5). These secreted proteins exert many biological effects by binding and activating specific tropomyosin-related kinase (Trk) receptors. NGF activates TrkA, BDNF and NT4/5 activate TrkB, and NT3 activates TrkC (Reichardt, 2006). Upon binding to BDNF, activated full-length TrkB triggers multiple intracellular signaling cascades through protein-protein interactions (Chao, 2003). The three major pathways, activated by TrkB include: (1) the PLC-γ pathway that leads to production of diacylglycerol and an increase in intracellular calcium, and as a result activation of CAM kinases and PKC; (2) PI-3-kinase pathway that activates AKT, which mediates anti-apoptotic effects; and (3) MAP/ERK pathway that activates regulators of protein translation (Segal, 2003). By activating these diverse signaling cascades in neurons, BDNF can regulate neuronal development and survival, initiation of neurite outgrowth and path-finding (Bhave et al., 1999; Encinas et al., 1999; Yamada et al., 1999, 2001). It can also mediate various synaptic reorganization processes, including formation and maintenance of dendrites and dendritic spines (McAllister et al., 1999; Orefice et al., 2013). Deletion of either the TrkB or Bdnf gene leads to cell atrophy, dendritic degeneration, and neuronal loss, as shown in the excitatory neurons of the dorsal forebrain (Xu et al., 2000a; Gorski et al., 2003). In addition, BDNF plays an important role in modulating synaptic function and plasticity such as long-term potentiation (LTP), a cellular substrate for learning and memory (McAllister et al., 1999; Poo, 2001).
The vital role of neurotrophins in survival of developing neurons in the peripheral nervous system (PNS) has been well established (Crowley et al., 1994; Smeyne et al., 1994). In the PNS, developing neurons at their final location compete for a limited amount of neurotrophic factors produced by their target tissues; neurons unable to obtain sufficient amounts of trophic factors die via programmed cell death (Zweifel et al., 2005). This mode of survival implies that peripheral target controls the final size of the innervating neuronal population through neurotrophic factors. In contrast, the role of neurotrophins in survival of developing neurons in the central nervous system (CNS) has not been determined until recently. In this review we will discuss our reports, demonstrating that in the developing striatum and LGE, BDNF and NT3 mediate survival of immature MSNs of the indirect and direct pathways, respectively, at the place of their origin before they migrate to their final destination (Baydyuk et al., 2013). Our findings support the idea that a single neurotrophin might be sufficient and necessary to support survival of developing neuronal populations in the CNS. Moreover, we propose a new mode of neurotrophic action in the brain, suggesting that innervating neurons may control the size of their target.
It has been shown that neurotrophins participate in the maintenance of adult neuronal populations in the brain (Xu et al., 2000b; Baquet et al., 2004). A modest increase in postnatal apoptosis was observed in hippocampal and cerebellar granule cells of TrkB and TrkC knockout mice, however these deletions do not appear to affect the size of these two neuronal populations (Minichiello and Klein, 1996; Alcántara et al., 1997). The redundancy of neurotrophin-mediated signaling pathways in brain regions where more than one Trk receptor is present can provide an explanation for the rather minor effect when one receptor or its ligands are removed. It is also difficult to assess the role of neurotrophins, BDNF in particular, in the postnatal CNS due to lethality of Bdnf-null mice in the first two postnatal weeks. Generation of several mouse lines with area-specific Bdnf and TrkB deletions allowed for the detailed examination of their roles in striatal postnatal growth and maturation. We will review several studies that demonstrate important functions of BDNF-TrkB signaling in promoting somatic growth, dendritic complexity, and spine density in striatal neurons (Baquet et al., 2004; Rauskolb et al., 2010; Li et al., 2012).
Deficiency in BDNF signaling has been linked with an increasing number of conditions that cause brain dysfunction, and the connection between BDNF loss in the striatum and Huntington’s disease (HD) pathology has been extensively investigated. HD is caused by the CAG trinucleotide repeat expansion in the first exon of the gene encoding huntingtin protein (htt; The Huntington’s Disease Collaborative Research Group, 1993). This mutation is translated into a polyglutamine (poly Q) stretch near the amino terminus of htt, which results in a toxic gain of function (Gusella and MacDonald, 2000). Although mutant htt is found throughout the HD brain, the striatum is affected early and more severely during the course of the disease. Striatal atrophy is due to selective degeneration of the MSNs with neuronal loss of 50–60% (Mann et al., 1993; Vonsattel and DiFiglia, 1998). Interestingly, the MSNs of the indirect pathway, responsible for inhibition of involuntary movement, are preferentially affected, causing motor symptoms of HD such as uncontrollable sequence of movements called chorea. The exact mechanism behind selective degeneration of striatal neurons remains to be elucidated, but it has been suggested that reduced trophic support renders striatal neurons more vulnerable to the toxic actions of mutant htt. In support of this view, reduced levels of BDNF protein are detected in the striatum of HD animal models (Spires et al., 2004; Apostol et al., 2008; Gharami et al., 2008) and HD patients (Ferrer et al., 2000). The changes in striatal gene expression profile are similar in HD patients and mice with BDNF deficiency (Strand et al., 2007). Moreover, lack of BDNF-mediated signaling alone is sufficient to cause dendritic abnormalities and neuronal loss in the striatum (Gorski et al., 2003; Baquet et al., 2004), and progression of HD is accelerated in Bdnf heterozygous mice (Canals et al., 2004). Importantly, it has been determined that mutant htt decreases striatal BDNF by interfering with BDNF synthesis and transport (Zuccato et al., 2001; Cattaneo, 2003; Gauthier et al., 2004). Furthermore, our recent study shows that the TrkB receptor is selectively expressed in striatal MSNs of the indirect pathway, which may explain why this population of neurons degenerates first in HD patients (Baydyuk et al., 2011). Taken together, these observations raise the possibility that reduced levels of striatal BDNF may significantly contribute to the HD pathogenesis and identify BDNF-TrkB signaling pathway as a potential therapeutic target for HD treatment.
BDNF and TrkB in the Adult and Developing Striatum: Expression Patterns
In the adult brain, BDNF protein is found in many regions, including the cerebral cortex, basal forebrain, striatum, hippocampus, hypothalamus, brainstem, and cerebellum (Conner et al., 1997). In most brain regions, such as cortex, both Bdnf mRNA and BDNF protein are present. In contrast, in the striatum Bdnf mRNA is virtually absent, whereas BDNF protein levels are high (Figure 1A; Spires et al., 2004; Apostol et al., 2008; Gharami et al., 2008). BDNF found in the adult striatum is synthesized and anterogradely transported from the cell bodies located in the cerebral cortex, substantia nigra pars compacta, amygdala, and thalamus (Altar et al., 1997; Baquet et al., 2004). Since the striatum does not produce BDNF but depends on it for its proper function, abnormalities in anterograde transport and reduced gene expression from brain regions supplying BDNF to the striatum might cause neuronal dysfunction and striatal atrophy.
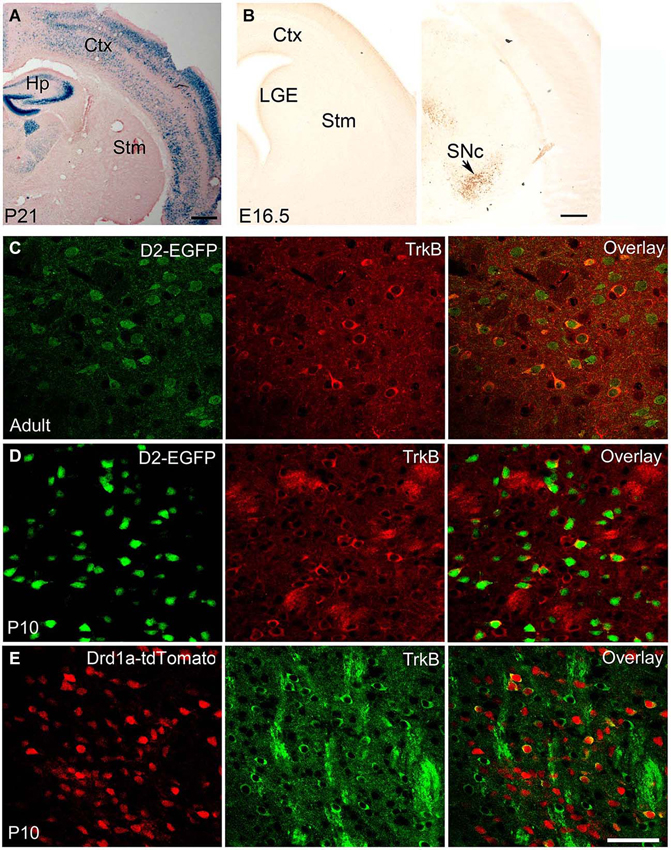
Figure 1. BDNF and TrkB expression in adult and developing striatum. (A) BDNF expression in BdnfLacZ/+ brain at P21, as revealed by X-gal staining for β-galactosidase (blue). Scale bars, 500 μm. (B) BDNF expression in BdnfLacZ/+ embryos at E16.5, as revealed by immunohistochemistry to β-galactosidase. The arrow denotes the substantia nigra. Scale bar, 250 μm. (C) Co-localization of TrkB with DRD2 in the striatum of adult TrkBLacZ/+; D2-EGFP mice in which β-galactosidase and EGFP serve as indicators for expression of TrkB and DRD2, respectively. Fluorescent immunohistochemistry with antibodies to β-galactosidase and EGFP shows that the majority of TrkB-expressing neurons also express DRD2 in the adult striatum. (D) High co-expression of TrkB and DRD2 in the young striatum. Nearly all EGFP+ cells expressed β-galactosidase in the striatum of TrkBLacZ/+; D2-EGFP mice at P10. (E) Low co-expression of TrkB and DRD1a in the striatum of TrkBLacZ/+; Drd1a-tdTomato mice at P10. Scale bar, 50 μm. Ctx, cerebral cortex; Hp, hippocampus; LGE, lateral ganglionic eminence; SNc, substantia nigra pars compacta; Stm, striatum. This figure is adapted from Baydyuk et al. (2011, 2013).
The expression levels of BDNF fluctuate during development. In rodents, BDNF is expressed by a number of cells in the hippocampus on embryonic day 15.5 (E15.5), and by E17 it is found in the piriform cortex, hippocampus, thalamus, hypothalamus and amygdala, but in only a few cells in the cortex and none in the striatum (Baydyuk et al., 2013). Cortical expression appears at postnatal day 4 (P4) and increases with age, reaching the adult levels by P21 (Baquet et al., 2004). The corticostriatal connections are also mainly formed during the first postnatal week (Nisenbaum et al., 1998; Christensen et al., 1999). Thus, it is unlikely that the cerebral cortex is the main source of BDNF for immature neurons in the LGE, in contrast to the adult striatum. Since BDNF is not expressed in the LGE and the nearby striatum (Figure 1B), but BDNF protein is already present in this region at E16.5 (Baydyuk et al., 2013), it likely comes from other brain regions that express BDNF during embryogenesis and form connections with the LGE and the developing striatum.
It is equally important to note that since immature neurons in the LGE have to migrate to the striatum before they send axons to striatal targets, it is impossible for them to obtain BDNF from their eventual targets through retrograde transport. Therefore, the only source of BDNF in the LGE is from axons of neurons that project to this region. In our recent study, we show that BDNF is expressed in the substantia nigra at P0 and E16.5 specifically in neurons positive for tyrosine hydroxylase (TH; Baydyuk et al., 2013; Figure 1B). It has also been shown that nigrostriatal projections are formed by E16.5 (Voorn et al., 1988; Hu et al., 2004). Furthermore, TH-positive axons are found in close proximity to dividing LGE progenitor cells as early as E13 (Ohtani et al., 2003). These expression and projection findings indicate that BDNF from mesencephalic dopaminergic neurons is the main source of BDNF in the developing striatum and the LGE.
The TrkB receptor is also widely expressed in the developing and adult brain. The TrkB mRNA is first detected in the neuroepithelium and in the neural crest at E9.5 (Klein et al., 1990). During development the TrkB is present at high levels throughout the brain with regional and cell type specific fluctuations. In adulthood, TrkB expression is also confined to specific regions or neuronal subtypes in a complex pattern. Both TrkB mRNA and protein are present in the developing and adult striatum (Yan et al., 1997). Interestingly, TrkB is not equally distributed in all striatal neurons. Instead, it is preferentially expressed by DRD2-expressing MSNs of the indirect pathway (Figures 1C,D), with 98% of DRD2 MSNs expressing TrkB at P10 and 43% in adult animal (Baydyuk et al., 2011). The observed preferential TrkB expression may provide insights into the selectivity of degeneration associated with HD, where a greater decrease in the number of DRD2 MSNs is observed (Reiner et al., 1988). These neurons act to terminate movement associated with the basal ganglia or suppress unwanted sequences of movements (Bolam et al., 2000). Hence, the loss of the indirect pathway neurons leads to disinhibition of the thalamus and increased facilitation of the motor cortex, producing hyperkinesias in HD patients (Calabresi et al., 2000). Direct pathway neurons that express DRD1a are less affected, and striatal interneurons are mostly spared in patients with HD (Ferrante et al., 1987a,b). In agreement with these facts, only 18% of DRD1a MSNs express TrkB at P10 (Figure 1E, Baydyuk et al., 2011). These findings indicate that decreased BDNF-TrkB signaling may preferentially affect indirect pathway MSNs, which express most of the TrkB in the striatum, therefore explaining the selective degeneration of this population and motor phenotype seen in HD.
Role of BDNF and NT3 in Survival of Developing Striatal Neurons at their Origin
Striatal projection MSNs originate in the VZ/SVZ of the LGE between E12 and P2 with a peak around E15, and subsequently migrate along guiding radial glia into the striatum (Marchand and Lajoie, 1986; Olsson et al., 1995, 1998). Several factors have been identified as regulators of striatal neurogenesis and fate determination. For instance, dopamine coming from the substantia nigra pars compacta and arriving in the LGE via nigrostriatal pathway as early as E13 (Voorn et al., 1988; Hu et al., 2004) has been shown to modulate striatal neurogenesis by influencing the cell cycle of progenitor cells in the LGE (Ohtani et al., 2003). Marked progress has also been made in understanding cell fate determination of striatal neurons. Several transcription factors have been identified to play a role in differentiation of neuronal precursors into striatal neurons, including Gsh-2, Mash1, and Dlx family members (Dlx1, Dlx2, Dlx5, and Dlx6) (Anderson et al., 1997; Casarosa et al., 1999; Corbin et al., 2000; Stenman et al., 2003). However, until recently it was unclear whether neurotrophins play any role in neuronal survival, target innervation, dendritic arborization, and synaptogenesis of developing striatal neurons.
In our recent studies, we demonstrate that neurotrophins are major players in determining striatal size by promoting survival of newborn striatal neurons. Selective deletion of TrkB in the striatal progenitors, using the Dlx5/6-Cre transgene (TrkBDlx), leads to a 50% loss of the MSNs, among which DRD2 population is most affected with up to 80% loss, while DRD1a MSNs suffer only 22% loss (Baydyuk et al., 2011). As we discussed above this selective loss of the MSNs in the indirect pathway is likely due to the preferential expression of the TrkB receptor by this population (Figures 1C–E). The magnitude of the observed neuronal loss in TrkBDlx mutants is similar at P0 and P21, ruling out the possibility of cell death during postnatal development. We also show that TrkB signaling does not affect striatal neurogenesis, in contrast to the cerebral cortex, where its role in promoting proliferation and differentiation of neuronal precursors is well established (Bartkowska et al., 2007). Instead, we observe numerous apoptotic cells in the striatal VZ/SVZ of TrkBDlx mice, indicating that TrkB signaling is essential for survival of developing striatal neurons at their origin (Baydyuk et al., 2011).
In agreement with our previous report, global deletion of BDNF produces similar phenotype, showing selective loss of DRD2 MSNs and the majority of cell death occurring within the LGE during embryogenesis (Baydyuk et al., 2013). These results confirm a crucial role of BDNF-TrkB signaling in survival of developing MSNs of the indirect pathway and indicate that developing MSNs need trophic support before they migrate to the striatum and send out axons.
MSNs of the direct pathway are not affected by deletion of TrkB or BDNF. Interestingly, signaling via another neurotrophin, NT3, plays an important role in survival of this neuronal population. Both NT3 and TrkC proteins are present in the developing striatum, where 86% of DRD1a-expressing neurons are positive for TrkC. Deletion of either TrkC or NT3 leads to ~30% loss of total number of striatal neurons, with 35% reduction in the number of the MSNs of the direct pathway, whereas MSNs of the indirect pathway are not significantly affected (Baydyuk et al., 2013). Thus, the DRD1a-expressing MSNs of the direct pathway are dependent on the NT3-TrkC signaling for their survival during development. Similar to BDNF, NT3 is not expressed in the LGE or the striatum. Both BDNF and NT3 are produced in the midbrain dopaminergic neurons and anterogradely transported via nigrostriatal pathway to the LGE and striatum (Figure 2A). Deletion of either BDNF or NT3 specifically in TH-positive dopaminergic neurons leads to selective loss of DRD2 or DRD1a MSNs, respectively (Baydyuk et al., 2013).
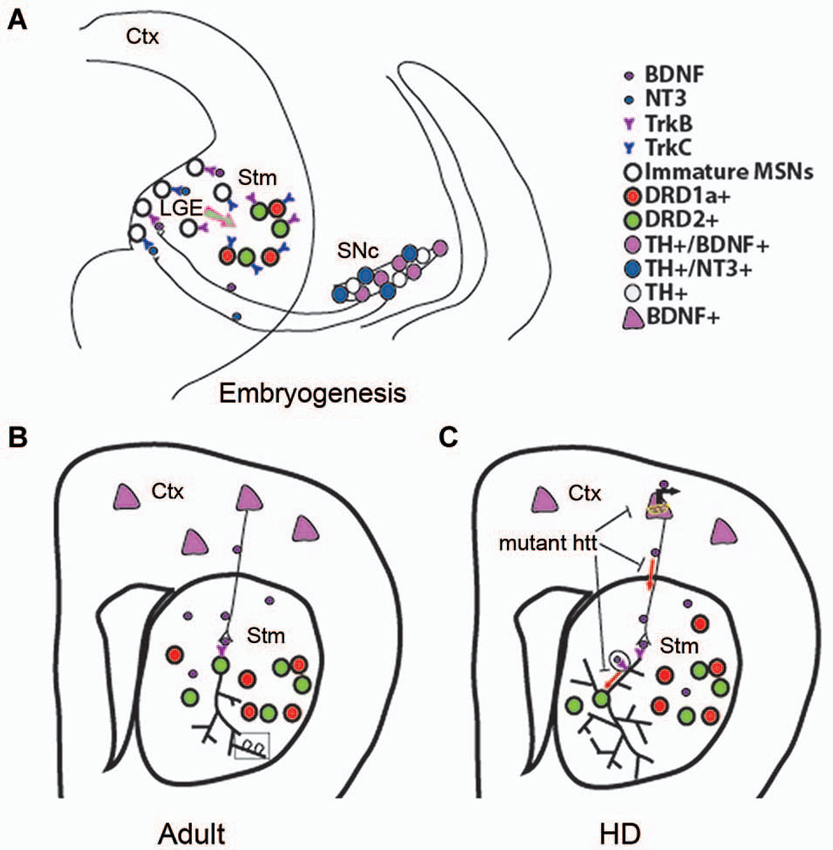
Figure 2. Role of BDNF in developing, adult, and HD striatum. (A) A proposed model showing that BDNF and NT3 anterogradely transported from mesencephalic dopaminergic neurons regulate survival of immature neurons in the indirect and direct pathways, respectively. Ctx, cerebral cortex; Stm, striatum; SN, substantia nigra. (B) Cortical BDNF in the adult striatum mediates dendritic complexity and spine number and morphology. (C) Mutant htt reduces BDNF-TrkB signaling by inhibiting BDNF gene transcription, axonal transport of vesicles containing BDNF, retrograde dendritic transport of TrkB-positive endosomes to the cell body. Panel A is adapted from Baydyuk et al. (2013).
Together, these findings establish a novel mode of neurotrophin actions in the CNS. It is distinct from the one that has been extensively studied in the PNS, where neurons depend on neurotrophins after they reach their final position and form connections with their targets, competing for a limiting amount of target-derived neurotrophins for their survival. These neurotrophin molecules are internalized at axonal terminals and retrogradely transported to the cell bodies to activate pro-survival signaling cascades (Zweifel et al., 2005). In this way, target tissue determines the final size of the innervating neuronal population. In some cases, PNS neurons also depend on neurotrophins during neurogenesis before target innervation is complete (Fariñas et al., 1998; Coppola et al., 2001). Notably, neurotrophins required for proper formation of these neurons are highly expressed in the close vicinity of developing neurons and growing axons (Fariñas et al., 1996, 2001). In contrast, in the developing striatum survival of immature MSNs at the place of their origin depends on neurotrophins, anterogradely transported from the midbrain dopaminergic neurons (Figure 2A). Thus, we propose that innervating neurons can regulate the size of their targeting neuronal population in the CNS by regulating their survival through neurotrophin release at axonal terminals during embryogenesis and early postnatal development. As a result, nigrostriatal dopaminergic neurons, a relatively small population, can regulate the size of their large target, the striatum, by controlling the survival of immature striatal neurons at their origin.
Role of BDNF in Striatal Postnatal Growth, Maturation, and Maintenance
To assess the role of BDNF-TrkB signaling in the postnatal development and maintenance of the striatum, several conditional BDNF knockouts have been generated. Since postnatal striatal BDNF arrives mainly from the cortex, where its expression starts during first postnatal week, cortical ablation of BDNF provides a tool to study a role of BDNF-TrkB signaling in the postnatal striatal growth. Using this approach, Baquet et al. (2004) demonstrate that the striatum of postnatal forebrain-specific Emx-Bdnf knockout mice is reduced in volume compared with controls, and the MSNs have shrunken cell somas, thinner dendrites, and fewer dendritic spines at P35. Although significant striatal neuronal loss is not detected at P35 or P120, 35% of striatal neurons are lost in Emx-Bdnf knockout mice aged beyond 1 year. Thus, cortical BDNF has a relatively minor role in determining the number of the MSNs early in life but is required for a long-term survival and is necessary for normal dendritic morphogenesis when BDNF expression rises in the cortex (Baquet et al., 2004).
Maturity of striatal neurons is determined by several factors, including expression levels of neuronal markers, complexity of dendritic arbor, and dendritic spine morphology. Studies using global BDNF deletion in the CNS show that BDNF ablation leads to reduction in striatal volume, dendritic complexity, and spine density, indicating impaired striatal maturation in these mutants (Rauskolb et al., 2010; Li et al., 2012). At the cellular level, DARPP32 (dopamine- and cAMP-regulated phosphoprotein, 32 kDa) is a well-established marker of differentiated striatal MSNs, and a key mediator of dopamine signaling (Svenningsson et al., 2004). Mutant mice with BDNF deletion show a marked reduction in DARPP32 expression levels and distribution (Baydyuk et al., 2011; Li et al., 2012). Decrease in dendritic spine number and size in BDNF mutant mice indicate deficits in formation of corticostriatal synaptic connections, as dendritic spines are the main targets of excitatory synapses formed by cortical afferents (Baquet et al., 2004; Rauskolb et al., 2010; Li et al., 2012). These observations indicate that BDNF is an important factor for establishing and maintaining corticostriatal synapses and can influence their strength (Figure 2B).
Morphological, cellular, and functional changes observed in the striatum of Bdnf and TrkB mutants are often accompanied by behavioral abnormalities. For instance, mutant mice with either forebrain-specific deletion of BDNF or TrkB deletion in striatal progenitors, display hindlimb and forelimb clasping phenotype, which has also been observed in transgenic lines with motor dysfunction or degeneration, including HD mouse models (Baquet et al., 2004; Baydyuk et al., 2011). Deletion of the Bdnf gene in the dopaminergic neurons causes cell deaths of the MSNs of the indirect pathway and leads to poor performance on the rotarod, reinforcing the importance of this pathway in motor coordination (Baydyuk et al., 2013). In agreement with this finding, ablation of TrkB selectively from these MSNs results in augmented spontaneous and drug-induced hyper-locomotion, further indicating the importance of BDNF-TrkB signaling in the MSNs of the indirect pathway, which control inhibition of locomotor behavior (Besusso et al., 2013). Importantly, a disrupted expression of Bdnf in the HD mouse models advances the onset of motor dysfunctions and produces more severe uncoordinated movements. In all the studies discussed above, the behavioral pathology correlates with specific degeneration of the MSNs of the indirect pathway (Canals et al., 2004).
Deficiency in BDNF-TrkB Signaling and HD: Molecular Mechanisms and Therapeutic Implications
Mutant Htt Alters BDNF Gene Expression
The pathogenic mechanisms of HD are not fully understood but are thought to involve the gain of toxic function and/or the loss of normal activities of htt protein (Borrell-Pagès et al., 2006). The combination of both mechanisms can lead to a decrease in striatal BDNF by interfering with BDNF synthesis and transport (Zuccato et al., 2001; Cattaneo, 2003; Gauthier et al., 2004). Wild-type htt is known to regulate transcription of multiple genes, including Bdnf (Zuccato et al., 2001). In rodents and humans, the Bdnf gene is transcribed from at least eight discrete promoters, producing different Bdnf mRNA species that encode the same protein (Aid et al., 2007). The transcripts are generated in different tissues in a stimulus- and development-specific manner and may have differential subcellular localizations and targets (Metsis et al., 1993; Timmusk et al., 1993; Pattabiraman et al., 2005). Zuccato et al. (2001) show that wild-type htt enhances Bdnf transcription from promoter II, whereas mutant htt suppresses Bdnf transcription from promoter II as well as two other Bdnf promoters in cultured cells and the cerebral cortex of YAC72 transgenic mice expressing mutant htt with an expanded tract of 72 glutamines. Wild-type htt promotes transcription of promoter II by sequestering the repressor element-1 transcription factor/neuron restrictive silencer factor (REST/NRSF) in the cytoplasm, thereby freeing the nucleus of the inhibitory complex and allowing transcription to occur (Zuccato et al., 2003). In contrast, mutant htt is unable to retain REST/NRSF complex in the cytoplasm, leading to aberrant accumulation of REST/NRSF in the nucleus and inhibition of Bdnf gene transcription.
In agreement with these findings, levels of Bdnf mRNA are reduced in the cerebral cortices of HD patients (Zuccato et al., 2008). It also has been shown that lower levels of BDNF are associated with higher numbers of CAG repeats in mutant htt alleles and correlate with the severity of the disease (Ciammola et al., 2007). However, this autopsy data should be interpreted with caution. As mutant htt alters electrophysiological properties of cortical neurons (Cummings et al., 2009) and neuronal activity regulates Bdnf gene expression (Aid et al., 2007), we should not exclude the possibility that the observed reduction in cortical Bdnf mRNA levels may be secondary to neurodegeneration.
Although most findings are in agreement with the notion that both mechanisms, suppressed Bdnf gene expression and deficient BDNF transport, might concomitantly contribute to reduced levels of BDNF in the striatum of HD patients and mouse models, several discrepancies still exist between reports on transcriptional regulation of BDNF by htt. A reduction in Bdnf transcription would predict reduced levels of BDNF protein in cerebral cortices of both HD patients and mouse models. This prediction has been confirmed in one study (Zuccato et al., 2008) but not in another study (Gauthier et al., 2004), which uses post-mortem tissues from multiple control subjects and HD patients. Furthermore, in situ hybridization revealed normal levels of cortical Bdnf mRNA in aging YAC128 mice that express the whole human htt gene with 128 CAG repeats (Xie et al., 2010). Consistent with this observation, levels of cortical BDNF in YAC128 mice and R6/1 mice, another HD model, were found to be similar to those in WT mice (Gharami et al., 2008; Xie et al., 2010). Despite the variations in determining cortical Bdnf mRNA levels, a significant reduction in striatal BDNF has been consistently shown in both HD patients and animal models, thus providing a strong evidence for BDNF as a crucial factor in the pathogenesis of HD.
Mutant Htt Inhibits BDNF and TrkB Transport
In addition to controlling Bdnf mRNA production in the cortex, wild-type htt also regulates BDNF transport along the corticostriatal axes (Figure 2C), the main supply line of BDNF in the adult striatum (Gauthier et al., 2004). Although present in the nucleus, htt is predominantly found in the cytoplasm, where it interacts with huntingtin-associated protein-1 (HAP1), a protein involved in axonal transport via association with p150glued subunit of dynactin, which is an essential part of the microtubule-based motor complex (Block-Galarza et al., 1997; Engelender et al., 1997; Li et al., 1998). Gauthier et al. (2004) show that under normal conditions wild-type htt promotes neuronal survival by facilitating the transport of BDNF-containing vesicles along microtubules. Consistent with a loss-of-function hypothesis, reduction in wild-type htt levels leads to attenuated BDNF trafficking. On the other hand, mutant htt binds tightly to p150glued via HAP1 and prevents efficient movement of BDNF-containing vesicles. Disruption of BDNF transport leads to decreased neurotrophic support and neurotoxicity, which can be rescued by wild-type htt (Gauthier et al., 2004).
Considering the important role of htt in axonal trafficking of BDNF-containing vesicles, it has been recently proposed that htt can also play a role in retrograde transport of the TrkB in striatal dendrites (Figure 2C). It has been shown that upon BDNF binding, TrkB-positive endosomes undergo dynein-dependent retrograde transport along microtubules to the cell body where TrkB induces survival signals (Watson et al., 1999; Heerssen et al., 2004; Ha et al., 2008), a mechanism studied primarily in axons. Recent technical advances that allow to selectively isolate dendrites from the cell bodies using microfluidic devices (Cohen et al., 2011), facilitated the investigation of the TrkB vesicular retrograde transport in control and HD striatal neurons. A study by Liot et al. shows that upon BDNF stimulation, TrkB binds to htt and dynein in dendrites of cultured striatal neurons, and wild-type htt promotes TrkB transport via dynein-dependent mechanism. In striatal neurons from HD mouse model, mutant htt alters binding of TrkB-containing vesicles to microtubules and reduces TrkB retrograde transport in dendrites, leading to reduced BDNF/TrkB signaling (Liot et al., 2013). These findings demonstrate that in addition to affecting BDNF axonal corticostriatal transport, mutant htt can also impair retrograde trafficking of the TrkB-positive endosomes from striatal dendrites to the cell bodies. This transport alteration may further impair BDNF-TrkB signaling, which plays a crucial role in the survival and maintenance of striatal neurons (Baydyuk et al., 2011).
BDNF Rescues HD Phenotype—Therapeutic Implications
The multiple lines of evidence discussed above indicate that reduction in striatal BDNF signaling plays a pivotal role in the pathogenesis of HD. As a result, efforts have been made to examine whether increasing BDNF expression may be a viable strategy for treating HD. Indeed, increasing striatal BDNF levels via pharmacological or behavioral stimulation that induces Bdnf gene expression (Duan et al., 2003; Spires et al., 2004; Peng et al., 2008; Simmons et al., 2009) or by viral delivery (Cho et al., 2007), improves disease phenotypes in several HD mouse models. Early symptoms in HD patients are manifested by cognitive and memory deficits that precede the characteristic motor dysfunction (Lawrence et al., 1998; Ho et al., 2003). Similarly, in HD mouse models, impaired learning and memory, measured as hippocampal LTP, occur prior to motor deficits and neuronal loss (Murphy et al., 2000; Mazarakis et al., 2005; Van Raamsdonk et al., 2005). Since BDNF has been shown to modulate LTP, the reduced levels of BDNF in HD patients and mice can disrupt synaptic changes important for learning and memory formation. Applying low concentrations of BDNF to hippocampal slices prepared from HD mice fully restores LTP (Lynch et al., 2007). Furthermore, up-regulation of endogenous BDNF levels with an ampakine, a positive modulator of AMPA-type glutamate receptors, rescues synaptic plasticity and reduces learning deficits in HD mice (Simmons et al., 2009).
In addition to promoting survival and inducing synaptic plasticity, BDNF also regulates adult neurogenesis (Scharfman et al., 2005; Henry et al., 2007; Bath et al., 2011), which appears to be altered in HD mouse models and human postmortem brains (Curtis et al., 2003; Gil et al., 2005; Phillips et al., 2005). Adenoviral delivery of BDNF to the striatum of R6/2 HD mice induces striatal neurogenesis (Cho et al., 2007). Majority of the newly born neurons differentiate to MSNs and become functional, delaying motor impairment and prolonging survival in R6/2 mice. Similar improvements have been seen in the same HD mouse model after administration of the antidepressant sertraline (Peng et al., 2008). By increasing BDNF levels and stimulating neurogenesis, sertraline treatment results in improved motor performance, reduced striatal atrophy, and prolonged survival.
To directly evaluate the effect of increased cortical BDNF supply to the striatum on the progression of HD, our group and others have examined the consequences of overexpression of BDNF in the cortex. In our studies, we employ a Bdnf transgene under the control of the promoter for Ca2+/calmodulin-dependent protein kinase II alpha (Gharami et al., 2008; Xie et al., 2010). This transgene starts to express BDNF in the cerebral cortex in the first postnatal week and reaches plateau in the third postnatal week, as does the endogenous Bdnf gene (Huang et al., 1999). It also expresses at low levels in the striatum where the endogenous Bdnf gene is mostly inactive (Gharami et al., 2008; Xie et al., 2010). We show that the Bdnf transgene is able to greatly increase BDNF levels in the striata of two HD mouse lines, R6/1 and YAC128, indicating that BDNF overexpressed in the cortex is efficiently transported to the striatum, despite the presence of mutant htt. Importantly, BDNF overexpression reverses brain atrophy, normalizes the expression of several important genes in the striatum, and ameliorates deficits in motor coordination in these two HD mouse models (Gharami et al., 2008; Xie et al., 2010). In addition, overexpression of BDNF in YAC128 mice prevents loss of striatal neurons, normalizes spine abnormalities of MSNs, and significantly improves procedural learning (Xie et al., 2010). In summary, these studies suggest that increasing striatal BDNF levels may have therapeutic potential for HD.
Concluding Remarks
We have described the important roles of neurotrophic signaling in the developing, adult, and diseased striatum. We have examined a novel model of neurotrophic signaling in the developing striatum, where neurotrophins, anterogradely transported from the midbrain dopaminergic neurons, provide a vital support for immature neurons at their origin. However, it remains unclear whether the mechanisms of action for neurotrophins derived from the target tissue as seen in the PNS or transported from the innervating neurons as seen in the striatum, differ from one another. A comprehensive examination of signaling cascades affected by abolishing BDNF-TrkB signaling in the developing and adult striatum could provide some insights into this question. Another major point raised in this review is that BDNF-mediated neuronal survival is occurring early in development, at the time when neurons are being generated, and before mature connections are formed. During postnatal development, BDNF arriving via anterograde axonal transport promotes striatal maturation and influences synaptic connectivity. Thus, BDNF signaling plays various roles at different stages of neuronal development.
Numerous studies presented in this review undoubtedly link BDNF loss in the striatum and HD pathogenesis. Currently, drugs used to treat HD act on the symptoms and do not slow or stop the disease progression. Attempting to restore striatal BDNF levels or activate downstream signaling pathways may have therapeutic potential in treating HD patients. Indeed, multiple lines of evidence, discussed above, suggest that restoring cortical expression, axonal transport, and release of BDNF in the striatum promotes neuronal survival and improves behavioral phenotypes in HD animal models. These findings indicate that increasing BDNF signaling may also overcome functional deficits observed in HD patients.
Conflict of Interest Statement
The Reviewer Dr. Tressarollo declares that, despite having collaborated with the authors, the review process was handled objectively. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the NIH Fellows Editorial Board for the editorial assistance. This work was supported by National Institutes of Health Grant R01 NS050596 (Baoji Xu) and NINDS Intramural Research Program (Maryna Baydyuk).
References
Aid, T., Kazantseva, A., Piirsoo, M., Palm, K., and Timmusk, T. (2007). Mouse and rat BDNF gene structure and expression revisited. J. Neurosci. Res. 85, 525–535. doi: 10.1002/jnr.21139
Alcántara, S., Frisén, J., Del Río, J. A., Soriano, E., Barbacid, M., and Silos-Santiago, I. (1997). TrkB signaling is required for postnatal survival of CNS neurons and protects hippocampal and motor neurons from axotomy-induced cell death. J. Neurosci. 17, 3623–3633.
Altar, C. A., Cai, N., Bliven, T., Juhasz, M., Conner, J. M., Acheson, A. L., et al. (1997). Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature 389, 856–860. doi: 10.1038/39885
Anderson, S. A., Qiu, M., Bulfone, A., Eisenstat, D. D., Meneses, J., Pedersen, R., et al. (1997). Mutations of the homeobox genes Dlx-1 and Dlx-2 disrupt the striatal subventricular zone and differentiation of late born striatal neurons. Neuron 19, 27–37. doi: 10.1016/s0896-6273(00)80345-1
Apostol, B. L., Simmons, D. A., Zuccato, C., Illes, K., Pallos, J., Casale, M., et al. (2008). CEP-1347 reduces mutant huntingtin-associated neurotoxicity and restores BDNF levels in R6/2 mice. Mol. Cell. Neurosci. 39, 8–20. doi: 10.1016/j.mcn.2008.04.007
Baquet, Z. C., Gorski, J. A., and Jones, K. R. (2004). Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J. Neurosci. 24, 4250–4258. doi: 10.1523/jneurosci.3920-03.2004
Bartkowska, K., Paquin, A., Gauthier, A. S., Kaplan, D. R., and Miller, F. D. (2007). Trk signaling regulates neural precursor cell proliferation and differentiation during cortical development. Development 134, 4369–4380. doi: 10.1242/dev.008227
Bath, K. G., Akins, M. R., and Lee, F. S. (2011). BDNF control of adult SVZ neurogenesis. Dev. Psychobiol. 54, 578–589. doi: 10.1002/dev.20546
Baydyuk, M., Russell, T., Liao, G. Y., Zang, K., An, J. J., Reichardt, L. F., et al. (2011). TrkB receptor controls striatal formation by regulating the number of newborn striatal neurons. Proc. Natl. Acad. Sci. U S A 108, 1669–1674. doi: 10.1073/pnas.1004744108
Baydyuk, M., Xie, Y., Tessarollo, L., and Xu, B. (2013). Midbrain-derived neurotrophins support survival of immature striatal projection neurons. J. Neurosci. 33, 3363–3369. doi: 10.1523/JNEUROSCI.3687-12.2013
Besusso, D., Geibel, M., Kramer, D., Schneider, T., Pendolino, V., Picconi, B., et al. (2013). BDNF-TrkB signaling in striatopallidal neurons controls inhibition of locomotor behavior. Nat. Commun. 4:2031. doi: 10.1038/ncomms3031
Bhave, S. V., Ghoda, L., and Hoffman, P. L. (1999). Brain-derived neurotrophic factor mediates the anti-apoptotic effect of NMDA in cerebellar granule neurons: signal transduction cascades and site of ethanol action. J. Neurosci. 19, 3277–3286.
Block-Galarza, J., Chase, K. O., Sapp, E., Vaughn, K. T., Vallee, R. B., DiFiglia, M., et al. (1997). Fast transport and retrograde movement of huntingtin and HAP 1 in axons. Neuroreport 8, 2247–2251. doi: 10.1097/00001756-199707070-00031
Bolam, J. P., Hanley, J. J., Booth, P. A., and Bevan, M. D. (2000). Synaptic organisation of the basal ganglia. J. Anat. 196, 527–542. doi: 10.1046/j.1469-7580.2000.19640527.x
Borrell-Pagès, M., Zala, D., Humbert, S., and Saudou, F. (2006). Huntington’s disease: from huntingtin function and dysfunction to therapeutic strategies. Cell. Mol. Life Sci. 63, 2642–2660. doi: 10.1007/s00018-006-6242-0
Calabresi, P., Centonze, D., Gubellini, P., Marfia, G. A., Pisani, A., Sancesario, G., et al. (2000). Synaptic transmission in the striatum: from plasticity to neurodegeneration. Prog. Neurobiol. 61, 231–265. doi: 10.1016/s0301-0082(99)00030-1
Canals, J. M., Pineda, J. R., Torres-Peraza, J. F., Bosch, M., Martin-Ibanez, R., Munoz, M. T., et al. (2004). Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington’s disease. J. Neurosci. 24, 7727–7739. doi: 10.1523/jneurosci.1197-04.2004
Casarosa, S., Fode, C., and Guillemot, F. (1999). Mash1 regulates neurogenesis in the ventral telencephalon. Development 126, 525–534.
Cattaneo, E. (2003). Dysfunction of wild-type huntingtin in Huntington disease. News Physiol. Sci. 18, 34–37. doi: 10.1152/nips.01410.2002
Chao, M. V. (2003). Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat. Rev. Neurosci. 4, 299–309. doi: 10.1038/nrn1078
Cho, S. R., Benraiss, A., Chmielnicki, E., Samdani, A., Economides, A., and Goldman, S. A. (2007). Induction of neostriatal neurogenesis slows disease progression in a transgenic murine model of Huntington disease. J. Clin. Invest. 117, 2889–2902. doi: 10.1172/jci31778
Christensen, J., Sorensen, J. C., Ostergaard, K., and Zimmer, J. (1999). Early postnatal development of the rat corticostriatal pathway: an anterograde axonal tracing study using biocytin pellets. Anat. Embryol. (Berl) 200, 73–80. doi: 10.1007/s004290050261
Ciammola, A., Sassone, J., Cannella, M., Calza, S., Poletti, B., Frati, L., et al. (2007). Low brain-derived neurotrophic factor (BDNF) levels in serum of Huntington’s disease patients. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 574–577. doi: 10.1002/ajmg.b.30501
Cohen, M. S., Bas Orth, C., Kim, H. J., Jeon, N. L., and Jaffrey, S. R. (2011). Neurotrophin-mediated dendrite-to-nucleus signaling revealed by microfluidic compartmentalization of dendrites. Proc. Natl. Acad. Sci. U S A 108, 11246–11251. doi: 10.1073/pnas.1012401108
Conner, J. M., Lauterborn, J. C., Yan, Q., Gall, C. M., and Varon, S. (1997). Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J. Neurosci. 17, 2295–2313.
Coppola, V., Kucera, J., Palko, M. E., Martinez-De Velasco, J., Lyons, W. E., Fritzsch, B., et al. (2001). Dissection of NT3 functions in vivo by gene replacement strategy. Development 128, 4315–4327.
Corbin, J. G., Gaiano, N., Machold, R. P., Langston, A., and Fishell, G. (2000). The Gsh2 homeodomain gene controls multiple aspects of telencephalic development. Development 127, 5007–5020.
Crowley, C., Spencer, S. D., Nishimura, M. C., Chen, K. S., Pitts-Meek, S., Armanini, M. P., et al. (1994). Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell 76, 1001–1011. doi: 10.1016/0092-8674(94)90378-6
Cummings, D. M., Andre, V. M., Uzgil, B. O., Gee, S. M., Fisher, Y. E., Cepeda, C., et al. (2009). Alterations in cortical excitation and inhibition in genetic mouse models of Huntington’s disease. J. Neurosci. 29, 10371–10386. doi: 10.1523/JNEUROSCI.1592-09.2009
Curtis, M. A., Penney, E. B., Pearson, A. G., van Roon-Mom, W. M., Butterworth, N. J., Dragunow, M., et al. (2003). Increased cell proliferation and neurogenesis in the adult human Huntington’s disease brain. Proc. Natl. Acad. Sci. U S A 100, 9023–9027. doi: 10.1073/pnas.1532244100
Day, M., Wang, Z., Ding, J., An, X., Ingham, C. A., Shering, A. F., et al. (2006). Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat. Neurosci. 9, 251–259. doi: 10.1038/nn1632
Duan, W., Guo, Z., Jiang, H., Ware, M., Li, X. J., and Mattson, M. P. (2003). Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression and increases survival in huntingtin mutant mice. Proc. Natl. Acad. Sci. U S A 100, 2911–2916. doi: 10.1073/pnas.0536856100
Encinas, M., Iglesias, M., Llecha, N., and Comella, J. X. (1999). Extracellular-regulated kinases and phosphatidylinositol 3-kinase are involved in brain-derived neurotrophic factor-mediated survival and neuritogenesis of the neuroblastoma cell line SH-SY5Y. J. Neurochem. 73, 1409–1421. doi: 10.1046/j.1471-4159.1999.0731409.x
Engelender, S., Sharp, A. H., Colomer, V., Tokito, M. K., Lanahan, A., Worley, P., et al. (1997). Huntingtin-associated protein 1 (HAP1) interacts with the p150Glued subunit of dynactin. Hum. Mol. Genet. 6, 2205–2212. doi: 10.1093/hmg/6.13.2205
Fariñas, I., Jones, K. R., Tessarollo, L., Vigers, A. J., Huang, E., Kirstein, M., et al. (2001). Spatial shaping of cochlear innervation by temporally regulated neurotrophin expression. J. Neurosci. 21, 6170–6180.
Fariñas, I., Wilkinson, G. A., Backus, C., Reichardt, L. F., and Patapoutian, A. (1998). Characterization of neurotrophin and Trk receptor functions in developing sensory ganglia: direct NT-3 activation of TrkB neurons in vivo. Neuron 21, 325–334. doi: 10.1016/s0896-6273(00)80542-5
Fariñas, I., Yoshida, C. K., Backus, C., and Reichardt, L. F. (1996). Lack of neurotrophin-3 results in death of spinal sensory neurons and premature differentiation of their precursors. Neuron 17, 1065–1078. doi: 10.1016/s0896-6273(00)80240-8
Ferrante, R. J., Beal, M. F., Kowall, N. W., Richardson, E. P. Jr., and Martin, J. B. (1987a). Sparing of acetylcholinesterase-containing striatal neurons in Huntington’s disease. Brain Res. 411, 162–166. doi: 10.1016/0006-8993(87)90694-9
Ferrante, R. J., Kowall, N. W., Beal, M. F., Martin, J. B., Bird, E. D., Richardson, E. P. Jr., et al. (1987b). Morphologic and histochemical characteristics of a spared subset of striatal neurons in Huntington’s disease. J. Neuropathol. Exp. Neurol. 46, 12–27. doi: 10.1097/00005072-198701000-00002
Ferrer, I., Goutan, E., Marin, C., Rey, M. J., and Ribalta, T. (2000). Brain-derived neurotrophic factor in Huntington disease. Brain Res. 866, 257–261. doi: 10.1016/s0006-8993(00)02237-x
Gauthier, L. R., Charrin, B. C., Borrell-Pages, M., Dompierre, J. P., Rangone, H., Cordelieres, F. P., et al. (2004). Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118, 127–138. doi: 10.1016/j.cell.2004.06.018
Gerfen, C. R., Engber, T. M., Mahan, L. C., Susel, Z., Chase, T. N., Monsma, F. J. Jr., et al. (1990). D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 250, 1429–1432. doi: 10.1126/science.2147780
Gerfen, C. R., Miyachi, S., Paletzki, R., and Brown, P. (2002). D1 dopamine receptor supersensitivity in the dopamine-depleted striatum results from a switch in the regulation of ERK1/2/MAP kinase. J. Neurosci. 22, 5042–5054.
Gharami, K., Xie, Y., An, J. J., Tonegawa, S., and Xu, B. (2008). Brain-derived neurotrophic factor over-expression in the forebrain ameliorates Huntington’s disease phenotypes in mice. J. Neurochem. 105, 369–379. doi: 10.1111/j.1471-4159.2007.05137.x
Gil, J. M., Mohapel, P., Araujo, I. M., Popovic, N., Li, J. Y., Brundin, P., et al. (2005). Reduced hippocampal neurogenesis in R6/2 transgenic Huntington’s disease mice. Neurobiol. Dis. 20, 744–751. doi: 10.1016/j.nbd.2005.05.006
Gorski, J. A., Zeiler, S. R., Tamowski, S., and Jones, K. R. (2003). Brain-derived neurotrophic factor is required for the maintenance of cortical dendrites. J. Neurosci. 23, 6856–6865.
Gusella, J. F., and MacDonald, M. E. (2000). Molecular genetics: unmasking polyglutamine triggers in neurodegenerative disease. Nat. Rev. Neurosci. 1, 109–115. doi: 10.1038/35039051
Ha, J., Lo, K. W., Myers, K. R., Carr, T. M., Humsi, M. K., Rasoul, B. A., et al. (2008). A neuron-specific cytoplasmic dynein isoform preferentially transports TrkB signaling endosomes. J. Cell Biol. 181, 1027–1039. doi: 10.1083/jcb.200803150
Hamasaki, T., Goto, S., Nishikawa, S., and Ushio, Y. (2003). Neuronal cell migration for the developmental formation of the mammalian striatum. Brain Res. Brain Res. Rev. 41, 1–12. doi: 10.1016/s0165-0173(02)00216-3
Heerssen, H. M., Pazyra, M. F., and Segal, R. A. (2004). Dynein motors transport activated Trks to promote survival of target-dependent neurons. Nat. Neurosci. 7, 596–604. doi: 10.1038/nn1242
Henry, R. A., Hughes, S. M., and Connor, B. (2007). AAV-mediated delivery of BDNF augments neurogenesis in the normal and quinolinic acid-lesioned adult rat brain. Eur. J. Neurosci. 25, 3513–3525. doi: 10.1111/j.1460-9568.2007.05625.x
Ho, A. K., Sahakian, B. J., Brown, R. G., Barker, R. A., Hodges, J. R., Ane, M. N., et al. (2003). Profile of cognitive progression in early Huntington’s disease. Neurology 61, 1702–1706. doi: 10.1212/01.wnl.0000098878.47789.bd
Hu, Z., Cooper, M., Crockett, D. P., and Zhou, R. (2004). Differentiation of the midbrain dopaminergic pathways during mouse development. J. Comp. Neurol. 476, 301–311. doi: 10.1002/cne.20230
Huang, Z. J., Kirkwood, A., Pizzorusso, T., Porciatti, V., Morales, B., Bear, M. F., et al. (1999). BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell 98, 739–755. doi: 10.1016/s0092-8674(00)81509-3
Kawaguchi, Y. (1997). Neostriatal cell subtypes and their functional roles. Neurosci. Res. 27, 1–8. doi: 10.1016/s0168-0102(96)01134-0
Klein, R., Martin-Zanca, D., Barbacid, M., and Parada, L. F. (1990). Expression of the tyrosine kinase receptor gene trkB is confined to the murine embryonic and adult nervous system. Development 109, 845–850.
Lawrence, A. D., Hodges, J. R., Rosser, A. E., Kershaw, A., Ffrench-Constant, C., Rubinsztein, D. C., et al. (1998). Evidence for specific cognitive deficits in preclinical Huntington’s disease. Brain 121(Pt. 7), 1329–1341. doi: 10.1093/brain/121.7.1329
Li, S. H., Gutekunst, C. A., Hersch, S. M., and Li, X. J. (1998). Interaction of huntingtin-associated protein with dynactin P150Glued. J. Neurosci. 18, 1261–1269.
Li, Y., Yui, D., Luikart, B. W., McKay, R. M., Li, Y., Rubenstein, J. L., et al. (2012). Conditional ablation of brain-derived neurotrophic factor-TrkB signaling impairs striatal neuron development. Proc. Natl. Acad. Sci. U S A 109, 15491–15496. doi: 10.1073/pnas.1212899109
Liot, G., Zala, D., Pla, P., Mottet, G., Piel, M., and Saudou, F. (2013). Mutant huntingtin alters retrograde transport of TrkB receptors in striatal dendrites. J. Neurosci. 33, 6298–6309. doi: 10.1523/jneurosci.2033-12.2013
Lynch, G., Kramar, E. A., Rex, C. S., Jia, Y., Chappas, D., Gall, C. M., et al. (2007). Brain-derived neurotrophic factor restores synaptic plasticity in a knock-in mouse model of Huntington’s disease. J. Neurosci. 27, 4424–4434. doi: 10.1523/jneurosci.5113-06.2007
Mann, D. M., Oliver, R., and Snowden, J. S. (1993). The topographic distribution of brain atrophy in Huntington’s disease and progressive supranuclear palsy. Acta Neuropathol. 85, 553–559. doi: 10.1007/BF00230496
Marchand, R., and Lajoie, L. (1986). Histogenesis of the striopallidal system in the rat. Neurogenesis of its neurons. Neuroscience 17, 573–590. doi: 10.1016/0306-4522(86)90031-x
Mazarakis, N. K., Cybulska-Klosowicz, A., Grote, H., Pang, T., Van Dellen, A., Kossut, M., et al. (2005). Deficits in experience-dependent cortical plasticity and sensory-discrimination learning in presymptomatic Huntington’s disease mice. J. Neurosci. 25, 3059–3066. doi: 10.1523/jneurosci.4320-04.2005
McAllister, A. K., Katz, L. C., and Lo, D. C. (1999). Neurotrophins and synaptic plasticity. Annu. Rev. Neurosci. 22, 295–318. doi: 10.1146/annurev.neuro.22.1.295
Metsis, M., Timmusk, T., Arenas, E., and Persson, H. (1993). Differential usage of multiple brain-derived neurotrophic factor promoters in the rat brain following neuronal activation. Proc. Natl. Acad. Sci. U S A 90, 8802–8806. doi: 10.1073/pnas.90.19.8802
Minichiello, L., and Klein, R. (1996). TrkB and TrkC neurotrophin receptors cooperate in promoting survival of hippocampal and cerebellar granule neurons. Genes Dev. 10, 2849–2858. doi: 10.1101/gad.10.22.2849
Mink, J. W., and Thach, W. T. (1993). Basal ganglia intrinsic circuits and their role in behavior. Curr. Opin. Neurobiol. 3, 950–957. doi: 10.1016/0959-4388(93)90167-w
Murphy, K. P., Carter, R. J., Lione, L. A., Mangiarini, L., Mahal, A., Bates, G. P., et al. (2000). Abnormal synaptic plasticity and impaired spatial cognition in mice transgenic for exon 1 of the human Huntington’s disease mutation. J. Neurosci. 20, 5115–5123.
Nisenbaum, L. K., Webster, S. M., Chang, S. L., McQueeney, K. D., and Loturco, J. J. (1998). Early patterning of prelimbic cortical axons to the striatal patch compartment in the neonatal mouse. Dev. Neurosci. 20, 113–124. doi: 10.1159/000017307
Ohtani, N., Goto, T., Waeber, C., and Bhide, P. G. (2003). Dopamine modulates cell cycle in the lateral ganglionic eminence. J. Neurosci. 23, 2840–2850.
Olsson, M., Bjorklund, A., and Campbell, K. (1998). Early specification of striatal projection neurons and interneuronal subtypes in the lateral and medial ganglionic eminence. Neuroscience 84, 867–876. doi: 10.1016/s0306-4522(97)00532-0
Olsson, M., Campbell, K., Wictorin, K., and Bjorklund, A. (1995). Projection neurons in fetal striatal transplants are predominantly derived from the lateral ganglionic eminence. Neuroscience 69, 1169–1182. doi: 10.1016/0306-4522(95)00325-d
Orefice, L. L., Waterhouse, E. G., Partridge, J. G., Lalchandani, R. R., Vicini, S., and Xu, B. (2013). Distinct roles for somatically and dendritically synthesized brain-derived neurotrophic factor in morphogenesis of dendritic spines. J. Neurosci. 33, 11618–11632. doi: 10.1523/JNEUROSCI.0012-13.2013
Parent, A., and Hazrati, L. N. (1995). Functional anatomy of the basal ganglia. I. The cortico-basal ganglia-thalamo-cortical loop. Brain Res. Brain Res. Rev. 20, 91–127. doi: 10.1016/0165-0173(94)00007-c
Pattabiraman, P. P., Tropea, D., Chiaruttini, C., Tongiorgi, E., Cattaneo, A., and Domenici, L. (2005). Neuronal activity regulates the developmental expression and subcellular localization of cortical BDNF mRNA isoforms in vivo. Mol. Cell. Neurosci. 28, 556–570. doi: 10.1016/j.mcn.2004.11.010
Peng, Q., Masuda, N., Jiang, M., Li, Q., Zhao, M., Ross, C. A., et al. (2008). The antidepressant sertraline improves the phenotype, promotes neurogenesis and increases BDNF levels in the R6/2 Huntington’s disease mouse model. Exp. Neurol. 210, 154–163. doi: 10.1016/j.expneurol.2007.10.015
Phillips, W., Morton, A. J., and Barker, R. A. (2005). Abnormalities of neurogenesis in the R6/2 mouse model of Huntington’s disease are attributable to the in vivo microenvironment. J. Neurosci. 25, 11564–11576. doi: 10.1523/jneurosci.3796-05.2005
Poo, M. M. (2001). Neurotrophins as synaptic modulators. Nat. Rev. Neurosci. 2, 24–32. doi: 10.1038/35049004
Rauskolb, S., Zagrebelsky, M., Dreznjak, A., Deogracias, R., Matsumoto, T., Wiese, S., et al. (2010). Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J. Neurosci. 30, 1739–1749. doi: 10.1523/JNEUROSCI.5100-09.2010
Reichardt, L. F. (2006). Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 1545–1564. doi: 10.1098/rstb.2006.1894
Reiner, A., Albin, R. L., Anderson, K. D., D’Amato, C. J., Penney, J. B., and Young, A. B. (1988). Differential loss of striatal projection neurons in Huntington disease. Proc. Natl. Acad. Sci. U S A 85, 5733–5737. doi: 10.1073/pnas.85.15.5733
Scharfman, H., Goodman, J., Macleod, A., Phani, S., Antonelli, C., and Croll, S. (2005). Increased neurogenesis and the ectopic granule cells after intrahippocampal BDNF infusion in adult rats. Exp. Neurol. 192, 348–356. doi: 10.1016/j.expneurol.2004.11.016
Segal, R. A. (2003). Selectivity in neurotrophin signaling: theme and variations. Annu. Rev. Neurosci. 26, 299–330. doi: 10.1146/annurev.neuro.26.041002.131421
Shuen, J. A., Chen, M., Gloss, B., and Calakos, N. (2008). Drd1a-tdTomato BAC transgenic mice for simultaneous visualization of medium spiny neurons in the direct and indirect pathways of the basal ganglia. J. Neurosci. 28, 2681–2685. doi: 10.1523/JNEUROSCI.5492-07.2008
Simmons, D. A., Rex, C. S., Palmer, L., Pandyarajan, V., Fedulov, V., Gall, C. M., et al. (2009). Up-regulating BDNF with an ampakine rescues synaptic plasticity and memory in Huntington’s disease knockin mice. Proc. Natl. Acad. Sci. U S A 106, 4906–4911. doi: 10.1073/pnas.0811228106
Smeyne, R. J., Klein, R., Schnapp, A., Long, L. K., Bryant, S., Lewin, A., et al. (1994). Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature 368, 246–249. doi: 10.1038/368246a0
Spires, T. L., Grote, H. E., Varshney, N. K., Cordery, P. M., Van Dellen, A., Blakemore, C., et al. (2004). Environmental enrichment rescues protein deficits in a mouse model of Huntington’s disease, indicating a possible disease mechanism. J. Neurosci. 24, 2270–2276. doi: 10.1523/jneurosci.1658-03.2004
Stenman, J., Toresson, H., and Campbell, K. (2003). Identification of two distinct progenitor populations in the lateral ganglionic eminence: implications for striatal and olfactory bulb neurogenesis. J. Neurosci. 23, 167–174.
Strand, A. D., Baquet, Z. C., Aragaki, A. K., Holmans, P., Yang, L., Cleren, C., et al. (2007). Expression profiling of Huntington’s disease models suggests that brain-derived neurotrophic factor depletion plays a major role in striatal degeneration. J. Neurosci. 27, 11758–11768. doi: 10.1523/jneurosci.2461-07.2007
Svenningsson, P., Nishi, A., Fisone, G., Girault, J. A., Nairn, A. C., and Greengard, P. (2004). DARPP-32: an integrator of neurotransmission. Annu. Rev. Pharmacol. Toxicol. 44, 269–296. doi: 10.1146/annurev.pharmtox.44.101802.121415
Timmusk, T., Palm, K., Metsis, M., Reintam, T., Paalme, V., Saarma, M., et al. (1993). Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron 10, 475–489. doi: 10.1016/0896-6273(93)90335-o
Van Raamsdonk, J. M., Pearson, J., Slow, E. J., Hossain, S. M., Leavitt, B. R., and Hayden, M. R. (2005). Cognitive dysfunction precedes neuropathology and motor abnormalities in the YAC128 mouse model of Huntington’s disease. J. Neurosci. 25, 4169–4180. doi: 10.1523/jneurosci.0590-05.2005
Vonsattel, J. P., and DiFiglia, M. (1998). Huntington disease. J. Neuropathol. Exp. Neurol. 57, 369–384.
Voorn, P., Kalsbeek, A., Jorritsma-Byham, B., and Groenewegen, H. J. (1988). The pre- and postnatal development of the dopaminergic cell groups in the ventral mesencephalon and the dopaminergic innervation of the striatum of the rat. Neuroscience 25, 857–887. doi: 10.1016/0306-4522(88)90041-3
Watson, F. L., Heerssen, H. M., Moheban, D. B., Lin, M. Z., Sauvageot, C. M., Bhattacharyya, A., et al. (1999). Rapid nuclear responses to target-derived neurotrophins require retrograde transport of ligand-receptor complex. J. Neurosci. 19, 7889–7900.
Xie, Y., Hayden, M. R., and Xu, B. (2010). BDNF overexpression in the forebrain rescues Huntington’s disease phenotypes in YAC128 mice. J. Neurosci. 30, 14708–14718. doi: 10.1523/JNEUROSCI.1637-10.2010
Xu, B., Gottschalk, W., Chow, A., Wilson, R. I., Schnell, E., Zang, K., et al. (2000a). The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. J. Neurosci. 20, 6888–6897.
Xu, B., Zang, K., Ruff, N. L., Zhang, Y. A., Mcconnell, S. K., Stryker, M. P., et al. (2000b). Cortical degeneration in the absence of neurotrophin signaling: dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron 26, 233–245. doi: 10.1016/S0896-6273(00)81153-8
Yamada, M., Ohnishi, H., Sano, S., Araki, T., Nakatani, A., Ikeuchi, T., et al. (1999). Brain-derived neurotrophic factor stimulates interactions of Shp2 with phosphatidylinositol 3-kinase and Grb2 in cultured cerebral cortical neurons. J. Neurochem. 73, 41–49. doi: 10.1046/j.1471-4159.1999.0730041.x
Yamada, M., Suzuki, K., Mizutani, M., Asada, A., Matozaki, T., Ikeuchi, T., et al. (2001). Analysis of tyrosine phosphorylation-dependent protein-protein interactions in trkb-mediated intracellular signaling using modified yeast two-hybrid system. J. Biochem. 130, 157–165. doi: 10.1093/oxfordjournals.jbchem.a002955
Yan, Q., Radeke, M. J., Matheson, C. R., Talvenheimo, J., Welcher, A. A., and Feinstein, S. C. (1997). Immunocytochemical localization of TrkB in the central nervous system of the adult rat. J. Comp. Neurol. 378, 135–157. doi: 10.1002/(SICI)1096-9861(19970203)378:1<135::AID-CNE8>3.0.CO;2-5
Zuccato, C., Ciammola, A., Rigamonti, D., Leavitt, B. R., Goffredo, D., Conti, L., et al. (2001). Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 293, 493–498. doi: 10.1126/science.1059581
Zuccato, C., Marullo, M., Conforti, P., Macdonald, M. E., Tartari, M., and Cattaneo, E. (2008). Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington’s disease. Brain Pathol. 18, 225–238. doi: 10.1111/j.1750-3639.2007.00111.x
Zuccato, C., Tartari, M., Crotti, A., Goffredo, D., Valenza, M., Conti, L., et al. (2003). Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet. 35, 76–83. doi: 10.1038/ng1219
Keywords: neurotrophins, BDNF, TrkB, striatum, Huntington’s disease, DRD1a, DRD2, dopaminergic neurons
Citation: Baydyuk M and Xu B (2014) BDNF signaling and survival of striatal neurons. Front. Cell. Neurosci. 8:254. doi: 10.3389/fncel.2014.00254
Received: 21 May 2014; Accepted: 11 August 2014;
Published online: 28 August 2014.
Edited by:
Marie-Christin Pauly, University Freiburg - Medical Center, GermanyReviewed by:
James M. Tepper, Rutgers, The State University of NJ, USALino Tessarollo, Mouse Cancer Genetics Program, Center for Cancer Research, National Cancer Institute, National Institutes of Health (NIH), USA
Copyright © 2014 Baydyuk and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Baoji Xu, Department of Neuroscience, The Scripps Research Institute Florida, 130 Scripps Way, #3C1, Jupiter, FL 33458, USA e-mail: bxu@scripps.edu