Synovial Fibrosis Involvement in Osteoarthritis
 Li Zhang1†
Li Zhang1†  Runlin Xing1,2† Zhengquan Huang1,2 Liang Ding1,2 Li Zhang1,2 Mingchao Li1 Xiaochen Li1 Peimin Wang1,2* Jun Mao1,2
Runlin Xing1,2† Zhengquan Huang1,2 Liang Ding1,2 Li Zhang1,2 Mingchao Li1 Xiaochen Li1 Peimin Wang1,2* Jun Mao1,2- 1Departments of Orthopedics, The Affiliated Hospital of Nanjing University of Chinese Medicine, Nanjing, China
- 2Jiangsu Province Hospital of Chinese Medicine, Nanjing, China
Bone changes have always been the focus of research on osteoarthritis, but the number of studies on synovitis has increased only over the last 10 years. Our current understanding is that the mechanism of osteoarthritis involves all the tissues that make up the joints, including nerve sprouting, pannus formation, and extracellular matrix environmental changes in the synovium. These factors together determine synovial fibrosis and may be closely associated with the clinical symptoms of pain, hyperalgesia, and stiffness in osteoarthritis. In this review, we summarize the consensus of clinical work, the potential pathological mechanisms, the possible therapeutic targets, and the available therapeutic strategies for synovial fibrosis in osteoarthritis to gain insight and provide a foundation for further study.
Rheumatology key Messages
Synovial fibrosis is closely associated with joint pain, hyperalgesia, and stiffness in osteoarthritis.
Sounder diagnostic criteria should be established for OA-related synovial fibrosis.
The mechanism of synovial fibrosis is being investigated, and available therapeutic strategies require further study.
Introduction
Osteoarthritis (OA) is the most common degenerative joint disease and is characterized by pain, stiffness, and limited function in the clinic (1). In 2017, OA affected nearly 303 million people worldwide, including ~263 million people with knee OA and 40 million people with hip OA (2). Bone changes, such as the progressive loss and destruction of articular cartilage, thickening of the subchondral bone, and the formation of osteophytes, reflect the pathogenesis of OA, so the study of cartilage and subchondral bone in OA has always been a priority (3). Such studies are highly consistent with the etiology of OA, which involves aging, mechanical stress, and environmental changes in the joints. Bone changes may be the determining factor for the eventual use of surgical treatment for OA; unexpectedly, the consistency of the bone structure with clinical symptoms remains unclear, at least in terms of pain (4, 5). This implies that further efforts are needed to discover the pathological mechanisms of OA, especially those related to OA symptoms.
As OA involves chronic low-grade inflammation, the presence of an inflammatory microenvironment is likely to affect all tissues constituting the joint (6). It is widely accepted that synovitis can occur in the early stage of OA, promoting the development of OA throughout the whole pathologic process. Therefore, non-steroidal anti-inflammatory drugs (NSAIDs) are strongly recommended for clinical treatment, and OA is considered to be a highly prevalent rheumatic musculoskeletal disorder (7, 8). As cartilage destruction partly depends on the effect of inflammation, which disrupts the balance between synthesis and degradation in the extracellular matrix (ECM) (7), it may also be valuable to evaluate the damage caused by inflammation in the synovium. Overall, one major outcome of inflammation or inflammatory exudation is fibrosis, especially in the lung, liver, and kidney. In OA, synovial fibrosis (SF) is an imbalance caused by fibroblast proliferation and the disturbance of collagen synthesis and degradation, ultimately leading to excessive collagen deposition in the ECM (9, 10).
Recent research has also revealed that the ECM plays multiple roles in OA (11, 12). This may indicate that SF is not only a pathological outcome but also a likely pathogenic factor. SF is often accompanied by angiogenesis in both OA and rheumatoid arthritis (RA) (13). Recent studies have also found evidence for increased sensory innervation in the synovium in knee OA, but there is still no direct evidence on whether SF is associated with pain (14, 15). Does joint stiffness due to fibrosis associate with OA pain? Is SF associated with increased sensory innervation? Can the progression of SF be blocked when synovitis is alleviated? Obtaining a narrative review of SF in OA is an interesting research direction; thus, we searched PubMed with the keywords “fibrosis,” “OA,” and “RA.” We reviewed the pertinent literature to answer the following questions: What do we know about SF in osteoarthritis?
Synovial Fibrosis and Synovitis
Synovitis is a typical chronic aseptic inflammation. Common symptoms caused by synovitis include pain, local temperature rise, swelling, joint movement limitation, and the severity of these symptoms is related to the degree of joint effusion (16). Synovitis is also known to produce a large number of pro-inflammatory factors, such as tumor necrosis factor (TNF), interleukin-1β (IL-1β), IL-6, IL-8, IL-15, IL-17, IL-21, inflammatory mediators, including PGE2, NO, adipokines, and matrix metalloproteinases (MMP-1, MMP-3, MMP-9, MMP-13), which lead to cartilage destruction, amplifying synovitis and ultimately creating a vicious cycle (17, 18). Besides, synovitis promotes the production of pain neurotransmitters, such as nerve growth factor and bradykinin (19). At the same time, synovitis promotes synovial angiogenesis, which in turn accelerates inflammation and leads to SF directly (20).
Usually, SF appears in the later stages of OA, which is different from synovitis. But in a study examining the effects of the intra-articular application of bupivacaine and levobupivacaine, inflammation and late fibrosis were found shortly after injection, suggesting that synovitis promotes fibrosis (21). On the other hand, as an aseptic chronic inflammatory disease, SF may be the inevitable outcome of “damage-repair,” and thus it can be emphasized that synovial inflammation drives the development of fibrosis. Notably, current studies cannot conclude that SF can cause synovitis independently, and whether synovial fibrosis can exist independently of synovitis, remains a topic of great interest to OA research. As for the relationship between synovitis and SF, maybe it is not well-understood what is the hen and what the egg, but this question is the one to inspire researchers' in-depth research.
Synovial Fibrosis in Osteoarthritis
Clinical Status of Synovial Fibrosis
Much evidence has shown that SF is one of the most important causes of joint stiffness, synovial hyperplasia, and limited function, which are common symptoms in moderate and severe OA; other evidence also confirmed that a higher SF score is correlated with lower scores for KL grade, which indicates that SF may be negatively associated with clinical symptoms of OA (22, 23). This is because generalized pain is a major claim in OA patients, while independent joint stiffness does not occur very often. When joint stiffness begins to bother OA patients and joint movement is limited, loss of function becomes a reality.
Surgical treatments for OA, such as total knee arthroplasty (TKA), can cause arthrofibrosis, a fibrosing pathology of the synovial membrane, and the infrapatellar fat pad (24). In contrast to moderate and severe OA, TKA eliminates the effects on cartilage and the meniscus, so post-operative pain and dysfunction derived from SF can be observed more easily. Kalson et al. attempted to establish criteria for the diagnosis, classification, and severity grading of soft-tissue fibrosis after TKA and suggested that the diagnosis of fibrosis after TKA should be based on the exclusion of other causes of stiffness, the range of movement of the knee, the pathological anatomy and histopathology (Table 1) (25). These recommendations may also be adapted for the diagnosis of SF associated with primary OA.
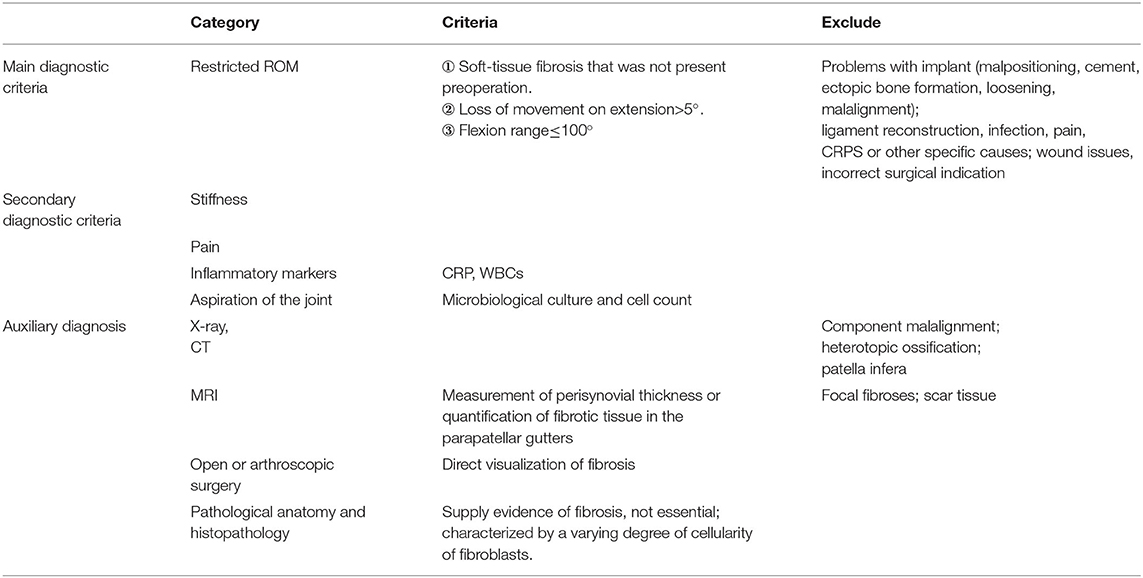
Table 1. Criteria for the diagnosis, classification and severity grading of soft-tissue fibrosis after TKA established by Kalson et al.
Recent developments in MRI and ultrasound have made it possible to investigate SF, but there is currently not enough evidence for routine use (26, 27). The degree of synovial thickening, not the volume, has been proven to be correlated with the level of SF on MRI, but others reported that the correlations between these factors were very weak (28, 29). This is likely because the MRI evaluation is based on the synovitis score, which fluctuates at different stages of OA, while the extent of fibrosis is relatively fixed. Ultrasound has also been shown to be useful in detecting and quantifying synovial abnormalities, especially for synovitis, as Doppler signals indicate active inflammation and vascularization in synovial arthritis but not fibrosis (30, 31). Laboratory tests of the synovium may be more advantageous for SF assessment than imaging evidence, and methods such as identification of cell phenotypes, quantitative detection of profibrotic markers, immunohistochemistry of collagen, and even HE staining can provide some guidance (32). According to the criteria established by Ruppert et al. for HE staining of sections, SF can be divided into three levels according to the ratio of the fibroblast-like synoviocyte (FLSs) length to the distance between FLSs (33). It seems that the “gold standard” evaluation for SF in OA is histology, although this requires an invasive biopsy that may not be applicable or acceptable to all patients.
Pathological Characteristics of Synovial Fibrosis
The normal synovium can be divided into the intima (synovial lining) and the subintima (outer layer). The intima comprises one to three layers of specialized columnar FLSs, which are interspersed with macrophages, while the subintima consists of multiple types of connective tissues, such as fibrous dense collagen, adipose tissue, or loose collagens. This layer is rich in type I and III collagen and microvascular blood supply, accompanied by lymphatic vessels and nerve fibers, but is relatively acellular (34, 35). From histological patterns, the synovium in OA patients is characterized by intima hyperplasia, subintima fibrosis, and stromal vascularization (36). In the latest report, scholars demonstrated increased innervation of the medial synovium after KOA surgical modeling, and the medial compartment of OA knees exhibited striking changes in NaV1.8+ innervation (7). Oehler et al. subtyped osteoarthritic synoviopathy and identified four patterns of OA-associated synoviopathy: hyperplastic, fibrotic, detritus-rich, and inflammatory synoviopathy (37). Interestingly, excluding hyperplastic synoviopathy, the remaining three subtypes are nearly all overlapping, with different emphases and degrees of inflammation. The fibrotic type has most of the characteristics of the other three types, but there is no macromolecular cartilage and bone debris, which is one of the typical features of detritus-rich cartilage. This indicates that OA induced by different factors could involve different subtypes of synoviopathy with varied features, and at least the fibrotic type originates more from inflammation than from cartilage debris. In summary, SF associated with OA should be identified as the accumulation of collagen under pathological conditions, dominated by abnormal remodeling of collagen types I and III in the subintima, together with angiogenesis and nerve invasion. Therefore, in the study of SF, angiogenesis and nerve invasion should also be considered.
The function of the normal synovium is mainly reflected by FLSs, as they are involved in the production of hyaluronan, collagens, and fibronectin in the intima and synovial fluid. This is essential for joint movement and cartilage nutrition (38). Macrophages make up a minority of cells in the normal intima, but their numbers increase dramatically in inflammatory arthritis (36, 38). Both types of cells are involved in SF associated with osteoarthritis. Surprisingly, these cells not only determine ECM changes but also dictate the functions of resident cells within tissues. The ECM supplies cells with proper chemical and mechanical signals to regulate cell proliferation, migration, and differentiation to maintain tissue homeostasis (36, 39). In SF, collagen I exhibit a disorganized structure and enhanced cross-linking, while collagen III is crucial for appropriate collagen I fibrillogenesis and tissue functionality (39). Petersen et al. believed that markers of type I or III collagen turnover may reflect the severity of synovitis and SF, which is highly correlated with OA pain sensitivity. Fragments of type I, II, and III collagens were then investigated in blood from OA patients compared with blood from control individuals, revealing increased degeneration of type I and II collagen and decreased degeneration of type III collagen, which was highly correlated with localized hyperalgesia in response to pressure stimulation (40). In conclusion, abnormal secretion of synovial cells forms the pathological basis of SF due to the inherent effects of the ECM as a pathogenic factor and biomechanical stimuli involved in OA. The subsequent collagen environment presents a fibrotic state with changes in synovial permeability and mechanical properties, which may cause pain and stiffness in OA joints.
Potential Etiology of Synovial Fibrosis
Fibrosis typically originates from abnormal tissue repair in response to wound healing. Fibroblasts activated by multiple diverse signals play a central role in this process, differentiate into myofibroblasts, and secrete matrix molecules to rebuild the ECM structure. Any risk factor for primary OA, such as aging, hypoxia, changes in the ECM environment, and mechanical stress, may play a similar role in SF (Table 2 and Figure 1). Although existing studies have not always targeted the synovium, evidence related to the balance of cartilage matrix degradation or fibrosis of other tissues can also provide some guidance for the study of SF.
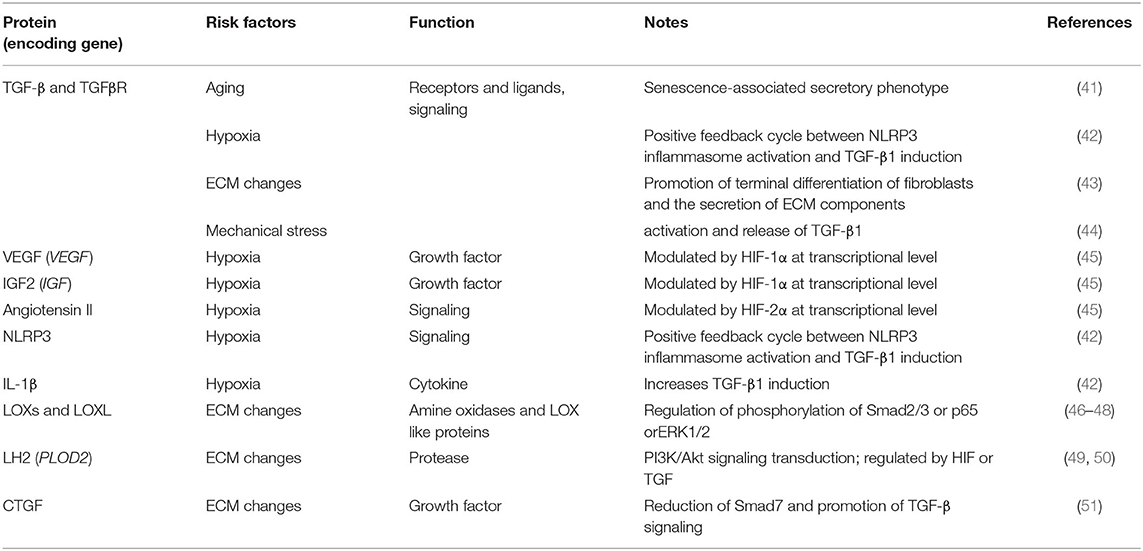
Table 2. Fibrogenic factors in OA.
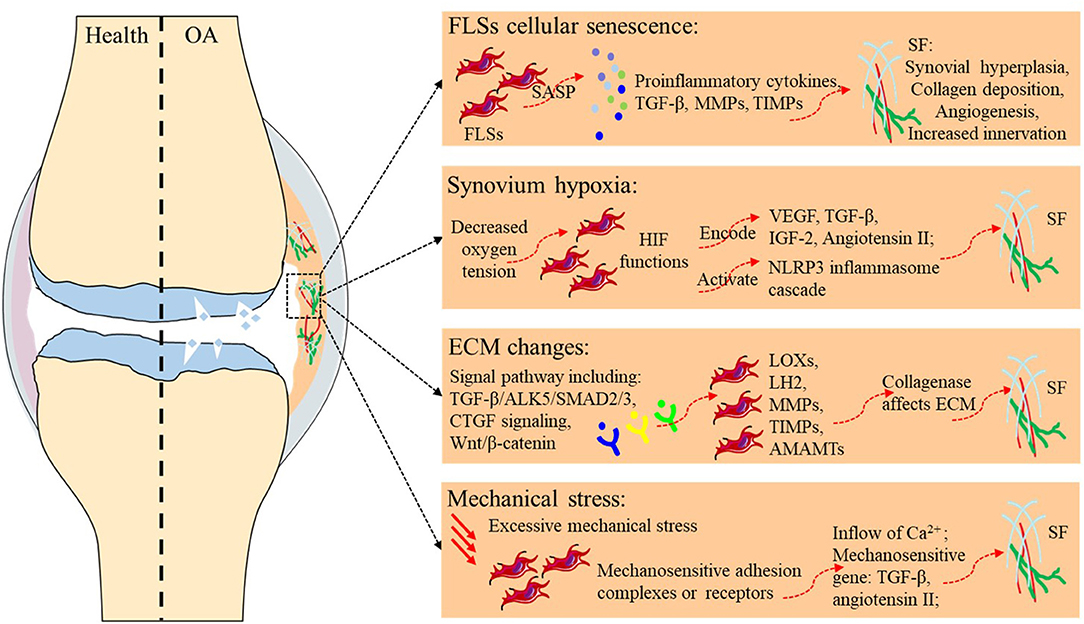
Figure 1. Potential etiology of synovial fibrosis in OA.
Aging
Directly relevant to aging is the study of cellular senescence, which refers to a state of cell cycle arrest, increased expression of cell cycle inhibitors, and enhanced production of proinflammatory cytokines, chemokines, and growth factors. A variety of stimuli and stresses, including telomere shortening, epigenetic changes, metabolic stresses, and mitochondrial dysfunction, can cause senescence. Markers for cellular senescence, including p16INK4A and p21, are upregulated in OA tissues, including cartilage, subchondral bone, and the synovium (41, 52, 53), suggesting cellular senescence in the FLSs of OA. Besides, senescence-associated secretory phenotype (SASP) is a pro-inflammatory secretory phenotype associated with cell senescence, including pro-inflammatory cytokines (such as IL-1α, IL-1β, IL-6, and IL-8), Tissue growth factors (TGF-β), MMPs, tissue inhibitors of metalloproteinases (TIMPs), and all these cytokines play important roles in SF. As cells in the synovium become proliferative and activated during SF, they may themselves become more susceptible to undergoing senescence. Thus, despite the lack of direct evidence, FLSs senescence is likely to promote the development of SF during aging.
Hypoxia
Hypoxia refers to a decrease in oxygen tension in tissues, and the central effector of the hypoxia response is the transcription factor hypoxia-inducible factor (HIF). In the hypoxic state, the alpha subunit in HIF is no longer hydroxylated but accumulates and translocates to the nucleus, where it binds to the beta subunit of HIF and exerts its function as a transcription factor (54). The genes encoding VEGF, TGF-β, and IGF-2, which are regulated by HIF-1α, and angiotensin II, which is modulated by HIF-2α, are all important profibrotic factors (45). Clinical studies have shown that HIF-1α levels in the serum, synovial fluid, and articular cartilage of knee OA patients are associated with progressive joint damage (55, 56). Hypoxic TGF-β1 induction increased succinate accumulation due to the reversal of succinate dehydrogenase activation and induced NLRP3 inflammasome activation in a manner dependent on HIF-1α induction. In response to NLRP3 inflammasome activation, the released IL-1β further increased TGF-β1 induction, suggesting the existence of a positive feedback cycle between inflammation and fibrosis in myofibroblast activation; this highlights the importance of studying SF associated with OA from the perspective of hypoxia (42).
Extracellular Matrix Changes
Remst et al. analyzed gene expression in TGF-β-stimulated human OA synovial fibroblasts and the synovium of mice with TGF-β-induced fibrosis, mice with experimental OA, and humans with end-stage OA. The genes encoding lysyl oxidase (LOX), pro-collagen-lysine, 2-oxoglutarate 5-dioxygenase 2 [PLOD2, also known as lysyl hydroxylase 2b (LH2b)], tissue inhibitor of metalloproteinase 1 (TIMP-1), collagen type I α1 chain (COL1A1), and collagen type V α1 chain (COL5A1) were upregulated under TGF-β stimulation, indicating that the signaling cascades of these key fibrotic factors were activated (57). Moreover, due to the vital role of matrix metalloproteinases, a disintegrin and metalloprotease (ADAMS) and a disintegrin and metalloproteinase with thrombospondin motif (ADAMTS) in ECM remodeling, the contributions of specific members in this family to SF should also be considered.
TGF-β plays a central role in the fibrotic cascade and is present as three isoforms (TGFβ1-3), all of which are elevated in OA patients and positively correlated with pain, loss of function, and radiographic staging (58). TGF-β signaling is initiated by binding to the TGF receptor, a heterodimer composed of TGFβR1 and TGFβR2. Further signal transduction is usually divided into SMAD-dependent classical pathways and non-classical pathways that are independent of SMAD. In the classical pathways, a phosphorylated TGFβR1, typically ALK5, can then transduce the TGF-β signal intracellularly to activate SMAD2/SMAD3, which complexes with SMAD4 to regulate gene expression. In contrast, the non-canonical pathway signals via other kinases, such as extracellular signal-regulated kinase, mitogen-activated protein kinase, nuclear factor-κB, and JUN amino-terminal kinase (43). TGF-β pathways promote the terminal differentiation of fibroblasts and the secretion of ECM components, especially collagen, fibronectin, and proteoglycans. A detailed description of the role of the TGF-β signaling pathway in OA is beyond the scope of this paper, but it is remarkable that TGF-β and its subfamily, bone morphogenetic proteins, play multiple roles in maintaining homeostasis of the cartilage and subchondral bone in OA. The TGF-β-mediated protective effects on cartilage matrix turnover rely not only on the production of ECM proteins such as type II collagen and aggrecan but also on the blockade of ECM protein degradation via increased production of protease inhibitors such as TIMP. Broeren et al. developed a 3-dimensional synovial membrane model involving micromasses made of either human primary synovial cell suspensions or a mixture of primary FLSs and CD14+ mononuclear cells. To recreate the synovial membrane in OA, the micromasses were exposed to TGF-β, which led to fibrosis-like changes in the membrane, including increased alpha smooth muscle actin (α-SMA) and increased expression of the fibrosis-related genes PLOD2 and COL1A1 (59). These results provide a detailed analysis of SF and show the suitability of this setup as a synovial membrane model for further research on RA and OA. Consistent with Broeren, Remst et al found that TGF-β induced PLOD2 expression in human FLSs via the ALK5/SMAD2/3 signaling pathway, thus aggravating SF in OA (60). In summary, high expression of TGF-β in the OA synovium accelerates OA progression, and inhibition of TGF-β in the synovium seems to be a favorable therapeutic strategy for SF. However, further research on TGF-β is still urgently needed due to its possible protective effects on cartilage.
The LOX family enzymes LOX and four lysyl oxidase-like proteins (LOXL1-4) are copper-dependent amine oxidases that catalyze the covalent cross-linking of collagen by oxidatively deaminating specific lysine and hydroxylysine residues in the telopeptide domains; this cross-linking increases collagen stiffness, which stiffens the ECM and promotes tissue fibrosis in the lung, myocardium, and liver. LOX may be induced by TGF-β1/Smad2/3 signaling, and knockdown of LOXL1 suppressed cell proliferation and fibrogenesis in TGF-β1-stimulated HSCs by regulating the phosphorylation of Smad2/3 (46, 47). Some research suggests that LOX expression was markedly elevated in OA-damaged regions of human cartilage and mouse OA cartilage induced by destabilization of the medial meniscus (DMM) surgery, and this elevated transcription caused cartilage destruction (61). Others have suggested that LOXL2 expression may be a protective response due to the inhibition of IL-1β-induced phospho-NF-κB/p65 and TGF-β1-induced ERK1/2 phosphorylation, although LOXL2 is upregulated in OA cartilage (48). These different results may be due to the varied expression of TGF-β and its receptors during different pathological stages of OA. Signal transduction in different environments may be a determinant of TGF-β and LOX function. Therefore, in the study of the OA synovium, researchers have observed that IL-1β simultaneously promotes LOX expression but has a depressing effect combined with TNF-α, while overexpressing LOX in the synovium exacerbates OA-related fibrosis (62, 63). Overall, LOX is closely related to tissue fibrosis through TGF signaling pathways, and the potential association with HIF-2α, mechanical conduction, and other OA-related factors may be a further direction for the study of SF.
PLOD2 encodes lysyl hydroxylase 2 (LH2), which catalyzes the hydroxylation of lysine intracellularly before the collagen is secreted. Then, LOX binds to hydroxylysine residues in the extracellular collagen fibers and induces cross-linking, the final step in the maturation of collagen, which is essential for the physical and mechanical properties of collagen fibrils (64). Aberrant lysyl hydroxylation and collagen cross-linking contribute to the progression of many collagen-related diseases, such as cancer and fibrosis. Wan et al demonstrated that PLOD2 expression was increased in endometrial carcinoma cells under hypoxic conditions and modulated the migration, invasion, and epithelial-mesenchymal transition of endometrial carcinoma cells via PI3K/Akt signaling (49). Other tumor diseases have also been reported to have similar pathological processes. In addition, PLOD2 is regulated by HIF-1 or TGF1 and mediates ECM remodeling, alignment, and mechanical properties through a transcriptionally mediated mechanism. Mia and Bank identified a selective inhibitor of IκB kinase, suppressed the expression of PLODs in dermal fibroblasts, and inhibited the TGFβ1-induced transition of fibroblasts into myofibroblasts, thus relieving excessive ECM synthesis (50). Gilkes et al. proved that HIF-1 activity in hypoxic fibroblasts promotes ECM remodeling by inducing the expression of the collagen hydroxylases P4HA1, P4HA2, and PLOD2 (65). In our most recent study, we explored the effect of inflammatory cascade amplification mediated by synovial macrophage pyroptosis on SF. High expression of TGF-β and PLOD2 in OA animals and FLSs was positively correlated with the degree of SF. Interestingly, TGFβ1, TGFβR1, LOX, PLOD1, and PLOD2 in the glenohumeral capsule of patients with shoulder instability may play a role in shoulder instability. We speculate that this correlation is closely related to SF of the shoulder joint, although the specific pathological mechanism still needs further study.
MMP1, which is also known as fibroblast collagenase, has mainly been implicated in mediating the degradation of type I collagen, which is most often mentioned in fibrosis as the major constituent of the fibrotic ECM. MMP1 cleaves collagen only between amino acids 775 and 776; thus, it is possible that hydroxylysylpyridinoline collagen cross-linked through aberrant PLOD2 and LOX is more difficult to degrade (66). It has been proven that ~0.1 Schiff base of LOX-mediated cross-linking per collagen molecule results in 2-3-fold higher resistance to human collagenase compared with that of un-cross-linked collagen (67). Therefore, despite the upregulation of both MMP1 and TIMP1 in the synovium in OA, the pathological changes of the synovial membrane continue to promote fibrosis, as indicated by not only the quantity of collagen but also the quality of collagen, as determined by its post-translational modifications, which actively drive the progression of fibrosis. In addition, MMP13 and ADAMTS-5 were also validated as drug targets that participate in the regulation of the ECM in OA, and ADAMTS-5 inhibitors were shown to reduce synovial joint damage in OA animal models.
Connective tissue growth factor (CTGF) is a well-known fibrogenic factor that has been shown to induce synovial fibrosis (60). It has been observed that both FLSs and chondrocytes were strongly induced to express CTGF after stimulation by TGF-β (60, 68). The main function of CTGF is to regulate proteoglycans on the cell surface, which can affect fibroblast proliferation, chemotaxis and accelerate ECM deposition (69). CTGF is thought to coordinate some fibrogenic effects through the TGF-β response element, but CTGF may also act independently of TGF-β (69, 70). Smad7, the inhibitory smad of TGF-β signaling, is reduced by CTGF, which in turn promotes TGF-β signaling, but the mechanism by which CTGF regulates Smad7 has not been fully elucidated (51). Therefore, it is valuable to further elucidate the induction effect of CTGF on synovial fibrosis in OA.
Wnt/β-catenin is closely associated with embryonic skeletal formation, tissue repair, fibrosis, and joint homeostasis (71). Wnt mediates several signaling cascades, especially the β-catenin–dependent (canonical) pathway (72), and β-catenin, as a transcriptional regulator, its stabilization or degradation is a central event in the Wnt signaling pathway. Existing studies show that the Wnt/β-catenin classical pathway has long been proven to be over-activated in the pathogenesis of OA (73). To be specific, increased expression of Wnt ligands and target genes was observed in both articular cartilage and synovium after injury, indicating Wnt signaling activation (74, 75). A recent study showed that XAV-939, a Wnt inhibitor, may reduce the proliferation of synovial fibroblasts and type I collagen levels by inhibiting the Wnt pathway, ultimately exerting a protective effect on synovial fibrosis (73). In conclusion, the Wnt/β-catenin signaling pathway may be a key molecular mechanism in the treatment of synovial fibrosis in OA, which may provide new ideas for the treatment of OA.
Mechanical Stress
Physical activity is one of the most frequently recommended non-pharmacological therapies for OA, but the duration and intensity of exercise vary widely. Moderate mechanical stress may reduce sensitization to the inflammatory response in the articular cartilage and chondrocytes and be beneficial for OA (76). However, excessive mechanical stress exacerbates OA progression by inducing chondrocyte apoptosis and osteophyte formation (77, 78). This indicates that mechanical stimulation can regulate the balance of synthesis-degradation in cartilage and osteogenesis-osteoclastogenesis in the subchondral bone. In vivo, mechanical stress is transduced into the cell from the sites at which the cells attach to the ECM. The cells may engage their ECM both via mechanosensitive adhesion complexes and via other surface receptors, including those for growth factors and inflammatory mediators, which cannot act as adhesive anchors but may modify the mechanical signals transduced at the cell/ECM interface (79). Under these conditions, activated mechanosensitive plasma membrane channels allow the inflow of Ca2+ that can act as a second messenger to regulate gene expression. TGF-β1 is a typical mechanosensitive gene, and previous studies have suggested that mechanical stretching activates and releases latent TGF-β1 in living tissues from fibrotic lungs (44). In cardiac fibrosis, mechanical stress is a major factor for cardiac hypertrophy in response to pressure or volume overload, and angiotensin II seems to be another mechanosensitive gene that promotes fibrosis (80). As the FLS response to mechanical stress is critical during the initial stages of OA, SF caused by excessive mechanical stimulation is likely to occur (81), and subsequent ECM stiffness may affect tissue delivery of mechanical signals and exacerbate OA progression.
Existing and Potential Treatments for Synovial Fibrosis
Open surgery for SF is undoubtedly the most direct and effective treatment, but it often requires large incisions with extensive exploration of the joint and surrounding extracapsular soft tissues. Unfortunately, the surgery itself induces a fibrotic process, and the outcomes of post-traumatic surgery are poor, with most patients unable to return to their presurgery level of function. Even if satisfactory results are achieved during the operation, SF is likely to recur within a certain period.
Recently, SF research has made some progress in conservative drug treatment. Numerous studies have consistently reported that PRG4 and HA attenuate profibrotic responses to TGF-β in OA animals or FLSs. Interestingly, FLSs themselves can synthesize and secrete PRG4 and HA. Correcting the pathological state of FLSs in OA seems to be of great significance for the treatment of SF. In this context, Qadri et al concluded that increasing intracellular cAMP levels in FLSs mitigates SF through enhanced production of HA and PRG4 (82). Plaas et al. proved that HA injection blocked all gait changes and protected joints from femoral cartilage erosion and tissue fibrosis in KOA mice, and they deduced that HA injection could mimic the protective effects of ADAMTS-5 ablation (83). Their further findings supported this hypothesis and demonstrated that ADAMTS-5 was blocked by a CD44-dependent mechanism (84). As PRG4 is a ligand of the CD44 receptor, Qadri et al examined the role of the PRG4-CD44 interaction in regulating SF in OA and demonstrated that PRG4 inhibited fibroblast-to-myofibroblast transition, thus downregulating the expression of fibrotic genes in the OA synovium (85).
Furthermore, regarding the balance between MMP and TIMP expression in FLSs from KOA with flexion contracture using adenovirus-mediated relaxin gene therapy, relaxin could serve as an alternative therapeutic agent during the initial stage of OA with flexion contracture by exerting antifibrogenic effects (86). In addition, methylene blue, NSAIDs, and salmon calcitonin were also reported to have therapeutic effects on SF, although their specific mechanisms are still unclear (87–89). The Wnt/β-catenin signaling pathway and senescent cells are potential targets for antifibrosis therapy, but the intervention procedure needs further exploration (73, 90).
Conclusion
Evidence from direct research on SF in OA and related studies suggests the following. (I) Fibrosis is the outcome of inflammation. It is not clear whether the ongoing process of SF has a pathogenic effect in OA, especially in relation to pain. If so, effective intervention to slow the progression of fibrosis is necessary even if fibrosis is irreversible, as the greatest advantage is the improvement of joint function and the relief of OA symptoms. (II) Although we have some understanding of ECM environmental changes and the mechanism by which they are involved in the pathological process of SF associated with OA, our knowledge of this pathological mechanism is still insufficient. Angiogenesis and nerve invasion are likely to have a strong correlation with the pathological process of SF and may be involved in the development of SF, which deserves further exploration. (III) Existing research on the pharmacodynamic targets and intervention effects of SF is not sufficient, and further exploration is still needed in the future. Notably, the combined use of antifibrotic drugs has high potential during anti-inflammatory therapy for OA.
Author Contributions
PW and JM conceptualized the current study. LZ (1st author) and RX drafted and revised the paper. ZH, LD, and LZ (5th author) provided the relevant literature. XL and ML were responsible for the proofreading. PW provided final approval of the version to be submitted. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81804123, No.81774334); the Leading Talents of Traditional Chinese Medicine Project (SLJ0207); and the Social Development Fund of Jiangsu Province (BE2017774).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hunter DJ, Bierma-Zeinstra S. Osteoarthritis. Lancet. (2019) 393:1745–59. doi: 10.1016/S0140-6736(19)30417-9
2. GBD. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. (2018) 392:1789–858. doi: 10.1016/S0140-6736(18)32279-7
3. Wang CJ, Cheng JH, Chou WY, Hsu SL, Chen JH, Huang CY. Changes of articular cartilage and subchondral bone after extracorporeal shockwave therapy in osteoarthritis of the knee. Int J Med Sci. (2017) 14:213–23. doi: 10.7150/ijms.17469
4. Hunter DJ, Guermazi A, Roemer F, Zhang Y, Neogi T. Structural correlates of pain in joints with osteoarthritis. Osteoarthritis Cartilage. (2013) 21:1170–8. doi: 10.1016/j.joca.2013.05.017
5. Shu CC, Zaki S, Ravi V, Schiavinato A, Smith MM, Little CB. The relationship between synovial inflammation, structural pathology, and pain in post-traumatic osteoarthritis: differential effect of stem cell and hyaluronan treatment. Arthritis Res Ther. (2020) 22:29. doi: 10.1186/s13075-020-2117-2
6. Herrero-Beaumont G, Largo R. Glucosamine and O-GlcNAcylation: a novel immunometabolic therapeutic target for OA and chronic, low-grade systemic inflammation? Ann Rheum Dis. (2020) 79:1261–3. doi: 10.1136/annrheumdis-2020-217454
7. Mathiessen A, Conaghan PG. Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthritis Res Ther. (2017) 19:18. doi: 10.1186/s13075-017-1229-9
8. Ruiz-Romero C, Lam M, Nilsson P, Onnerfjord P, Utz PJ, Van Eyk JE, et al. Mining the proteome associated with rheumatic and autoimmune diseases. J Proteome Res. (2019) 18:4231–9. doi: 10.1021/acs.jproteome.9b00360
9. Remst DF, Blaney DE, van der Kraan PM. Unravelling osteoarthritis-related synovial fibrosis: a step closer to solving joint stiffness. Rheumatology (Oxford). (2015) 54:1954–63. doi: 10.1093/rheumatology/kev228
10. Remst DF, Blaney DE, Vitters EL, Blom AB, Stoop R, Snabel JM, et al. Osteoarthritis-related fibrosis is associated with both elevated pyridinoline cross-link formation and lysyl hydroxylase 2b expression. Osteoarthritis Cartilage. (2013) 21:157–64. doi: 10.1016/j.joca.2012.10.002
11. Li SH, Wu QF. MicroRNAs target on cartilage extracellular matrix degradation of knee osteoarthritis. Eur Rev Med Pharmacol Sci. (2021) 25:1185–97. doi: 10.26355/eurrev_202102_24821
12. Xu Z, Shen ZH, Wu B, Gong SL, Chen B. Small molecule natural compound targets the NF-kappaB signaling and ameliorates the development of osteoarthritis. J Cell Physiol. (2021). doi: 10.1002/jcp.30392
13. Chen Y, Qiu F, Zhu X, Mo H, Wu Z, Xiao C. Pannus does not occur only in rheumatoid arthritis: a pathological observation of pannus of knee osteoarthritis. Nan Fang Yi Ke Da Xue Xue Bao. (2019) 39:747–50. doi: 10.12122/j.issn.1673-4254.2019.06.19
14. Ashraf S, Wibberley H, Mapp PI, Hill R, Wilson D, Walsh DA. Increased vascular penetration and nerve growth in the meniscus: a potential source of pain in osteoarthritis. Ann Rheum Dis. (2011) 70:523–9. doi: 10.1136/ard.2010.137844
15. Obeidat AM, Miller RE, Miller RJ, Malfait AM. The nociceptive innervation of the normal and osteoarthritic mouse knee. Osteoarthritis Cartilage. (2019) 27:1669–79. doi: 10.1016/j.joca.2019.07.012
16. Wang T, He C. Pro-inflammatory cytokines: the link between obesity and osteoarthritis. Cytokine Growth Factor Rev. (2018) 44:38–50. doi: 10.1016/j.cytogfr.2018.10.002
17. Benito MJ, Veale DJ, FitzGerald O, van den Berg WB, Bresnihan B. Synovial tissue inflammation in early and late osteoarthritis. Ann Rheum Dis. (2005) 64:1263–7. doi: 10.1136/ard.2004.025270
18. Smith MD, Triantafillou S, Parker A, Youssef PP, Coleman M. Synovial membrane inflammation and cytokine production in patients with early osteoarthritis. J Rheumatol. (1997) 24:365–71.
19. Farinelli L, Barba M, Beltrami B, Baranzini M, Milani D, Lattanzi W, et al. Effects of NGF and BDNF on chondrocytes: a microarray analysis. J Biol Regul Homeost Agents. (2020) 34(Suppl. 3):83–9.
20. Li W, Lin J, Wang Z, Ren S, Wu X, Yu F, et al. Bevacizumab tested for treatment of knee osteoarthritis via inhibition of synovial vascular hyperplasia in rabbits. J Orthop Translat. (2019) 19:38–46. doi: 10.1016/j.jot.2019.04.002
21. Kurkcuoglu BS, Donmez M, Altinel S, Akinci SB, Saricaoglu F, Aypar U. Comparison of intraarticular bupivacaine and levobupivacaine injection in rat synovial inflammation. Turk J Med Sci. (2014) 44:540–5. doi: 10.3906/sag-1302-32
22. Minten M, Blom A, Snijders GF, Kloppenburg M, van den Hoogen F, den Broeder AA, et al. Exploring longitudinal associations of histologically assessed inflammation with symptoms and radiographic damage in knee osteoarthritis: combined results of three prospective cohort studies. Osteoarthritis Cartilage. (2019) 27:71–9. doi: 10.1016/j.joca.2018.10.014
23. Song C, Xu X, Wu Y, Ji B, Zhou X, Qin L. Study of the mechanism underlying hsa-miR338-3p downregulation to promote fibrosis of the synovial tissue in osteoarthritis patients. Mol Biol Rep. (2019) 46:627–37. doi: 10.1007/s11033-018-4518-8
24. Abdul N, Dixon D, Walker A, Horabin J, Smith N, Weir DJ, et al. Fibrosis is a common outcome following total knee arthroplasty. Sci Rep. (2015) 5:16469. doi: 10.1038/srep16469
25. Kalson NS, Borthwick LA, Mann DA, Deehan DJ, Lewis P, Mann C, et al. International consensus on the definition and classification of fibrosis of the knee joint. Bone Joint J. (2016) 98-B:1479–88. doi: 10.1302/0301-620X.98B10.37957
26. Park HJ, Lee SS, Park B, Yun J, Sung YS, Shim WH, et al. Radiomics analysis of gadoxetic acid-enhanced MRI for staging liver fibrosis. Radiology. (2019) 290:380–7. doi: 10.1148/radiol.2018181197
27. Berchtold L, Friedli I, Crowe LA, Martinez C, Moll S, Hadaya K, et al. Validation of the corticomedullary difference in magnetic resonance imaging-derived apparent diffusion coefficient for kidney fibrosis detection: a cross-sectional study. Nephrol Dial Transplant. (2020) 35:937–45. doi: 10.1093/ndt/gfy389
28. Loeuille D, Chary-Valckenaere I, Champigneulle J, Rat AC, Toussaint F, Pinzano-Watrin A, et al. Macroscopic and microscopic features of synovial membrane inflammation in the osteoarthritic knee: correlating magnetic resonance imaging findings with disease severity. Arthritis Rheum. (2005) 52:3492–501. doi: 10.1002/art.21373
29. Ostergaard M, Stoltenberg M, Lovgreen-Nielsen P, Volck B, Jensen CH, Lorenzen I. Magnetic resonance imaging-determined synovial membrane and joint effusion volumes in rheumatoid arthritis and osteoarthritis: comparison with the macroscopic and microscopic appearance of the synovium. Arthritis Rheum. (1997) 40:1856–67. doi: 10.1002/art.1780401020
30. Terslev L, Christensen R, Aga AB, Sexton J, Haavardsholm EA, Hammer HB. Assessing synovitis in the hands in patients with rheumatoid arthritis by ultrasound: an agreement study exploring the most inflammatory active side from two Norwegian trials. Arthritis Res Ther. (2019) 21:166. doi: 10.1186/s13075-019-1930-y
31. Lai KL, Chen DY, Wen MC, Chen YM, Hung WT, Chen YH, et al. What does power Doppler signal indicate in rheumatoid synovitis? A point of view from synovial histopathology. J Chin Med Assoc. (2018) 81:383–6. doi: 10.1016/j.jcma.2016.07.008
32. Najm A, Le Goff B, Orr C, Thurlings R, Canete JD, Humby F, et al. Standardisation of synovial biopsy analyses in rheumatic diseases: a consensus of the EULAR synovitis and OMERACT synovial tissue biopsy groups. Arthritis Res Ther. (2018) 20:265. doi: 10.1186/s13075-018-1795-5
33. Ruppert M, Theiss C, Knoss P, Kendoff D, Krukemeyer MG, Schroder N, et al. Histopathological, immunohistochemical criteria and confocal laser-scanning data of arthrofibrosis. Pathol Res Pract. (2013) 209:681–8. doi: 10.1016/j.prp.2013.05.009
34. Smith MD. The normal synovium. Open Rheumatol J. (2011) 5:100–6. doi: 10.2174/1874312901105010100
35. Wei Q, Kong N, Liu X, Tian R, Jiao M, Li Y, et al. Pirfenidone attenuates synovial fibrosis and postpones the progression of osteoarthritis by anti-fibrotic and anti-inflammatory properties in vivo and in vitro. J Transl Med. (2021) 19:157. doi: 10.1186/s12967-021-02823-4
36. Prieto-Potin I, Largo R, Roman-Blas JA, Herrero-Beaumont G, Walsh DA. Characterization of multinucleated giant cells in synovium and subchondral bone in knee osteoarthritis and rheumatoid arthritis. BMC Musculoskelet Disord. (2015) 16:226. doi: 10.1186/s12891-015-0664-5
37. Oehler S, Neureiter D, Meyer-Scholten C, Aigner T. Subtyping of osteoarthritic synoviopathy. Clin Exp Rheumatol. (2002) 20:633–40.
38. Huang J, Ye Z, Wang J, Chen Q, Huang D, Liu H. USP13 mediates PTEN to ameliorate osteoarthritis by restraining oxidative stress, apoptosis and inflammation via AKT-dependent manner. Biomed Pharmacother. (2021) 133:111089. doi: 10.1016/j.biopha.2020.111089
39. Walker CJ, Crocini C, Ramirez D, Killaars AR, Grim JC, Aguado BA, et al. Nuclear mechanosensing drives chromatin remodelling in persistently activated fibroblasts. Nat Biomed Eng. (2021). doi: 10.1038/s41551-021-00709-w
40. Petersen KK, Siebuhr AS, Graven-Nielsen T, Simonsen O, Boesen M, Gudbergsen H, et al. Sensitization and serological biomarkers in knee osteoarthritis patients with different degrees of synovitis. Clin J Pain. (2016) 32:841–8. doi: 10.1097/AJP.0000000000000334
41. Malaise O, Tachikart Y, Constantinides M, Mumme M, Ferreira-Lopez R, Noack S, et al. Mesenchymal stem cell senescence alleviates their intrinsic and seno-suppressive paracrine properties contributing to osteoarthritis development. Aging (Albany NY). (2019) 11:9128–46. doi: 10.18632/aging.102379
42. Li Y, Zheng JY, Liu JQ, Yang J, Liu Y, Wang C, et al. Succinate/NLRP3 inflammasome induces synovial fibroblast activation: therapeutical effects of clematichinenoside AR on arthritis. Front Immunol. (2016) 7:532. doi: 10.3389/fimmu.2016.00532
43. Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. (2003) 113:685–700. doi: 10.1016/S0092-8674(03)00432-X
44. Froese AR, Shimbori C, Bellaye PS, Inman M, Obex S, Fatima S, et al. Stretch-induced activation of transforming growth factor-beta1 in pulmonary fibrosis. Am J Respir Crit Care Med. (2016) 194:84–96. doi: 10.1164/rccm.201508-1638OC
45. Zhu WJ, Chang BY, Wang XF, Zang YF, Zheng ZX, Zhao HJ, et al. FBW7 regulates HIF-1alpha/VEGF pathway in the IL-1beta induced chondrocytes degeneration. Eur Rev Med Pharmacol Sci. (2020) 24:5914–24. doi: 10.26355/eurrev_202006_21484
46. Lu M, Qin Q, Yao J, Sun L, Qin X. Induction of LOX by TGF-beta1/Smad/AP-1 signaling aggravates rat myocardial fibrosis and heart failure. Iubmb Life. (2019) 71:1729–39. doi: 10.1002/iub.2112
47. Ma L, Zeng Y, Wei J, Yang D, Ding G, Liu J, et al. Knockdown of LOXL1 inhibits TGF-beta1-induced proliferation and fibrogenesis of hepatic stellate cells by inhibition of Smad2/3 phosphorylation. Biomed Pharmacother. (2018) 107:1728–35. doi: 10.1016/j.biopha.2018.08.156
48. Huang ZM, Du SH, Huang LG, Li JH, Xiao L, Tong P. Leptin promotes apoptosis and inhibits autophagy of chondrocytes through upregulating lysyl oxidase-like 3 during osteoarthritis pathogenesis. Osteoarthritis Cartilage. (2016) 24:1246–53. doi: 10.1016/j.joca.2016.02.009
49. Wan J, Qin J, Cao Q, Hu P, Zhong C, Tu C. Hypoxia-induced PLOD2 regulates invasion and epithelial-mesenchymal transition in endometrial carcinoma cells. Genes Genomics. (2020) 42:317–24. doi: 10.1007/s13258-019-00901-y
50. Mia MM, Bank RA. The IkappaB kinase inhibitor ACHP strongly attenuates TGFbeta1-induced myofibroblast formation and collagen synthesis. J Cell Mol Med. (2015) 19:2780–92. doi: 10.1111/jcmm.12661
51. Qi W, Chen X, Twigg S, Zhang Y, Gilbert RE, Kelly DJ, et al. The differential regulation of Smad7 in kidney tubule cells by connective tissue growth factor and transforming growth factor-beta1. Nephrology (Carlton). (2007) 12:267–74. doi: 10.1111/j.1440-1797.2007.00788.x
52. Del RM, Valin A, Usategui A, Ergueta S, Martin E, Municio C, et al. Senescent synovial fibroblasts accumulate prematurely in rheumatoid arthritis tissues and display an enhanced inflammatory phenotype. Immun Ageing. (2019) 16:29. doi: 10.1186/s12979-019-0169-4
53. Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. (2017) 23:775–81. doi: 10.1038/nm.4324
54. Zhang Y, Zhou S, Cai W, Han G, Li J, Chen M, et al. Hypoxia/reoxygenation activates the JNK pathway and accelerates synovial senescence. Mol Med Rep. (2020) 22:265–76. doi: 10.3892/mmr.2020.11102
55. Chu H, Xu ZM, Yu H, Zhu KJ, Huang H. Association between hypoxia-inducible factor-1a levels in serum and synovial fluid with the radiographic severity of knee osteoarthritis. Genet Mol Res. (2014) 13:10529–36. doi: 10.4238/2014.December.12.15
56. Qing L, Lei P, Liu H, Xie J, Wang L, Wen T, et al. Expression of hypoxia-inducible factor-1alpha in synovial fluid and articular cartilage is associated with disease severity in knee osteoarthritis. Exp Ther Med. (2017) 13:63–8. doi: 10.3892/etm.2016.3940
57. Remst DF, Blom AB, Vitters EL, Bank RA, van den Berg WB, Blaney DE, et al. Gene expression analysis of murine and human osteoarthritis synovium reveals elevation of transforming growth factor beta-responsive genes in osteoarthritis-related fibrosis. Arthritis Rheumatol. (2014) 66:647–56. doi: 10.1002/art.38266
58. Kapetanakis S, Drygiannakis I, Kazakos K, Papanas N, Kolios G, Kouroumalis E, et al. Serum TGF-beta2 and TGF-beta3 are increased and positively correlated to pain, functionality, and radiographic staging in osteoarthritis. Orthopedics. (2010) 33. doi: 10.3928/01477447-20100625-09
59. Broeren M, Waterborg C, Wiegertjes R, Thurlings RM, Koenders MI, Van Lent P, et al. A three-dimensional model to study human synovial pathology. ALTEX. (2019) 36:18–28. doi: 10.14573/altex.1804161
60. Remst DF, Blaney DE, Vitters EL, Bank RA, van den Berg WB, van der Kraan PM. TGF-ss induces Lysyl hydroxylase 2b in human synovial osteoarthritic fibroblasts through ALK5 signaling. Cell Tissue Res. (2014) 355:163–71. doi: 10.1007/s00441-013-1740-5
61. Kim JH, Lee G, Won Y, Lee M, Kwak JS, Chun CH, et al. Matrix cross-linking-mediated mechanotransduction promotes posttraumatic osteoarthritis. Proc Natl Acad Sci USA. (2015) 112:9424–9. doi: 10.1073/pnas.1505700112
62. Zhang Y, Jiang J, Xie J, Xu C, Wang C, Yin L, et al. Combined effects of tumor necrosis factor-alpha and interleukin-1beta on lysyl oxidase and matrix metalloproteinase expression in human knee synovial fibroblasts in vitro. Exp Ther Med. (2017) 14:5258–66. doi: 10.3892/etm.2017.5264
63. Li H, Lei M, Yu C, Lv Y, Song Y, Yang L. Mechano growth factor-E regulates apoptosis and inflammatory responses in fibroblast-like synoviocytes of knee osteoarthritis. Int Orthop. (2015) 39:2503–9. doi: 10.1007/s00264-015-2974-5
64. Piersma B, Bank RA. Collagen cross-linking mediated by lysyl hydroxylase 2: an enzymatic battlefield to combat fibrosis. Essays Biochem. (2019) 63:377–87. doi: 10.1042/EBC20180051
65. Gilkes DM, Bajpai S, Chaturvedi P, Wirtz D, Semenza GL. Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J Biol Chem. (2013) 288:10819–29. doi: 10.1074/jbc.M112.442939
66. van der Slot AJ, Zuurmond AM, van den Bogaerdt AJ, Ulrich MM, Middelkoop E, Boers W, et al. Increased formation of pyridinoline cross-links due to higher telopeptide lysyl hydroxylase levels is a general fibrotic phenomenon. Matrix Biol. (2004) 23:251–7. doi: 10.1016/j.matbio.2004.06.001
67. Avery NC, Bailey AJ. Enzymic and non-enzymic cross-linking mechanisms in relation to turnover of collagen: relevance to aging and exercise. Scand J Med Sci Sports. (2005) 15:231–40. doi: 10.1111/j.1600-0838.2005.00464.x
68. Cao Z, Liu W, Qu X, Bi H, Sun X, Yu Q, et al. miR-296-5p inhibits IL-1beta-induced apoptosis and cartilage degradation in human chondrocytes by directly targeting TGF-beta1/CTGF/p38MAPK pathway. Cell Cycle. (2020) 19:1443–53. doi: 10.1080/15384101.2020.1750813
69. Shi-Wen X, Leask A, Abraham D. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine Growth Factor Rev. (2008) 19:133–44. doi: 10.1016/j.cytogfr.2008.01.002
70. Frazier K, Williams S, Kothapalli D, Klapper H, Grotendorst GR. Stimulation of fibroblast cell growth, matrix production, and granulation tissue formation by connective tissue growth factor. J Invest Dermatol. (1996) 107:404–11. doi: 10.1111/1523-1747.ep12363389
71. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. (2012) 149:1192–205. doi: 10.1016/j.cell.2012.05.012
72. Miller JR, Hocking AM, Brown JD, Moon RT. Mechanism and function of signal transduction by the Wnt/beta-catenin and Wnt/Ca2+ pathways. Oncogene. (1999) 18:7860–72. doi: 10.1038/sj.onc.1203245
73. Lietman C, Wu B, Lechner S, Shinar A, Sehgal M, Rossomacha E, et al. Inhibition of Wnt/beta-catenin signaling ameliorates osteoarthritis in a murine model of experimental osteoarthritis. JCI Insight. (2018) 3:e96308. doi: 10.1172/jci.insight.96308
74. Dell'Accio F, De Bari C, El TN, Barone F, Mitsiadis TA, O'Dowd J, et al. Activation of WNT and BMP signaling in adult human articular cartilage following mechanical injury. Arthritis Res Ther. (2006) 8:R139. doi: 10.1186/ar2029
75. Dell'Accio F, De Bari C, Eltawil NM, Vanhummelen P, Pitzalis C. Identification of the molecular response of articular cartilage to injury, by microarray screening: Wnt-16 expression and signaling after injury and in osteoarthritis. Arthritis Rheum. (2008) 58:1410–21. doi: 10.1002/art.23444
76. Yang Y, Wang Y, Kong Y, Zhang X, Zhang H, Gang Y, et al. Mechanical stress protects against osteoarthritis via regulation of the AMPK/NF-kappaB signaling pathway. J Cell Physiol. (2019) 234:9156–67. doi: 10.1002/jcp.27592
77. Xu B, Xing R, Huang Z, Yin S, Li X, Zhang L, et al. Excessive mechanical stress induces chondrocyte apoptosis through TRPV4 in an anterior cruciate ligament-transected rat osteoarthritis model. Life Sci. (2019) 228:158–66. doi: 10.1016/j.lfs.2019.05.003
78. Venne G, Tse MY, Pang SC, Ellis RE. Mechanically-induced osteophyte in the rat knee. Osteoarthritis Cartilage. (2020) 28:853–64. doi: 10.1016/j.joca.2020.02.834
79. Viji BP, Rianna C, Mirastschijski U, Radmacher M. Nano-mechanical mapping of interdependent cell and ECM mechanics by AFM force spectroscopy. Sci Rep. (2019) 9:12317. doi: 10.1038/s41598-019-48566-7
80. Yamazaki T, Yazaki Y. Role of tissue angiotensin II in myocardial remodelling induced by mechanical stress. J Hum Hypertens. (1999) 13(Suppl. 1):S43–7, S49–50. doi: 10.1038/sj.jhh.1000747
81. Schroder A, Nazet U, Muschter D, Grassel S, Proff P, Kirschneck C. Impact of mechanical load on the expression profile of synovial fibroblasts from patients with and without osteoarthritis. Int J Mol Sci. (2019) 20:585. doi: 10.3390/ijms20030585
82. Qadri MM, Jay GD, Ostrom RS, Zhang LX, Elsaid KA. cAMP attenuates TGF-beta's profibrotic responses in osteoarthritic synoviocytes: involvement of hyaluronan and PRG4. Am J Physiol Cell Physiol. (2018) 315:C432–43. doi: 10.1152/ajpcell.00041.2018
83. Plaas A, Li J, Riesco J, Das R, Sandy JD, Harrison A. Intraarticular injection of hyaluronan prevents cartilage erosion, periarticular fibrosis and mechanical allodynia and normalizes stance time in murine knee osteoarthritis. Arthritis Res Ther. (2011) 13:R46. doi: 10.1186/ar3286
84. Li J, Gorski DJ, Anemaet W, Velasco J, Takeuchi J, Sandy JD, et al. Hyaluronan injection in murine osteoarthritis prevents TGFbeta 1-induced synovial neovascularization and fibrosis and maintains articular cartilage integrity by a CD44-dependent mechanism. Arthritis Res Ther. (2012) 14:R151. doi: 10.1186/ar3887
85. Qadri M, Jay GD, Zhang LX, Richendrfer H, Schmidt TA, Elsaid KA. Proteoglycan-4 regulates fibroblast to myofibroblast transition and expression of fibrotic genes in the synovium. Arthritis Res Ther. (2020) 22:113. doi: 10.1186/s13075-020-02207-x
86. Ko JH, Kang YM, Yang JH, Kim JS, Lee WJ, Kim SH, et al. Regulation of MMP and TIMP expression in synovial fibroblasts from knee osteoarthritis with flexion contracture using adenovirus-mediated relaxin gene therapy. Knee. (2019) 26:317–29. doi: 10.1016/j.knee.2019.01.010
87. Namazi H, Emami MJ, Nazhvani FD, Dehghani NA, Kargarshouroki Z. Effectiveness of methylene blue in the prevention of stifle joint arthrofibrosis in rabbit models. Arch Bone Jt Surg. (2019) 7:269–77.
88. Haraoui B, Pelletier JP, Cloutier JM, Faure MP, Martel-Pelletier J. Synovial membrane histology and immunopathology in rheumatoid arthritis and osteoarthritis. In vivo effects of antirheumatic drugs. Arthritis Rheum. (1991) 34:153–63. doi: 10.1002/art.1780340205
89. Yang R, Deng H, Hou J, Li W, Zhang C, Yu M, et al. Investigation of salmon calcitonin in regulating fibrosis-related molecule production and cell-substrate adhesion in frozen shoulder synovial/capsular fibroblasts. J Orthop Res. (2020) 38:1375–85. doi: 10.1002/jor.24571
Keywords: osteoarthritis, fibrosis, synovitis, fibroblast-like synoviocytes, extracellular matrix
Citation: Zhang L, Xing R, Huang Z, Ding L, Zhang L, Li M, Li X, Wang P and Mao J (2021) Synovial Fibrosis Involvement in Osteoarthritis. Front. Med. 8:684389. doi: 10.3389/fmed.2021.684389
Received: 23 March 2021; Accepted: 03 May 2021;
Published: 26 May 2021.
Edited by:
Ali Mobasheri, University of Oulu, FinlandReviewed by:
Frank Zaucke, Orthopaedic University Hospital Friedrichsheim, GermanyMartijn H. Van Den Bosch, Radboud University Nijmegen Medical Centre, Netherlands
Copyright © 2021 Zhang, Xing, Huang, Ding, Zhang, Li, Li, Wang and Mao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peimin Wang, drwpm@163.com
†These authors have contributed equally to this work and share first authorship