Airway Remodeling in Asthma
 Kenneth P. Hough
Kenneth P. Hough  Miranda L. Curtiss
Miranda L. Curtiss  Trevor J. Blain
Trevor J. Blain  Rui-Ming Liu Jennifer Trevor
Rui-Ming Liu Jennifer Trevor  Jessy S. Deshane*
Jessy S. Deshane*  Victor J. Thannickal*
Victor J. Thannickal*- Division of Pulmonary Allergy and Critical Care Medicine, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL, United States
Asthma is an inflammatory disease of the airways that may result from exposure to allergens or other environmental irritants, resulting in bronchoconstriction, wheezing, and shortness of breath. The structural changes of the airways associated with asthma, broadly referred to as airway remodeling, is a pathological feature of chronic asthma that contributes to the clinical manifestations of the disease. Airway remodeling in asthma constitutes cellular and extracellular matrix changes in the large and small airways, epithelial cell apoptosis, airway smooth muscle cell proliferation, and fibroblast activation. These pathological changes in the airway are orchestrated by crosstalk of different cell types within the airway wall and submucosa. Environmental exposures to dust, chemicals, and cigarette smoke can initiate the cascade of pro-inflammatory responses that trigger airway remodeling through paracrine signaling and mechanostimulatory cues that drive airway remodeling. In this review, we explore three integrated and dynamic processes in airway remodeling: (1) initiation by epithelial cells; (2) amplification by immune cells; and (3) mesenchymal effector functions. Furthermore, we explore the role of inflammaging in the dysregulated and persistent inflammatory response that perpetuates airway remodeling in elderly asthmatics.
Introduction
Asthma is a widely prevalent disease. In the United States, 13.4% of adults aged 18 years and older, and 11.6% of children are diagnosed with asthma. These rates are higher in adult females (15.2%) and male children (13%); in general, asthma prevalence is higher in minority populations and populations with low socioeconomic status1 (1). According to the most recent Global Initiative for Asthma (GINA) guidelines, asthma is defined as “a heterogeneous disease, usually characterized by chronic airway inflammation.” It is characterized by respiratory symptoms such as wheezing, shortness of breath, chest tightness, and cough that vary over time and in intensity, together with variable expiratory airflow limitation2. The variable airflow limitation in asthmatics is due to a combination of bronchoconstriction, airway edema, mucus secretion, airway hyper-responsiveness, and airway remodeling (2)1. However, variable airflow limitation may progress to persistent airflow limitation or fixed airway obstruction in a subset of patients (3).
Epidemiology
Among the general population, asthma accounts for 30–50% of those individuals with fixed airway obstruction (4–6); in severe or difficult-to-treat adult asthmatics, 55–60% have fixed airway obstruction (7, 8). Airway remodeling may explain persistent airflow obstruction present in some asthmatic patients, attributed to goblet cell hyperplasia, decreased epithelial cell and cartilage integrity, subepithelial collagen deposition with increased thickness of the reticular basement membrane, increased airway smooth muscle mass and angiogenesis of the airways (9–15). This is present in asthmatics with mild disease (16), but tends to worsen in parallel with increasing disease severity (13, 17). Importantly, onset of airway remodeling has been identified in pre-school children as young as 1-year-old (18, 19) and in school-age children, persisting through adulthood. Thus, airway remodeling in some patients may occur early in the disease process (20–22). Conversely, adult asthma patients with minimal airway remodeling similar to healthy controls have also been identified (23, 24), and adult mild asthmatics acutely increase parameters of airway remodeling with exposure to asthma triggers (25). Thus, while airway remodeling may be a consequence of inflammation, the heterogeneity in its presentation suggests that it should not be assumed to occur downstream of a single (or central) mechanism. Ultimately, these varying mechanisms will illuminate our understanding of asthma endotypes.
Asthma Endotypes
The field of personalized medicine in asthma care has benefitted greatly from the recognition that “asthma” refers to an umbrella term encompassing a range of clinical presentations (26). An aspirational goal of this phenotyping process is to eventually link clinical phenotype to molecular mechanisms, defining an “endotype” that would predict response to therapy (27).
One of the first approaches to phenotyping asthmatics was to evaluate sputum cellularity as an indirect readout of airway inflammation. Four subgroups of adult asthmatics were identified: eosinophilic asthma, neutrophilic asthma, mixed granulocytic asthma with both sputum eosinophils and neutrophils, and paucigranulocytic asthma with neither (28, 29). In multiple cohorts, the distribution of asthma favors eosinophilic (40–50%) and paucigranulocytic (30–50%) airway inflammation, with only 10–20% of patients manifesting neutrophilic asthma (28–32).
Unbiased identification of phenotypic clusters of asthmatics have incorporated objective clinical and morphometric parameters, including peripheral blood eosinophil counts, sputum cellularity, history of atopy, age of onset of asthmatic disease, body mass index, asthma control questionnaire, and presence of fixed vs. variable airflow obstruction (33, 34). These parameters when applied with unbiased clustering algorithms to large patient cohorts [SARP (Severe Asthma Research Program), ADEPT (Airways Disease Endotyping for Personalized Therapeutics), and U-BIOPRED (Unbiased BIOmarkers in PREDiction of respiratory disease outcomes)] consistently identify four to six clinically defined clusters (35–37). From these studies, a consensus has arisen that, at a minimum, asthma includes “Th2-high” and “Th2-low” disease subclusters. Th2-high includes: early-onset allergic asthma, late-onset steroid-resistant eosinophilic asthma, and aspirin-exacerbated respiratory disease (AERD). Th2-low tends to be steroid-resistant with either neutrophilic or paucigranulocytic inflammation and it is further classified by obesity, smoking, or onset after 50 years of age (38).
In contrast to unbiased clustering methods utilized above, the most widely used and easily implemented parameters in clinical use to assess asthma phenotype are the presence of sensitization to perennial aeroallergens, peripheral eosinophil count, fixed airway obstruction, and fractional exhaled nitric oxide (FENO) (39). This classification scheme differentiates Th2-high asthma, which is amenable to treatment with anti-IgE, anti-IL-5 or anti-IL-5Rα, and anti-IL-4Rα therapy (40, 41), from Th2-low asthma (42). In the context of cigarette use history and associated onset of asthma symptoms, patients with fixed airway obstruction may also be classified as “asthma-COPD (chronic obstructive pulmonary disease) overlap” (43).
Airway Remodeling Endotypes
Airway remodeling may provide further specification of asthma endotypes. Post-mortem studies of asthmatic patients reveal that airway remodeling can affect both large and small airways (44, 45). However, invasive assessments of remodeling predominantly evaluated endobronchial biopsies of proximal airways rather than small airways (46), limiting our understanding of the small airway changes that are relevant to air trapping and may be relevant to airway remodeling (6). Non-invasive means of evaluating airway remodeling are needed that correlate with airway morphometric analyses. The gold standard of airway remodeling requires bronchial biopsy and direct assessment of lung tissue (6). Quantitative Computerized Tomography (CT) imaging of lung is a non-invasive means of assessing airway remodeling. In adult asthmatics, comparison of airway biopsies with CT morphometrics indicates a good correlation between airway wall volume and increased reticular basement membrane thickness (47). However, this association was not reproduced in a pediatric severe asthma cohort undergoing endobronchial biopsy (48) in spite of consistent identification of bronchial wall thickening on CT in cohorts of children with difficult-to-treat asthma (49). Regardless, CT imaging is now increasingly used to assess airway remodeling in adult asthmatics (50–52).
Asthma is often characterized as childhood-onset or adult-onset (53, 54). Childhood-onset asthma typically occurs prior to 12 years of age, whereas 40% of adult asthmatics report symptoms after 40 years of age (55, 56). Childhood-onset asthma is predominantly atopic and eosinophilic, even into adulthood with marked airway remodeling, increased reticular basement membrane thickness and airway smooth muscle mass (23, 57). Airway remodeling in adult-onset asthma is less well-characterized but prevalence appears to be lower than childhood-onset asthma (58). When unbiased clustering of CT airway wall thickness or airway lumen thickness was applied to an adult asthma cohort, all asthma patients manifested air trapping but one cluster with less air trapping and lacking changes consistent with airway remodeling correlated with clinically mild disease (59). Two other cohorts of adult onset asthmatics with severe disease were found to contain a subpopulation (25–30%) lacking both eosinophilic inflammation and airway remodeling (24, 50). Unbiased clustering analysis has also revealed an adult severe asthma population, identified as “paucigranulocytic” asthma, with airway remodeling in the absence of airway inflammation (28, 29, 60). In paucigranulocytic asthma, airway remodeling is thought to occur in a manner “uncoupled” from airway inflammation, perhaps as a direct consequence of airway smooth muscle hypertrophy or neurogenic factors contributing to bronchospasm (61). In most cases, however, airway remodeling seems to behave as a function of either disease severity and/or chronicity of inflammation.
Extracellular Matrix in Asthmatic Airway Remodeling
Increased deposition of extracellular matrix (ECM) proteins in the reticular basement membrane region, lamina propria, and submucosa is a characteristic of asthmatic airways and contributes to the airway wall thickening and airflow obstruction. The ECM is composed of a diverse group of proteins and glycoproteins, including (a) structural proteins, including collagen and elastin, (b) adhesion proteins, including fibronectin and tenascin, etc, and (c) glycosaminoglycans (GAGs) and proteoglycans (62). Collagen fibers are the most abundant elements of the ECM in the lung. Fibrillar collagens, including type I, II, III, V, and XI collagens, have great tensile strength but low elasticity and contribute to the overarching architecture of the lung. Overproduction and deposition of collagen leads to lung stiffness. Elastic fibers, on the other hand, have high elasticity and provide the lung with compliance and elastic recoil. ECM adhesion proteins, such as fibronectin, provide binding sites for cell adhesion receptors including integrins. Therefore, the ECM proteins provide structural and mechanical support for lung tissue and a substratum for cell adhesion, migration, activation, and proliferation. Aberrant accumulation of ECM may, however, lead to changes in tissue structure and function that contribute to airway remodeling.
It has been well-documented that the deposition of various ECM molecules is increased in asthmatic airways, including structural proteins collagens I, III, and V, adhesion proteins fibronectin and tenascin, as well as proteoglycans such as lumican and biglycan (63–67). Fibroblasts are the major producer of ECM. Fibroblasts in asthmatic airways are activated and produce large amounts of ECM (66, 68, 69). It has also been shown that asthmatic airway epithelial cells stimulate naïve lung fibroblasts to produce collagens, fibronectin, and the pro-fibrotic mediator, TGF-β (68). Airway smooth muscle (ASM) hypertrophy and hyperplasia are characteristic features of asthmatic airways. Besides fibroblasts, smooth muscle cells in asthmatic airways also produce increased amounts of ECM, including collagens and fibronectin (70–72). Known environmental risk factors such as biomass fuels, cigarette smoke, and rhinovirus have been shown to stimulate the productions of ECM proteins by airway epithelial cells, ASM and fibroblasts (73–77). Additionally, it has been reported that the degradation products of matrix proteins, sometimes referred to as “matrikines,” regulates the remodeling process; for example, tumstatin, a type IV collagen-derived matrikine, modulates ASM production of ECM proteins (71). These matrikines, which are increased in asthmatic airways, interact with ECM proteins to regulate the composition of the matrix and modulate airway hyper-responsiveness (78, 79). Together, emerging data indicate that the deposition of ECM proteins in asthmatic airways is increased and may be post-translationally modified, which lead to specific endotypes of airway remodeling in asthma.
Aging and Airway Remodeling
Asthma mortality has declined in the United States, but not in elderly patients. The probability of death from asthma is more than five times higher in elderly asthmatics (80–82). Aging affects the lung and chest wall, reducing FEV1 (forced expiratory volume), FEV1/FVC (forced vital capacity), and FVC (with minimal change in lung volume), and increasing residual volume (83). Age-dependent decrements in FEV1 proceed linearly from 25 to 30 years of age through adulthood, then accelerate with increasing age (84). This deterioration is further accelerated in asthmatic patients (85). Lung parenchyma structural changes affecting elastic recoil are postulated to underlie peripheral airway narrowing with reduced airway surface-to-volume ratio observed in the elderly (3, 86). Elderly patients with no known underlying lung disease also manifest alveolar dilation and ductal ectasia without emphysema or fibrosis (87). The chest wall compliance of elderly patients is reduced by costochondral joint calcification, degenerative joint disease of the spine, and kyphosis (83). Diaphragmatic weakness and skeletal muscle weakness reduce maximum inspiratory and expiratory pressures (83, 88). Comorbidities, frailty, and poor nutrition result in respiratory muscle weakness (89, 90). However, airway remodeling does not appear to occur as an intrinsic feature of aging. Rather, it is an intrinsic feature of asthma, manifesting in a subset of adult asthmatic patients.
Airway Epithelial Cells as “Initiators” of Airway Remodeling
The airway epithelium is subject to airborne particles and infectious agents and represents the frontline barrier between the host and environment in the airways (91) (schematic in Figure 1). Epithelial cells are armed with pattern recognition receptors (PRR) which detect pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) that are derived from pathogens, allergens, and injured cells due to environmental insults (92–95). Triggering of PRRs by allergens, results in the recruitment of dendritic cells (DCs) through the secretion of chemokines and cytokines, such as CCL2, CCL20, IL-12, IL-12p40, TSLP, and GMCSF (94, 96, 97).
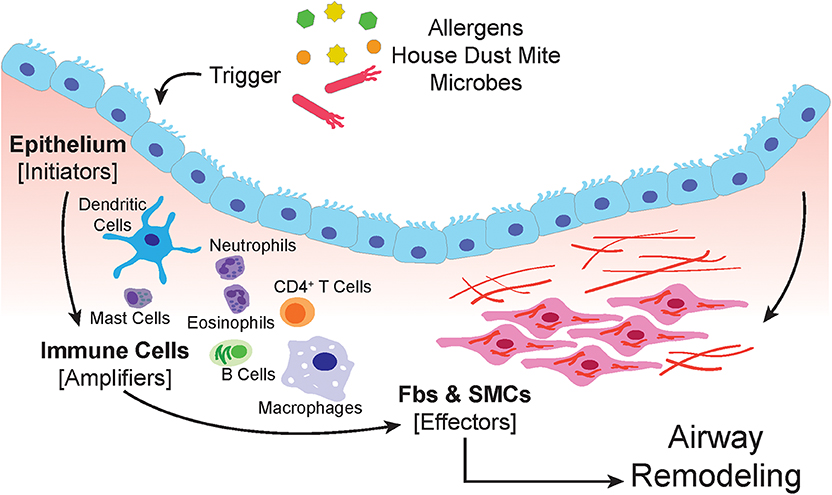
Figure 1. The airway epithelium serves as the primary interface between the environment and the lung. When triggered by allergens, house dust mite or microbes, epithelial cells respond by secreting soluble factors that recruit, and activate immune cells. The amplification of the immune response involves macrophages, dendritic cells, neutrophils, mast cells, eosinophils, and lymphocytes. Both the epithelium and immune cells produce paracrine signals that induce proliferation, expansion, and activation of the submucosal mesenchyme that include resident airway smooth muscle cells and fibroblasts.
The environmental insult on airway epithelial cells may also induce apoptosis, or programmed cell death (98). Apoptosis of the epithelium, accompanied by soluble paracrine factors such as TGF-β can initiate the tissue regenerative process in an attempt to restore homeostasis (99, 100). However, persistent damage and prolonged stimulation by growth factors, can lead to aberrant tissue repair and remodeling of the airways that leads to the pathophysiological conditions seen in asthma. For example, thickening of the airway smooth muscle is a result of mesenchymal differentiation into myofibroblast (101).
As the lung expands with air, the accompanying morphological changes also affects the cellular constituents of the lung (102–104). The mechanical forces experienced by cells during regular respiration is minimal; however, during bronchoconstriction and bronchospasms, epithelial cells are subject to compressive forces at a magnitude higher than normal physiological conditions (103, 105). These compressive forces may often result in mechanostimulation, which can activate epithelial cells to produce TGF-β and GMCSF, which can then recruit DCs and other immune cells (106). Recruited DCs orchestrate the activation of both innate and adaptive arms of the immune system, facilitating the inflammatory process in the airways of asthmatics (107). Mechanostimulation also increases gene expression of early growth response 1 (Egr-1), endothelin 1, transforming growth factor β1 (TGF-β1), and epidermal growth factors (EGF) (108). Secreted EGF can bind epithelial epidermal growth factor receptors (EGFR), which in turn mediates a positive feedback loop to increase EGFR ligand production (109). EGF promotes goblet-cell metaplasia (110), which also contribute to the physiological changes observed in patients exposed to repeated bronchoconstriction by methacholine challenge (25). Repeated bronchoconstriction induces goblet cell proliferation, sub-epithelial thickening, and mucus secretion which can lead to airway obstruction (25). Compressive stress also increases YKL-40 expression, encoded by the gene CHI3L1, and its secretion (111); this stimulates angiogenesis, smooth muscle cell proliferation, and migration (112).
Cytokines involved in asthma, such as IL-6, IL-8, and TSLP are known drivers of cellular senescence (113, 114). Cellular senescence, or the irreversible arrest of the cell cycle, may also contribute to airway remodeling and its detrimental effects on the airways of asthmatics (115). Senescent epithelial cells induced by cigarette smoke have been shown to destroy the alveoli resulting in progression of disease (116). In normal physiological setting, cellular senescence is usually a protective mechanism to prevent cells that have undergone telomere erosion and stress from proliferation and transformation (117). However, accumulation of senescent fibroblasts can contribute to reduced pulmonary compliance and remodeling of the airways (118, 119).
Immune Cells as “Amplifiers” of Airway Remodeling
The lung, like many other organs and tissues, contain a diverse population of immune cells that protect us from pathogens invading the airways (120–122). Immune surveillance and phagocytosis facilitates the resolution of the inflammatory process back to homeostasis. However, aberrant activation and prolonged immune responses are key drivers in asthma and have detrimental effects to the airways. Notably, T helper type 2 (Th2) and Th17 cells produce cytokines that promote airway inflammation and remodeling in allergic asthma (123, 124). Th2 cytokines, such as IL-4 and IL-13 enhance subepithelial fibrosis, mucous hyperplasia, and collagen deposition (125–127). Despite the controversial role of Th17 cells in airway remodeling and inflammation, the synergistic effect of DCs together with Th17 cytokines promote accumulation of fibrotic matrix components that correlate with TGF-β expression (123).
Alveolar macrophages (AM) are important lung-resident immune cells involved in immune response and tissue repair in the lung (128). They have a protective role in maintaining pulmonary tissue homeostasis as well as phagocytosis and host defense like many other macrophages. Generally, AM dampen the inflammatory responses in the airways through phagocytosis of apoptotic bodies and clearance of innate immune cell infiltrates. In asthmatics, however, these mechanisms are impaired; thus amplified and prolonged inflammation is present in the airways. Alveolar macrophages also contribute to airway remodeling through activation by TGF-β and release of matrix metalloproteinases that alter the extracellular matrix (ECM) and airway structure.
TGF-β is strongly implicated in airway remodeling and is released by eosinophils at the site of allergic inflammation (129, 130). TGF-β promotes metalloproteinase-9 (MMP-9) production, also known as gelatinase B; a metalloproteinase found in BAL fluid as well as plasma from asthmatics (131). MMP-9 is activated by tryptase secreted from mast cells, which have been tied to hypersensitivity and allergic inflammatory responses (132). Tryptase also induces fibroblast, endothelial, and epithelial cell proliferation further fueling remodeling of the airways in asthmatics (133, 134). Neutrophils have also been shown to produce MMP-9 and are associated with severe forms of asthma (135–137). In particular, neutrophils are associated with non-allergic and steroid resistant asthma (138, 139). In non-allergic asthma, epithelial cells initiate the inflammatory process through the release of IL-6, TGF-β, and IL1-β, which stimulates the production of IL-17 (140, 141). Th17 cytokines such as IL-17 and IL-22 facilitate neutrophilic recruitment, and TGF-β production, further amplifying the inflammatory and airway remodeling responses (142–144).
In addition to cellular mediators discussed above, the complement cascade can drive the same inflammatory and remodeling responses seen in asthma. Both the classical and alternative complement pathways promote airway inflammation through recruitment of proinflammatory immune cells, such as Th2 cells, mast cells, eosinophils, and macrophages. The recruitment of these immune cells helps amplify the magnitude of airway remodeling through the release of TGF-β, IL-13, and PDGF by both immune and epithelial cells. Specifically, C3a and C5a complement molecules have been reported to stimulate pro-airway remodeling factors such as TGF-β by epithelial cells (145). These pro-remodeling factors help drive fibroblast-to-myofibroblast differentiation and the production of metalloproteinases that drive structural changes in the airways of asthmatics (145).
Mesenchymal Cells as “Effectors” of Airway Remodeling
It is now well-recognized that resident airway smooth muscle (ASM) cells and fibroblasts drive key cellular and structural features of asthmatic airway remodeling, specifically the increase in ASM mass and subepithelial fibrosis (146). Paracrine signals from epithelial cells and immune cells may sustain mesenchymal cell activation in the airway wall (147). Bidirectional crosstalk between the epithelium and the mesenchyme is critical for normal lung development including branching morphogenesis; reactivation of this epithelial-mesenchymal tropic unit (EMTU) has been proposed as a driving mechanism in the repair response to chronic injury (148). Cytokines such as transforming growth factor-β (TGF-β) and fibroblast growth factors (FGFs) secreted by the mesenchyme instruct the growth and differentiation of epithelial cells, while epithelial growth factor (EGF), TGF-β, sonic hedgehog (SHH), and Wnt proteins from the epithelium direct the proliferation, differentiation, and fate of mesenchymal cells. An aberrantly activated EMTU in combination with inflammatory stimuli, such as the Th2 cytokines IL-4 and IL-13, may sustain the sub-mucosal mesenchymal response by ASM and fibroblasts to execute pathological airway remodeling.
Increased ASM mass has been recognized as a hallmark of airway remodeling in asthma (149, 150). There is abundant evidence for ASM plasticity, and the regulation of its proliferative, synthetic, and contractile properties (151). In support of the concept of an EMTU that recapitulates lung development, ADAM33, a membrane-anchored metalloprotease that is developmentally regulated, was identified as an asthma susceptibility gene by positional cloning in an outbred population (152). Several ADAM33 protein isoforms are expressed in human embryonic bronchi and surrounding mesenchyme, and its “reactivation” in adult ASM may explain its genetic association with asthma and bronchial hyper-responsiveness (153). In addition to pro-inflammatory/pro-fibrotic cytokines and contractile agonists that regulate ASM mass, cell intrinsic properties are also important. For example, the mitochondrial Bcl-2 adenovirus E1B 19 kDa-interacting protein, Bnip3, regulates the expression of adhesion proteins that control ASM adhesion, migration, and proliferation (154). ASM responses to β2 agonists is decreased by TGF-β1 signaling via the modulation of intracellular cAMP levels and a Smad2/3-dependent mechanism (155, 156). The TGF-β-induced activation of the reactive oxygen species (ROS)-generating enzyme, NADPH oxidase 4 (Nox4) that induces myofibroblast differentiation (157), is implicated in ASM proliferation and hypercontractility in asthma (158, 159), as well as in epithelial ciliary dysfunction in neutrophilic asthma.
Another distinct hallmark of airway remodeling in asthma is subepithelial fibrosis that is primarily mediated by submucosal resident fibroblasts that proliferate and differentiate into myofibroblasts. In addition to airway resident fibroblasts, the number, activation, and differentiation of circulating bone marrow-derived fibrocytes have been correlated with asthma severity (160). Consistently, TGF-β is recognized as a key mediator of this response, a number of paracrine mediators secreted by epithelial cells and immune cells are capable of activating submucosal fibroblasts (101). The SHH pathway has been implicated in induction of epithelial-mesenchymal transition (EMT) induced in bronchial epithelial cells by house dust mite exposure (161). The ECM itself and related proteases may serve to sustain these fibrogenic activities. For example, eosinophil-mediated fibroblast-to-myofibroblast transition and increased migration of fibroblasts is dependent on expression of matrix metalloproteinase-2 (162). In addition to pro-fibrotic cytokines such as TGF-β and Wnt proteins secreted by the epithelium, there is also evidence that epithelial-derived factors may mitigate fibrogenic responses in subepithelial fibroblasts. Club cell secretory protein-16 (CCSP-16), a member of the secretoglobin family, is decreased in serum of severe asthmatics and animal studies support a protective role of this protein against airway fibrosis and airway remodeling (162). Extracellular vesicle (EV)-mediated transfer of inositol polyphosphate 4-phosphatase type I A (INPP4A), a lipid phosphatase and an asthma candidate gene, functions to restrain the proliferative capacity of fibroblasts by dampening PI3K/Akt signaling (163). In contrast, fibroblast-derived EVs that carry fibronectin on its surface promotes invasion in recipient fibroblasts (164). Unique lipid signatures of EVs have been identified in the airways of human asthmatic subjects (165); in this study, lipidomics analysis revealed that phosphatidylglycerol, ceramide-phosphates, and ceramides were significantly reduced in exosomes from asthmatics exposed to tobacco smoke, while sphingomyelin 34:1 was more abundant in this group compared to healthy controls.
In addition to the traditional concept of targeting inflammatory responses in asthma, recent studies support potential utility in targeting the mesenchymal remodeling component. Although corticosteroids can mitigate chronic inflammation which secondarily contribute to airway remodeling, there is growing interest in developing therapies that more directly target airway fibrosis. None of the currently approved biologics, with the potential exception of IL-4/IL-13 targeted therapies, directly target cellular components of airway remodeling. However, bronchial thermoplasty is a non-pharmacological approach that may target the ASM component of airway remodeling. Bronchial thermoplasty involves the application of radiofrequency energy to the airway wall during bronchoscopy, and is thought to selectively ablate ASM; there is evidence that this procedure reduces asthma exacerbations and improves quality of life in patients with severe uncontrolled asthma (166). However, many questions remain as to its utility in severe asthma, specifically as it relates to mechanism(s) of action, patient selection, and predictors of response (167). There is likely to be advances in development of anti-fibrotic therapies for asthmatic airway remodeling. Although targeting Th2 cytokines are not particularly novel, there is continued interest in targeting this pathway in selected asthma endotypes. Recent studies suggest that epithelial cell responses to IL-4/IL-13 increases the IL-4Rα-dependent smooth muscle contribution to airway hyper-responsiveness, supporting IL-4Rα-targeted therapy in asthma (168). Activation of estrogen receptor-β signaling has been shown to downregulate airway hyper-responsiveness and airway remodeling (169). Agonists of the bitter taste receptors (TAS2Rs) promote bronchodilation, restrict allergen-induced inflammatory responses, and ASM proliferation and mitigate features of airway reactivity in vitro and in animal models (169, 170). Increased sphingosine kinase 2 (SPHK2) levels in proliferating ASM cells may be exploited to alleviate airway smooth muscle thickening with synthetic substrates (171, 172). When bronchodilatory responses to β-receptor agonists are blunted, the synthetic peroxisome proliferator activated receptor (PPAR)-γ agonist, rosiglitazone, may have benefit in eliciting ASM relaxation in ex vivo mouse lung slice models (173). Antagonism of prostaglandin D2 type 2 with fevipiprant reduced ASM mass in patients with asthma by decreasing airway eosinophilia in concert with reduced recruitment of myofibroblasts (174). Targeting another TGF-β-inducible gene, plasminogen activator inhibitor 1 (PAI-1), with a small molecule inhibitor has been shown to suppress eosinophilic allergic responses and ameliorate airway remodeling in an ovalbumin-sensitized murine model of chronic asthma (175).
Aging and Inflammaging
The repercussions and effects of the aging immune system are wide-ranging and diverse. Aging, as a whole, produces complex and ubiquitous physiological alterations across nearly all organ and tissue systems leading to many age-related diseases (176–181). The human immune system relies on an intricate interplay between the innate arm of the immune system, comprised of primary sentinel immune cell populations, and adaptive responses that rely on immunologic memory (176–181). A major consequence of immune senescence is an impaired capacity to repopulate naive B cells from the bone marrow and T cells from the thymus, which involutes over time. In addition to the loss of such cellularity, there is progressive loss of functional competency of both innate and adaptive immune cells (176–182). The biological and physiological nature of immune senescence remains largely unexplored, and there is growing interest in defining mechanisms and developing novel therapeutic approaches.
Human lungs are an intricate fractal network of airways composed of the trachea, bronchioles, alveolar ducts, and terminal alveolar regions. During aging, the lung undergoes a remarkable transformation with structural and functional alterations (183–185). Concurrent with aging, a loss of bone density (osteoporosis) and muscle atrophy produce physical alterations to the spine, chest wall, and thoracic cavity. The resultant physiological and functional changes include the reduction in lung elasticity of lung tissue, forced expiratory volume (FEV), forced vital capacity (FVC), and tidal volume (TV) (183–185). Collectively, the cellular, structural, and functional changes in the aging lung results in impaired host responses to respiratory infections, higher rates of autoimmunity, and diminished capacity to repair and regenerate (183–185).
Inflammaging
Described in 2000 by Franceschi et al., “inflammaging” is the process by which immune senescence is accompanied by low-level, chronic inflammation (186). Immune senescence is characterized by a decreased proliferative ability of cells and secretion of pro-inflammatory cytokines/mediators that is referred to as senescence-associated secretory phenotype (SASP). Replicative senescence results from a shortening of telomeres, which ultimately triggers DNA damage responses. In addition to replicative senescence, oxidative stress, epigenetic alterations, oncogene activation, and other stressors that induce DNA damage can induce cellular senescence (187). SASP activation leads to the production of pro-inflammatory cytokines such as IL-12, IL-6, IL-1β, TNFα, IFNγ, and other factors such as C-reactive protein and prostaglandins (187). The pervasive inflammatory milieu can be local or systemic in nature and is proposed to result from an accumulation of self-antigens produced by age-associated damage to tissues (186). The resultant accumulation of endogenous cellular matter induces an inflammatory environment leading to tissue destruction and injury (186). Inflammaging is well-recognized as an integral contributor to age-related disease pathology. This important recognition has led to growing interest in understanding the mechanisms that govern the inflammaging process and the subsequent effects on lung pathophysiology and age-related lung disorders. Elderly adults are more susceptible to pulmonary disorders such as asthma, Idiopathic Pulmonary Fibrosis (IPF), and COPD, however, the age-related mechanisms that drive disease pathology remains largely unresolved (90). Asthma is a disease characterized by airway inflammation, elevated mucus production, and airway obstruction (90). The disease phenotype of early-onset asthma has been well-defined and is consistent with a pattern of allergic inflammation including eosinophilia, and a Th2 bias (188). In contrast, the pathophysiology of late-onset asthma which affects the elderly is less well-understood, although inflammaging may represent one mechanism for disease susceptibility, progression, and relative obstinate responsiveness to therapy.
Cells Implicated in Inflammaging
Macrophages resident within the alveolar compartment are a first-line defense in innate immune responses, and are implicated in inflammaging (189). Macrophages elicit varied responses depending on microenvironmental cues, and can initiate inflammation (classically activated/M1) or attenuate inflammation and promote wound healing (alternatively activated/M2) (190). In contrast to this concept of polarized macrophage phenotypes, it is now appreciated that varied phenotypes can emerge along this differentiation spectrum. Macrophages play a central role in lung homeostasis by clearing surfactant and cellular debris from apoptotic cells (191–194). During inflammaging, macrophages lose plasticity and are unable to alternate between the pro- and anti-inflammatory phenotypes (189). The age-induced alterations in macrophage function include decreased production of pro-inflammatory cytokines (IL-6, IL-1β, TNFα), deficiency in phagocytosis and cellular debris clearance, and decreased Toll-like receptor (TLR) expression in mice (195–198). This functional transformation may, in part, be responsible for creating a pervasive cycle that sustains inflammaging in the aging lung.
Naïve T helper cells, after activation, differentiate into a multitude of specialized effector subtypes of which CD4+ Th2 and Th17 T cells have been associated with asthma. Th2 cells and type 2 immune responses are predominately associated with early-onset asthma, although other endotypes are observed in children, highlighting the heterogeneous nature of the disease (188). The Th17 subtype is generated from naïve CD4+ T cells in response to IL-1β, IL-6, TGFβ, and IL-23 (199–202). They are characterized by the fate-determining transcription factor RORγt and produce IL-17a, IL-17F, and IL-22 (203, 204). Th17 cells are associated with mucosal barriers and involved in pathogen clearance (205) A new non-canonical role for Th17 cells is emerging in infection-induced asthma. IL-17 produced by Th17 cells recruit and activate neutrophils via crosstalk with airway epithelial cells (206). Additionally, it has been shown that IL-17a, IL-17f, and IL-23 promote increased mucous production, airway remodeling, and inflammation (207). Molet et al. (208) reported an increase in IL-17 in the sputum and bronchoalveolar lavage of asthmatics; they showed that IL-17 activation of macrophages and fibroblasts promoted the secretion of IL-6, IL-1β, and TNFα in vitro. As highlighted previously, a curious paradox exists with respect to inflammaging, an increase in immune cell senescence concurrent with chronic inflammation.
We have focused, thus far, on immune cell senescence as a regulator of immune effector cell function, chronic inflammation, and airway remodeling. An alternative view suggests that immune senescence is propagated by activation of regulatory cells possessing immunosuppressive properties. A plethora of myeloid and lymphoid-derived regulatory cells with such properties has described regulatory T cells (Tregs), regulatory B cells (Bregs), and myeloid-derived suppressor cells (MDSCs) (209–214). Regulatory T cells, defined by expression of the fate-determining transcription factor Forkhead Box P3 (Foxp3), are paramount to the maintenance of immune homeostasis (209, 210). After infectious challenge and its resolution, Tregs promote tissue repair and restrain immune hyper-reactivity through the secretion of anti-inflammatory cytokines, IL-10, and TGF-β (209–212). Tregs also express high levels of CD25 (IL-2Ra) and are thought to compete with immune effector cells for IL-2 in local inflammatory environments (205). A number of modalities of Treg-induced immunosuppression have been proposed (215). Similar to other immune cell types, Tregs precipitously decline in aged adults, the mechanism of which is incompletely understood; the role of cellular senescence in this process is likely. Traditional CD4+ Foxp3+ regulatory T cells develop in the thymus and are designated natural Tregs (nTregs) (209–212). Additionally, Tregs can be induced in the periphery from conventional CD4+ T cells (iTregs) (205). The frequency and total numbers of CD4+ Tregs are elevated during the aging process in humans (205). However, murine studies have revealed that the proportion of nTregs to iTregs increases during aging, suggesting a defect in the inducibility of Tregs from the conventional T-cell pool (205). These seemingly paradoxical observations underscore the complex nature of immune network remodeling and phenotypic switching that occurs with age.
As discussed earlier, fibroblasts represent a specialized mesenchymal cell population that produce collagen, fibronectin, and proteoglycans which comprise major components of the ECM and are found in the stroma of virtually all tissue types (216). In response to injury, fibroblasts deposit ECM components providing the physical architecture and matrix-generated signals that promote wound healing (217, 218). In age-induced pathologies such as asthma, COPD, and IPF, a reorganization of airway architecture has been consistently observed. Fibroblasts produce a number of cytokines and respond to an assortment of cytokines from neighboring cells, primarily epithelial cells and immune cells. Cross-talk between fibroblasts and immune cells has been shown to be central to disease pathologies (219). The low-level, chronic inflammation that characterizes inflammaging may alter the cytokine microenvironment in aging lung tissues; the resulting dysregulation in stromal cell-immune cell cross-talk may contribute to disease progression. The precise mechanisms driving inflammaging and its link to fibrosis and airway remodeling requires further investigation. Given the dynamism involved in the progression and pathophysiology of inflammaging, it is not surprising that the mechanistic underpinnings have yet to be fully defined. In the ensuing section, we will highlight several emerging and proposed mechanisms. Cellular senescence of ASM induced by hyperoxia leads to secretion of pro-inflammatory and pro-fibrotic mediators factors that has been proposed to contribute to pediatric airway disease in the context of sequelae of preterm birth (220, 221).
The Role of Oxidative Stress in Inflammaging
Oxidative stress participates in myriad disease pathologies, and is particularly relevant for diseases of aging (222, 223). This biological phenomenon results from an imbalance between the production and clearance of ROS (222). ROS are produced during normal cellular processes such as oxidative metabolism, responses to bacterial infections, and during signaling events by Nox enzymes (222). ROS are important signaling mediators and during redox homeostasis participate in many cellular and physiologic processes such as proliferation, differentiation, migration, and apoptosis (222, 224). Conversely, during redox imbalance, ROS are implicated in disease pathogenesis due to excess cellular atrophy and death, macromolecule damage, and exacerbated inflammation (222) A specific source of ROS, mitochondrial ROS (mtROS), has gained considerable attention recently given the ubiquity of cellular respiration, even during homeostatic conditions. In healthy individuals, the oxidant/antioxidant balance is maintained. The oxidative stress theory of aging, proposed by Denham Harman in 1950 (225), was the first to implicate ROS in age-induced molecular alterations. This theory posits that an accumulation of reactive species (ROS/RNS) during normal cellular metabolism promotes the aging process by disturbing the redox balance in favor of pro-oxidants (225). Metabolic dysfunction and damage induces inflammaging through an innate immune sensing mechanism (226). DAMPs released following necrotic and apoptotic cell death are recognized by innate immune cells bearing PRRs such as TLRs and NOD-like receptors (NLRs) (226). Necroptosis, a form of regulated cell death, is regulated via a multiprotein signaling complex called the necrosome consisting of receptor-interacting kinase 1 (RIPK1), receptor-interacting kinase 3 (RIPK3), and mixed lineage kinase domain-like pseudokinase (MLKL) (227). This regulated cell death is also called inflammatory cell death, as necroptosis induced cell membrane rupture releases endogenous DAMPs [mitochondrial DNA (mtDNA), high-mobility group box 1 (HMGB1), genomic DNA and RNA] to the surrounding tissue microenvironment, leading to “sterile” inflammation (228). Pinti et al. (229) noted an increase in circulating mtDNA in aged adults correlated with elevated pro-inflammatory cytokines. These observations, along with previous studies, collectively suggests that maintenance of inflammaging in the aging lung may be mediated by age-induced tissue damage, release of endogenous DAMPs, and senescence of immune and non-immune cells.
Inflammaging and the Epigenome
In addition to genetic factors, environmental and other factors may contribute to inflammaging by epigenetic mechanisms. Several epigenetic mechanisms, including direct DNA methylation, non-coding RNAs, and histone modifications, that regulate chromatin remodeling may participate in this process (230). The initiation of epigenetic modulation is triggered by diverse environment stimuli including diet, infection exposure, toxins, and the microbiome (222). Chemical modifications to histone tails leads to three-dimensional chromatin remodeling. Euchromatin, a loose, uncoiled chromatin structure, is transcriptionally permissive, while heterochromatin with a tightly packed 3-D structure is transcriptionally silent (222) While not fully understood, it is well-accepted that chemical modifications of histones such as methylation, acetylation, phosphorylation, and ubiquitination alter chromatin structure. Methylation and acetylation of histone tails, the most studied of the modifications, are associated with permissive (H3K4me3, H3K9me1, H3K9ac, and H3K27ac) in addition to, repressive (H3K9me3 and H3K9me3) histone signatures (222). Ultimately, these chemical modifications influence gene expression in a dynamic interplay that also requires specific transcription factors and co-factors. Age-dependent and ubiquitous hypomethylation of DNA and heterochromatin loss have been broadly reported (222, 231). Furthermore, age-related alterations in post-translational modifications of histone tails have been detailed and a ubiquitous loss of nucleosome density has been observed (231). The direct relationship between age-driven epigenetic remodeling and inflammaging in pulmonary tissues is incompletely understood. It has been proposed that, in aging human cells, loss of heterochromatin is due to diminished nucleosome occupancy (231). In fact, (232), first observed this phenomenon in human skin fibroblasts (231). It was reported by Agrawal et al. (233) that DNA from elderly adults is more immunogenic relative to aged controls. Moreover, it was speculated that DNA from aged adults is a more potent DAMP due to hypomethylation, thus indistinguishable from microbial DNA by PRRs expressed by innate immune cells (222).
Metabolism and Inflammaging
Metabolic dysfunction is implicated in many age-associated chronic diseases and has been suggested to not only be a result of inflammaging, but also directly contributes to the aging process (234). Both innate and adaptive immune responses have been demonstrated to be regulated via the metabolism of multiple amino acids including tryptophan, arginine, phenylalanine, cysteine, and glutamine (235). Arginine has been shown to play a critical role in the pathogenesis of allergen-driven asthma (236–238). The catabolism of arginine via arginase is not only a biomarker for the onset of asthma, but it has been suggested to be directly involved in the manifestation of allergic airway disorders (236–238). Arginine is the substrate for both nitric oxide synthase (NOS) and arginase. These enyzmes regulate each other by controlling the availability of the substrate arginine (236–238). Given the opposing regulatory functions of NOS and arginase, a better understanding of this dynamic equilibrium in allergy-driven asthma is required (239, 240). In a 2015 study, Comhair et al. (241) identified 25 metabolic intermediates that were significantly different in the plasma of asthmatics vs healthy controls. Furthermore, their work revealed that severe asthmatics demonstrated lower levels of steroid metabolism intermediates. Alternatively, increased plasma levels of taurine, bile acids, nicotinamide, arachidonate, and adenosine-5-phosphate were observed (241). Collectively, these studies and others, underscore how the alterations in tightly regulated metabolic and homeostatic processes in aging individuals may increase susceptibility to allergic asthma.
The role of lipid metabolism in age-related diseases is not well-understood. However, it has been reported that plasma triglyceride levels increase, while phosphatidylethanolamine (PE) and phosphatidylcholine (PC), which are generally associated with membranes, decrease with age (242). Moreover, Lawton et al. (243) reported the age-related modulation of lipid composition including elevated levels of fatty acids, beta-hydroxybutyrate, carnitine, and cholesterol in older individuals. The relationship between disturbed lipid metabolism dynamics and inflammatory responses remain poorly understood. Three isoforms of the nuclear receptor family of transcription factors- peroxisome proliferation-activated receptors exist: PPARa, PPARb, and PPARg (244, 245). Fatty acid metabolism is facilitated ultimately through alterations in transcription of PPAR sensitive genes, thus PPARs function as fatty acid sensors (244, 245). PPARg has been shown to down-modulate gene expression in both monocytes and macrophages, inhibiting their activation and production of pro-inflammatory cytokines (246–248). Lipid metabolism can also regulate chromatin remodeling by direct modification of histone tails (242). Fatty acid beta-oxidation produces multiple intermediates including acetyl CoA (242). Acetyl CoA is a required cofactor used by histone acetyltransferases in order to acetylate histone tails (242). S-adenosyl methionine (SAM) is a common co-substrate and acts primarily as a methyl donor for many physiological processes such as histone and DNA methylation, as well as lipid methylation (249). The role of fatty acid metabolism in inflammaging is likely complex and may involve coordinated regulation of the epigenome with diverse biological processes altered during aging.
Loss of homeostasis in aging tissues, including the lung, is characterized by progressive metabolic dysfunction and physiological decline in aged populations (250). Aging-related alterations in metabolic function include mitochondrial dysfunction, hyperlipidemia, and increased production of ROS, all of which are implicated in inflammatory processes as well. The relationship between aging pulmonary tissues, metabolic function, and inflammation is nuanced and complex, underscoring the need for additional research into the mechanistic underpinnings of inflammaging in the lung.
Conclusion
The development of asthma, its progression and associated physiological declines in lung function is dependent on a number of genetic, environmental, and host-related factors that include age (251, 252) (Figure 2). Our understanding of the epidemiology, clinical behavior/prognosis, and the cellular/molecular pathogenesis of asthma have advanced over the past decade. Most notably, the recognition and improved understanding of disease subphenotypes and pathological endotypes has informed the need for greater precision in both the diagnosis and treatment of these heterogeneous group of clinical “syndromes.” Airway remodeling may be viewed as a specific endotype of asthma pathology that is relatively refractory to conventional anti-inflammatory therapies. It has been proposed that corticosteroid refractory asthma may represent a sub-phenotype characterized by a heightened neutrophilic airway inflammatory response in the presence or absence of eosinophils, with evidence of increased tissue injury and remodeling (253). Asthma in the aging population appears to share several features with this group of corticosteroid-refractory asthma. Recognizing that airway remodeling may occur in parallel with chronic inflammation, and not simply as a (serial) consequence of the inflammatory response, will be critical to developing novel therapeutic strategies. Over the past several years, we have witnessed intensified efforts by both academia and industry to more specifically target airway remodeling events in disease pathogenesis. It is our hope that such efforts will lead to the discovery and development of more effective therapies for severe, steroid-resistant asthma.
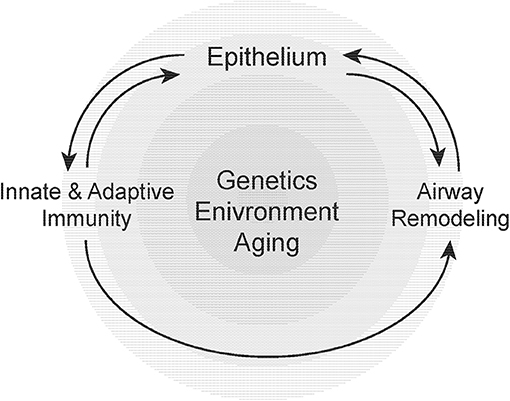
Figure 2. Susceptibility to airway remodeling in individual hosts is dependent on genetic susceptibility, environmental exposures, and aging. These risk factors regulate the cross-talk between the epithelium, innate, and adaptive immunity and mesenchymal stromal cells that contribute to airway remodeling.
Author Contributions
KH wrote sections and drew figures for manuscript. JD and VT formulated the outline of the review, wrote sections of the manuscript, and edited the manuscript. MC and TB wrote sections of the manuscript. JT and R-ML edited the manuscript. All authors contributed to the review.
Funding
JD is supported by R01HL128502, P01HL114470, U54 ES030246-01, and P30 AI027767-31 and from the National Institute of Health. VT is supported by NIH grants, P01 HL114470 and R01 AG046210, and a VA Merit Award I01BX003056.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
1. ^Available online at: http://www.nhlbi.nih.gov/health-pro/guidelines/current/asthma-guidelines
2. ^Available online at: www.ginathma.org
References
1. Sumi Y, Foley S, Daigle S, L'Archeveque J, Olivenstein R, Letuve S, et al. Structural changes and airway remodelling in occupational asthma at a mean interval of 14 years after cessation of exposure. Clin Exp Allergy. (2007) 37:1781–7. doi: 10.1111/j.1365-2222.2007.02828.x
2. Lemanske RF Jr, Busse WW. 6. Asthma. J Allergy Clin Immunol. (2003). 111(2 Suppl):S502–19. doi: 10.1067/mai.2003.94
3. Dunn RM, Busse PJ, Wechsler ME. Asthma in the elderly and late-onset adult asthma. Allergy. (2018) 73:284–94. doi: 10.1111/all.13258
4. Guerra S, Sherrill DL, Kurzius-Spencer M, Venker C, Halonen M, Quan SF, et al. The course of persistent airflow limitation in subjects with and without asthma. Respir Med. (2008) 102:1473–82. doi: 10.1016/j.rmed.2008.04.011
5. Marsh SE, Travers J, Weatherall M, Williams MV, Aldington S, Shirtcliffe PM, et al. Proportional classifications of COPD phenotypes. Thorax. (2008) 63:761–7. doi: 10.1136/thx.2007.089193
6. Saglani S, Lloyd CM. Novel concepts in airway inflammation and remodelling in asthma. Eur Respir J. (2015) 46:1796–804. doi: 10.1183/13993003.01196-2014
7. Lee JH, Haselkorn T, Borish L, Rasouliyan L, Chipps BE, Wenzel SE. Risk factors associated with persistent airflow limitation in severe or difficult-to-treat asthma: insights from the TENOR study. Chest. (2007) 132:1882–9. doi: 10.1378/chest.07-0713
8. Guerra S, Martinez FD. Epidemiology of the origins of airflow limitation in asthma. Proc Am Thorac Soc. (2009) 6:707–11. doi: 10.1513/pats.200908-085DP
9. Tai A, Tran H, Roberts M, Clarke N, Wilson J, Robertson CF. The association between childhood asthma and adult chronic obstructive pulmonary disease. Thorax. (2014) 69:805–10. doi: 10.1136/thoraxjnl-2013-204815
10. Wang YC, Jaakkola MS, Lajunen TK, Lai CH, Jaakkola JJK. Asthma-COPD Overlap Syndrome among subjects with newly diagnosed adult-onset asthma. Allergy. (2018) 73:1554–7. doi: 10.1111/all.13459
11. Holgate ST. Epithelium dysfunction in asthma. J Allergy Clin Immunol. (2007) 120:1233–44; quiz 45–6. doi: 10.1016/j.jaci.2007.10.025
12. Liesker JJ, Ten Hacken NH, Zeinstra-Smith M, Rutgers SR, Postma DS, Timens W. Reticular basement membrane in asthma and COPD: similar thickness, yet different composition. Int J Chron Obstruct Pulmon Dis. (2009) 4:127–35. doi: 10.2147/COPD.S4639
13. Bourdin A, Neveu D, Vachier I, Paganin F, Godard P, Chanez P. Specificity of basement membrane thickening in severe asthma. J Allergy Clin Immunol. (2007) 119:1367–74. doi: 10.1016/j.jaci.2007.01.055
14. Joubert P, Hamid Q. Role of airway smooth muscle in airway remodeling. J Allergy Clin Immunol. (2005) 116:713–6. doi: 10.1016/j.jaci.2005.05.042
15. McDonald DM. Angiogenesis and remodeling of airway vasculature in chronic inflammation. Am J Respir Crit Care Med. (2001) 164(10 Pt 2):S39–45. doi: 10.1164/ajrccm.164.supplement_2.2106065
16. Wilson SJ, Rigden HM, Ward JA, Laviolette M, Jarjour NN, Djukanovic R. The relationship between eosinophilia and airway remodelling in mild asthma. Clin Exp Allergy. (2013) 43:1342–50. doi: 10.1111/cea.12156
17. Bonsignore MR, Profita M, Gagliardo R, Riccobono L, Chiappara G, Pace E, et al. Advances in asthma pathophysiology: stepping forward from the Maurizio Vignola experience. Eur Respir Rev. (2015) 24:30–9. doi: 10.1183/09059180.10011114
18. Barbato A, Turato G, Baraldo S, Bazzan E, Calabrese F, Panizzolo C, et al. Epithelial damage and angiogenesis in the airways of children with asthma. Am J Respir Crit Care Med. (2006) 174:975–81. doi: 10.1164/rccm.200602-189OC
19. Saglani S, Payne DN, Zhu J, Wang Z, Nicholson AG, Bush A, et al. Early detection of airway wall remodeling and eosinophilic inflammation in preschool wheezers. Am J Respir Crit Care Med. (2007) 176:858–64. doi: 10.1164/rccm.200702-212OC
20. Payne DN, Rogers AV, Adelroth E, Bandi V, Guntupalli KK, Bush A, et al. Early thickening of the reticular basement membrane in children with difficult asthma. Am J Respir Crit Care Med. (2003) 167:78–82. doi: 10.1164/rccm.200205-414OC
21. Jenkins HA, Cool C, Szefler SJ, Covar R, Brugman S, Gelfand EW, et al. Histopathology of severe childhood asthma: a case series. Chest. (2003) 124:32–41. doi: 10.1378/chest.124.1.32
22. Barbato A, Turato G, Baraldo S, Bazzan E, Calabrese F, Tura M, et al. Airway inflammation in childhood asthma. Am J Respir Crit Care Med. (2003) 168:798–803. doi: 10.1164/rccm.200305-650OC
23. Bossley CJ, Fleming L, Gupta A, Regamey N, Frith J, Oates T, et al. Pediatric severe asthma is characterized by eosinophilia and remodeling without T(H)2 cytokines. J Allergy Clin Immunol. (2012) 129:974–82.e13. doi: 10.1016/j.jaci.2012.01.059
24. Miranda C, Busacker A, Balzar S, Trudeau J, Wenzel SE. Distinguishing severe asthma phenotypes: role of age at onset and eosinophilic inflammation. J Allergy Clin Immunol. (2004) 113:101–8. doi: 10.1016/j.jaci.2003.10.041
25. Grainge CL, Lau LC, Ward JA, Dulay V, Lahiff G, Wilson S, et al. Effect of bronchoconstriction on airway remodeling in asthma. N Engl J Med. (2011) 364:2006–15. doi: 10.1056/NEJMoa1014350
26. Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. (2012) 18:716–25. doi: 10.1038/nm.2678
27. Agache IO. Endotype driven treatment of asthma. Curr Treat Options Allergy. (2014) 1:198–212. doi: 10.1007/s40521-014-0014-0
28. Ntontsi P, Loukides S, Bakakos P, Kostikas K, Papatheodorou G, Papathanassiou E, et al. Clinical, functional and inflammatory characteristics in patients with paucigranulocytic stable asthma: comparison with different sputum phenotypes. Allergy. (2017) 72:1761–7. doi: 10.1111/all.13184
29. Simpson JL, Scott R, Boyle MJ, Gibson PG. Inflammatory subtypes in asthma: assessment and identification using induced sputum. Respirology. (2006) 11:54–61. doi: 10.1111/j.1440-1843.2006.00784.x
30. Brooks CR, van Dalen CJ, Zacharasiewicz A, Simpson JL, Harper JL, Le Gros G, et al. Absence of airway inflammation in a large proportion of adolescents with asthma. Respirology. (2016) 21:460–6. doi: 10.1111/resp.12701
31. Moore WC, Hastie AT, Li X, Li H, Busse WW, Jarjour NN, et al. Sputum neutrophil counts are associated with more severe asthma phenotypes using cluster analysis. J Allergy Clin Immunol. (2014) 133:1557–63.e5. doi: 10.1016/j.jaci.2013.10.011
32. Schleich FN, Manise M, Sele J, Henket M, Seidel L, Louis R. Distribution of sputum cellular phenotype in a large asthma cohort: predicting factors for eosinophilic vs neutrophilic inflammation. BMC Pulm Med. (2013) 13:11. doi: 10.1186/1471-2466-13-11
33. Ray A, Oriss TB, Wenzel SE. Emerging molecular phenotypes of asthma. Am J Physiol Lung Cell Mol Physiol. (2015) 308:L130–40. doi: 10.1152/ajplung.00070.2014
34. Wenzel S. Severe asthma: from characteristics to phenotypes to endotypes. Clin Exp Allergy. (2012) 42:650–8. doi: 10.1111/j.1365-2222.2011.03929.x
35. Moore WC, Meyers DA, Wenzel SE, Teague WG, Li H, Li X, et al. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med. (2010) 181:315–23. doi: 10.1164/rccm.200906-0896OC
36. Loza MJ, Djukanovic R, Chung KF, Horowitz D, Ma K, Branigan P, et al. Validated and longitudinally stable asthma phenotypes based on cluster analysis of the ADEPT study. Respir Res. (2016) 17:165. doi: 10.1186/s12931-016-0482-9
37. Lefaudeux D, De Meulder B, Loza MJ, Peffer N, Rowe A, Baribaud F, et al. U-BIOPRED clinical adult asthma clusters linked to a subset of sputum omics. J Allergy Clin Immunol. (2017) 139:1797–807. doi: 10.1016/j.jaci.2016.08.048
38. Kuruvilla ME, Lee FE, Lee GB. Understanding asthma phenotypes, endotypes, and mechanisms of disease. Clin Rev Allergy Immunol. (2019) 56:219–33. doi: 10.1007/s12016-018-8712-1
39. Robinson D, Humbert M, Buhl R, Cruz AA, Inoue H, Korom S, et al. Revisiting type 2-high and type 2-low airway inflammation in asthma: current knowledge and therapeutic implications. Clin Exp Allergy. (2017) 47:161–75. doi: 10.1111/cea.12880
40. Svenningsen S, Nair P. Asthma endotypes and an overview of targeted therapy for asthma. Front Med (Lausanne). (2017) 4:158. doi: 10.3389/fmed.2017.00158
41. Fajt ML, Wenzel SE. Asthma phenotypes and the use of biologic medications in asthma and allergic disease: the next steps toward personalized care. J Allergy Clin Immunol. (2015) 135:299–310; quiz 1. doi: 10.1016/j.jaci.2014.12.1871
42. Gelfand EW, Alam R. The other side of asthma: steroid-refractory disease in the absence of TH2-mediated inflammation. J Allergy Clin Immunol. (2015) 135:1196–8. doi: 10.1016/j.jaci.2015.01.032
43. Sin DD, Miravitlles M, Mannino DM, Soriano JB, Price D, Celli BR, et al. What is asthma-COPD overlap syndrome? Towards a consensus definition from a round table discussion. Eur Respir J. (2016) 48:664–73. doi: 10.1183/13993003.00436-2016
44. Elliot JG, Jones RL, Abramson MJ, Green FH, Mauad T, McKay KO, et al. Distribution of airway smooth muscle remodelling in asthma: relation to airway inflammation. Respirology. (2015) 20:66–72. doi: 10.1111/resp.12384
45. James AL, Elliot JG, Jones RL, Carroll ML, Mauad T, Bai TR, et al. Airway smooth muscle hypertrophy and hyperplasia in asthma. Am J Respir Crit Care Med. (2012) 185:1058–64. doi: 10.1164/rccm.201110-1849OC
46. Jeffery P, Holgate S, Wenzel S, Endobronchial Biopsy W. Methods for the assessment of endobronchial biopsies in clinical research: application to studies of pathogenesis and the effects of treatment. Am J Respir Crit Care Med. (2003) 168(6 Pt 2):S1–17. doi: 10.1164/rccm.200202-150WS
47. Kasahara K, Shiba K, Ozawa T, Okuda K, Adachi M. Correlation between the bronchial subepithelial layer and whole airway wall thickness in patients with asthma. Thorax. (2002) 57:242–6. doi: 10.1136/thorax.57.3.242
48. Saglani S, Papaioannou G, Khoo L, Ujita M, Jeffery PK, Owens C, et al. Can HRCT be used as a marker of airway remodelling in children with difficult asthma? Respir Res. (2006) 7:46. doi: 10.1186/1465-9921-7-46
49. Marchac V, Emond S, Mamou-Mani T, Le Bihan-Benjamin C, Le Bourgeois M, De Blic J, et al. Thoracic CT in pediatric patients with difficult-to-treat asthma. AJR Am J Roentgenol. (2002) 179:1245–52. doi: 10.2214/ajr.179.5.1791245
50. Kim S, Lee CH, Jin KN, Cho SH, Kang HR. Severe asthma phenotypes classified by site of airway involvement and remodeling via chest CT scan. J Investig Allergol Clin Immunol. (2018) 28:312–20. doi: 10.18176/jiaci.0265
51. Berair R, Hartley R, Mistry V, Sheshadri A, Gupta S, Singapuri A, et al. Associations in asthma between quantitative computed tomography and bronchial biopsy-derived airway remodelling. Eur Respir J. (2017) 49:1601507. doi: 10.1183/13993003.01507-2016
52. Zhang X, Xia T, Lai Z, Zhang Q, Guan Y, Zhong N. Uncontrolled asthma phenotypes defined from parameters using quantitative CT analysis. Eur Radiol. (2019) 29:2848–58. doi: 10.1007/s00330-018-5913-1
53. Amelink M, de Groot JC, de Nijs SB, Lutter R, Zwinderman AH, Sterk PJ, et al. Severe adult-onset asthma: a distinct phenotype. J Allergy Clin Immunol. (2013) 132:336–41. doi: 10.1016/j.jaci.2013.04.052
54. Amelink M, de Nijs SB, de Groot JC, van Tilburg PM, van Spiegel PI, Krouwels FH, et al. Three phenotypes of adult-onset asthma. Allergy. (2013) 68:674–80. doi: 10.1111/all.12136
55. Burrows B, Barbee RA, Cline MG, Knudson RJ, Lebowitz MD. Characteristics of asthma among elderly adults in a sample of the general population. Chest. (1991) 100:935–42. doi: 10.1378/chest.100.4.935
56. Braman SS, Kaemmerlen JT, Davis SM. Asthma in the elderly. A comparison between patients with recently acquired and long-standing disease. Am Rev Respir Dis. (1991) 143:336–40. doi: 10.1164/ajrccm/143.2.336
57. Regamey N, Ochs M, Hilliard TN, Muhlfeld C, Cornish N, Fleming L, et al. Increased airway smooth muscle mass in children with asthma, cystic fibrosis, and non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med. (2008) 177:837–43. doi: 10.1164/rccm.200707-977OC
58. Tan DJ, Walters EH, Perret JL, Lodge CJ, Lowe AJ, Matheson MC, et al. Age-of-asthma onset as a determinant of different asthma phenotypes in adults: a systematic review and meta-analysis of the literature. Expert Rev Respir Med. (2015) 9:109–23. doi: 10.1586/17476348.2015.1000311
59. Gupta S, Hartley R, Khan UT, Singapuri A, Hargadon B, Monteiro W, et al. Quantitative computed tomography-derived clusters: redefining airway remodeling in asthmatic patients. J Allergy Clin Immunol. (2014) 133:729–38.e18. doi: 10.1016/j.jaci.2013.09.039
60. Desai M, Oppenheimer J. Elucidating asthma phenotypes and endotypes: progress towards personalized medicine. Ann Allergy Asthma Immunol. (2016) 116:394–401. doi: 10.1016/j.anai.2015.12.024
61. Tliba O, Panettieri RA Jr. Paucigranulocytic asthma: Uncoupling of airway obstruction from inflammation. J Allergy Clin Immunol. (2019) 143:1287–94. doi: 10.1016/j.jaci.2018.06.008
62. Olczyk P, Mencner L, Komosinska-Vassev K. The role of the extracellular matrix components in cutaneous wound healing. BioMed Res Int. (2014) 2014:747584. doi: 10.1155/2014/747584
63. Roche WR, Beasley R, Williams JH, Holgate ST. Subepithelial fibrosis in the bronchi of asthmatics. Lancet. (1989) 1:520–4. doi: 10.1016/S0140-6736(89)90067-6
64. Araujo BB, Dolhnikoff M, Silva LF, Elliot J, Lindeman JH, Ferreira DS, et al. Extracellular matrix components and regulators in the airway smooth muscle in asthma. Eur Respir J. (2008) 32:61–9. doi: 10.1183/09031936.00147807
65. Royce SG, Cheng V, Samuel CS, Tang ML. The regulation of fibrosis in airway remodeling in asthma. Mol Cell Endocrinol. (2012) 351:167–75. doi: 10.1016/j.mce.2012.01.007
66. Mostaco-Guidolin LB, Osei ET, Ullah J, Hajimohammadi S, Fouadi M, Li X, et al. Defective fibrillar collagen organization by fibroblasts contributes to airway remodeling in asthma. Am J Respir Crit Care Med. (2019) 200:431–43. doi: 10.1164/rccm.201810-1855OC
67. Ito JT, Lourenco JD, Righetti RF, Tiberio I, Prado CM, Lopes F. Extracellular matrix component remodeling in respiratory diseases: what has been found in clinical and experimental studies? Cells. (2019) 8:342. doi: 10.3390/cells8040342
68. Reeves SR, Kolstad T, Lien TY, Elliott M, Ziegler SF, Wight TN, et al. Asthmatic airway epithelial cells differentially regulate fibroblast expression of extracellular matrix components. J Allergy Clin Immunol. (2014) 134:663–70.e1. doi: 10.1016/j.jaci.2014.04.007
69. Fang CL, Yin LJ, Sharma S, Kierstein S, Wu HF, Eid G, et al. Resistin-like molecule-β (RELM-β) targets airways fibroblasts to effect remodelling in asthma: from mouse to man. Clin Exp Allergy. (2015) 45:940–52. doi: 10.1111/cea.12481
70. Cheng W, Yan K, Xie LY, Chen F, Yu HC, Huang YX, et al. MiR-143-3p controls TGF-β1-induced cell proliferation and extracellular matrix production in airway smooth muscle via negative regulation of the nuclear factor of activated T cells 1. Mol Immunol. (2016) 78:133–9. doi: 10.1016/j.molimm.2016.09.004
71. Harkness LM, Weckmann M, Kopp M, Becker T, Ashton AW, Burgess JK. Tumstatin regulates the angiogenic and inflammatory potential of airway smooth muscle extracellular matrix. J Cell Mol Med. (2017) 21:3288–97. doi: 10.1111/jcmm.13232
72. Koopmans T, Crutzen S, Menzen MH, Halayko AJ, Hackett TL, Knight DA, et al. Selective targeting of CREB-binding protein/β-catenin inhibits growth of and extracellular matrix remodelling by airway smooth muscle. Br J Pharmacol. (2016) 173:3327–41. doi: 10.1111/bph.13620
73. Kuo C, Lim S, King NJ, Bartlett NW, Walton RP, Zhu J, et al. Rhinovirus infection induces expression of airway remodelling factors in vitro and in vivo. Respirology. (2011) 16:367–77. doi: 10.1111/j.1440-1843.2010.01918.x
74. Kuo C, Lim S, King NJ, Johnston SL, Burgess JK, Black JL, et al. Rhinovirus infection induces extracellular matrix protein deposition in asthmatic and nonasthmatic airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. (2011) 300:L951–7. doi: 10.1152/ajplung.00411.2010
75. Wang B, Chen H, Chan YL, Wang G, Oliver BG. Why do intrauterine exposure to air pollution and cigarette smoke increase the risk of asthma? Front Cell Dev Biol. (2020) 8:38. doi: 10.3389/fcell.2020.00038
76. Liu G, Cooley MA, Jarnicki AG, Hsu AC, Nair PM, Haw TJ, et al. Fibulin-1 regulates the pathogenesis of tissue remodeling in respiratory diseases. JCI Insight. (2016) 1:e86380. doi: 10.1172/jci.insight.86380
77. Krimmer D, Ichimaru Y, Burgess J, Black J, Oliver B. Exposure to biomass smoke extract enhances fibronectin release from fibroblasts. PLoS One. (2013) 8:e83938. doi: 10.1371/journal.pone.0083938
78. O'Dwyer DN, Moore BB. The role of periostin in lung fibrosis and airway remodeling. Cell Mol Life Sci. (2017) 74:4305–14. doi: 10.1007/s00018-017-2649-z
79. Patel DF, Peiro T, Shoemark A, Akthar S, Walker SA, Grabiec AM, et al. An extracellular matrix fragment drives epithelial remodeling and airway hyperresponsiveness. Sci Transl Med. (2018) 10:eaaq0693. doi: 10.1126/scitranslmed.aaq0693
80. Moorman JE, Mannino DM. Increasing U.S. asthma mortality rates: who is really dying? J Asthma. (2001) 38:65–71. doi: 10.1081/JAS-100000023
81. Tsai CL, Lee WY, Hanania NA, Camargo CA Jr. Age-related differences in clinical outcomes for acute asthma in the United States, 2006-2008. J Allergy Clin Immunol. (2012) 129:1252–8.e1. doi: 10.1016/j.jaci.2012.01.061
82. Moorman JE, Akinbami LJ, Bailey CM, Zahran HS, King ME, Johnson CA, et al. National surveillance of asthma: United States, 2001–2010. Vital Health Stat 3. (2012) 1–58.
83. Janssens JP. Aging of the respiratory system: impact on pulmonary function tests and adaptation to exertion. Clin Chest Med. (2005) 26:469–84, vi–vii. doi: 10.1016/j.ccm.2005.05.004
84. Kerstjens HA, Rijcken B, Schouten JP, Postma DS. Decline of FEV1 by age and smoking status: facts, figures, and fallacies. Thorax. (1997) 52:820–7. doi: 10.1136/thx.52.9.820
85. Lange P, Parner J, Vestbo J, Schnohr P, Jensen G. A 15-year follow-up study of ventilatory function in adults with asthma. N Engl J Med. (1998) 339:1194–200. doi: 10.1056/NEJM199810223391703
86. Niewoehner DE, Knoke JD, Kleinerman J. Peripheral airways as a determinant of ventilatory function in the human lung. J Clin Invest. (1977) 60:139–51. doi: 10.1172/JCI108750
87. Verbeken EK, Cauberghs M, Mertens I, Clement J, Lauweryns JM, Van de Woestijne KP. The senile lung. Comparison with normal and emphysematous lungs. 2. Functional aspects. Chest. (1992) 101:800–9. doi: 10.1378/chest.101.3.800
88. Enright PL, Kronmal RA, Manolio TA, Schenker MB, Hyatt RE. Respiratory muscle strength in the elderly. Correlates and reference values. Cardiovascular Health Study Research Group. Am J Respir Crit Care Med. (1994) 149(2 Pt 1):430–8. doi: 10.1164/ajrccm.149.2.8306041
89. Yanez A, Cho SH, Soriano JB, Rosenwasser LJ, Rodrigo GJ, Rabe KF, et al. Asthma in the elderly: what we know and what we have yet to know. World Allergy Organ J. (2014) 7:8. doi: 10.1186/1939-4551-7-8
90. Lowery EM, Brubaker AL, Kuhlmann E, Kovacs EJ. The aging lung. Clin Interv Aging. (2013) 8:1489–96. doi: 10.2147/CIA.S51152
91. Swindle EJ, Collins JE, Davies DE. Breakdown in epithelial barrier function in patients with asthma: identification of novel therapeutic approaches. J Allergy Clin Immunol. (2009) 124:23–34; quiz 5–6. doi: 10.1016/j.jaci.2009.05.037
92. Hertz CJ, Wu Q, Porter EM, Zhang YJ, Weismuller KH, Godowski PJ, et al. Activation of Toll-like receptor 2 on human tracheobronchial epithelial cells induces the antimicrobial peptide human β defensin-2. J Immunol. (2003) 171:6820–6. doi: 10.4049/jimmunol.171.12.6820
93. Sha Q, Truong-Tran AQ, Plitt JR, Beck LA, Schleimer RP. Activation of airway epithelial cells by toll-like receptor agonists. Am J Respir Cell Mol Biol. (2004) 31:358–64. doi: 10.1165/rcmb.2003-0388OC
94. Nathan AT, Peterson EA, Chakir J, Wills-Karp M. Innate immune responses of airway epithelium to house dust mite are mediated through β-glucan-dependent pathways. J Allergy Clin Immunol. (2009) 123:612–8. doi: 10.1016/j.jaci.2008.12.006
95. Pichavant M, Charbonnier AS, Taront S, Brichet A, Wallaert B, Pestel J, et al. Asthmatic bronchial epithelium activated by the proteolytic allergen Der p 1 increases selective dendritic cell recruitment. J Allergy Clin Immunol. (2005) 115:771–8. doi: 10.1016/j.jaci.2004.11.043
96. Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat Med. (2009) 15:410–6. doi: 10.1038/nm.1946
97. Walter MJ, Kajiwara N, Karanja P, Castro M, Holtzman MJ. Interleukin 12 p40 production by barrier epithelial cells during airway inflammation. J Exp Med. (2001) 193:339–51. doi: 10.1084/jem.193.3.339
98. White SR. Apoptosis and the airway epithelium. J Allergy (Cairo). (2011) 2011:948406. doi: 10.1155/2011/948406
99. Thompson HG, Mih JD, Krasieva TB, Tromberg BJ, George SC. Epithelial-derived TGF-β2 modulates basal and wound-healing subepithelial matrix homeostasis. Am J Physiol Lung Cell Mol Physiol. (2006) 291:L1277–85. doi: 10.1152/ajplung.00057.2006
100. Torrego A, Hew M, Oates T, Sukkar M, Fan Chung K. Expression and activation of TGF-β isoforms in acute allergen-induced remodelling in asthma. Thorax. (2007) 62:307–13. doi: 10.1136/thx.2006.063487
101. Zhang S, Smartt H, Holgate ST, Roche WR. Growth factors secreted by bronchial epithelial cells control myofibroblast proliferation: an in vitro co-culture model of airway remodeling in asthma. Lab Invest. (1999) 79:395–405.
102. Mead J, Takishima T, Leith D. Stress distribution in lungs: a model of pulmonary elasticity. J Appl Physiol. (1970) 28:596–608. doi: 10.1152/jappl.1970.28.5.596
103. Yager D, Butler JP, Bastacky J, Israel E, Smith G, Drazen JM. Amplification of airway constriction due to liquid filling of airway interstices. J Appl Physiol (1985). (1989). 66:2873–84. doi: 10.1152/jappl.1989.66.6.2873
104. Macklem PT. Respiratory mechanics. Annu Rev Physiol. (1978) 40:157–84. doi: 10.1146/annurev.ph.40.030178.001105
105. Wiggs BR, Hrousis CA, Drazen JM, Kamm RD. On the mechanism of mucosal folding in normal and asthmatic airways. J Appl Physiol (1985). (1997) 83:1814–21. doi: 10.1152/jappl.1997.83.6.1814
106. Grainge C, Dennison P, Lau L, Davies D, Howarth P. Asthmatic and normal respiratory epithelial cells respond differently to mechanical apical stress. Am J Respir Crit Care Med. (2014) 190:477–80. doi: 10.1164/rccm.201401-0107LE
107. Gill MA. The role of dendritic cells in asthma. J Allergy Clin Immunol. (2012) 129:889–901. doi: 10.1016/j.jaci.2012.02.028
108. Ressler B, Lee RT, Randell SH, Drazen JM, Kamm RD. Molecular responses of rat tracheal epithelial cells to transmembrane pressure. Am J Physiol Lung Cell Mol Physiol. (2000) 278:L1264–72. doi: 10.1152/ajplung.2000.278.6.L1264
109. Tschumperlin DJ, Dai G, Maly IV, Kikuchi T, Laiho LH, McVittie AK, et al. Mechanotransduction through growth-factor shedding into the extracellular space. Nature. (2004) 429:83–6. doi: 10.1038/nature02543
110. Takeyama K, Dabbagh K, Lee HM, Agusti C, Lausier JA, Ueki IF, et al. Epidermal growth factor system regulates mucin production in airways. Proc Natl Acad Sci U S A. (1999) 96:3081–6. doi: 10.1073/pnas.96.6.3081
111. Park JA, Drazen JM, Tschumperlin DJ. The chitinase-like protein YKL-40 is secreted by airway epithelial cells at base line and in response to compressive mechanical stress. J Biol Chem. (2010) 285:29817–25. doi: 10.1074/jbc.M110.103416
112. Bara I, Ozier A, Girodet PO, Carvalho G, Cattiaux J, Begueret H, et al. Role of YKL-40 in bronchial smooth muscle remodeling in asthma. Am J Respir Crit Care Med. (2012) 185:715–22. doi: 10.1164/rccm.201105-0915OC
113. Ortiz-Montero P, Londono-Vallejo A, Vernot JP. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun Signal. (2017) 15:17. doi: 10.1186/s12964-017-0172-3
114. Wu J, Dong F, Wang RA, Wang J, Zhao J, Yang M, et al. Central role of cellular senescence in TSLP-induced airway remodeling in asthma. PLoS One. (2013) 8:e77795. doi: 10.1371/journal.pone.0077795
115. Parikh P, Wicher S, Khandalavala K, Pabelick CM, Britt RD Jr, Prakash YS. Cellular senescence in the lung across the age spectrum. Am J Physiol Lung Cell Mol Physiol. (2019) 316:L826–L42. doi: 10.1152/ajplung.00424.2018
116. Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence in patients with pulmonary emphysema. Am J Respir Crit Care Med. (2006) 174:886–93. doi: 10.1164/rccm.200509-1374OC
117. Bernadotte A, Mikhelson VM, Spivak IM. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging (Albany NY). (2016) 8:3–11. doi: 10.18632/aging.100871
118. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. (2017) 8:14532. doi: 10.1038/ncomms14532
119. Waters DW, Blokland KEC, Pathinayake PS, Burgess JK, Mutsaers SE, Prele CM, et al. Fibroblast senescence in the pathology of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. (2018) 315:L162–L72. doi: 10.1152/ajplung.00037.2018
120. Chen K, Kolls JK. T cell-mediated host immune defenses in the lung. Annu Rev Immunol. (2013) 31:605–33. doi: 10.1146/annurev-immunol-032712-100019
121. Martin TR, Frevert CW. Innate immunity in the lungs. Proc Am Thorac Soc. (2005) 2:403–11. doi: 10.1513/pats.200508-090JS
122. Guida G, Riccio AM. Immune induction of airway remodeling. Semin Immunol. (2019) 46:101346. doi: 10.1016/j.smim.2019.101346
123. Peters M, Kohler-Bachmann S, Lenz-Habijan T, Bufe A. Influence of an allergen-specific Th17 response on remodeling of the airways. Am J Respir Cell Mol Biol. (2016) 54:350–8. doi: 10.1165/rcmb.2014-0429OC
124. Choy DF, Hart KM, Borthwick LA, Shikotra A, Nagarkar DR, Siddiqui S, et al. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med. (2015) 7:301ra129. doi: 10.1126/scitranslmed.aab3142
125. Wen FQ, Liu XD, Terasaki Y, Fang QH, Kobayashi T, Abe S, et al. Interferon-γ reduces interleukin-4- and interleukin-13-augmented transforming growth factor-β2 production in human bronchial epithelial cells by targeting Smads. Chest. (2003) 123(3 Suppl):372S−3S. doi: 10.1016/S0012-3692(15)35217-X
126. Tukler Henriksson J, Coursey TG, Corry DB, De Paiva CS, Pflugfelder SC. IL-13 stimulates proliferation and expression of mucin and immunomodulatory genes in cultured conjunctival goblet cells. Invest Ophthalmol Vis Sci. (2015) 56:4186–97. doi: 10.1167/iovs.14-15496
127. Oeser K, Schwartz C, Voehringer D. Conditional IL-4/IL-13-deficient mice reveal a critical role of innate immune cells for protective immunity against gastrointestinal helminths. Mucosal Immunol. (2015) 8:672–82. doi: 10.1038/mi.2014.101
128. Balhara J, Gounni AS. The alveolar macrophages in asthma: a double-edged sword. Mucosal Immunol. (2012) 5:605–9. doi: 10.1038/mi.2012.74
129. Wong DT, Elovic A, Matossian K, Nagura N, McBride J, Chou MY, et al. Eosinophils from patients with blood eosinophilia express transforming growth factor β1. Blood. (1991) 78:2702–7. doi: 10.1182/blood.V78.10.2702.bloodjournal78102702
130. Januskevicius A, Vaitkiene S, Gosens R, Janulaityte I, Hoppenot D, Sakalauskas R, et al. Eosinophils enhance WNT-5a and TGF-β1 genes expression in airway smooth muscle cells and promote their proliferation by increased extracellular matrix proteins production in asthma. BMC Pulm Med. (2016) 16:94. doi: 10.1186/s12890-016-0254-9
131. Kobayashi T, Kim H, Liu X, Sugiura H, Kohyama T, Fang Q, et al. Matrix metalloproteinase-9 activates TGF-β and stimulates fibroblast contraction of collagen gels. Am J Physiol Lung Cell Mol Physiol. (2014) 306:L1006–15. doi: 10.1152/ajplung.00015.2014
132. Fang KC, Raymond WW, Lazarus SC, Caughey GH. Dog mastocytoma cells secrete a 92-kD gelatinase activated extracellularly by mast cell chymase. J Clin Invest. (1996) 97:1589–96. doi: 10.1172/JCI118583
133. Cairns JA, Walls AF. Mast cell tryptase is a mitogen for epithelial cells. Stimulation of IL-8 production and intercellular adhesion molecule-1 expression. J Immunol. (1996) 156:275–83.
134. Compton SJ, Cairns JA, Holgate ST, Walls AF. The role of mast cell tryptase in regulating endothelial cell proliferation, cytokine release, and adhesion molecule expression: tryptase induces expression of mRNA for IL-1 β and IL-8 and stimulates the selective release of IL-8 from human umbilical vein endothelial cells. J Immunol. (1998) 161:1939–46.
135. Ardi VC, Kupriyanova TA, Deryugina EI, Quigley JP. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc Natl Acad Sci U S A. (2007) 104:20262–7. doi: 10.1073/pnas.0706438104
136. Ventura I, Vega A, Chacon P, Chamorro C, Aroca R, Gomez E, et al. Neutrophils from allergic asthmatic patients produce and release metalloproteinase-9 upon direct exposure to allergens. Allergy. (2014) 69:898–905. doi: 10.1111/all.12414
137. Cundall M, Sun Y, Miranda C, Trudeau JB, Barnes S, Wenzel SE. Neutrophil-derived matrix metalloproteinase-9 is increased in severe asthma and poorly inhibited by glucocorticoids. J Allergy Clin Immunol. (2003) 112:1064–71. doi: 10.1016/j.jaci.2003.08.013
138. Jatakanon A, Uasuf C, Maziak W, Lim S, Chung KF, Barnes PJ. Neutrophilic inflammation in severe persistent asthma. Am J Respir Crit Care Med. (1999) 160(5 Pt 1):1532–9. doi: 10.1164/ajrccm.160.5.9806170
139. Louis R, Lau LC, Bron AO, Roldaan AC, Radermecker M, Djukanovic R. The relationship between airways inflammation and asthma severity. Am J Respir Crit Care Med. (2000) 161:9–16. doi: 10.1164/ajrccm.161.1.9802048
140. Huang G, Wang Y, Chi H. Regulation of TH17 cell differentiation by innate immune signals. Cell Mol Immunol. (2012) 9:287–95. doi: 10.1038/cmi.2012.10
141. Jevnikar Z, Ostling J, Ax E, Calven J, Thorn K, Israelsson E, et al. Epithelial IL-6 trans-signaling defines a new asthma phenotype with increased airway inflammation. J Allergy Clin Immunol. (2019) 143:577–90. doi: 10.1016/j.jaci.2018.05.026
142. Flannigan KL, Ngo VL, Geem D, Harusato A, Hirota SA, Parkos CA, et al. IL-17A-mediated neutrophil recruitment limits expansion of segmented filamentous bacteria. Mucosal Immunol. (2017) 10:673–84. doi: 10.1038/mi.2016.80
143. Leyva-Castillo JM, Yoon J, Geha RS. IL-22 promotes allergic airway inflammation in epicutaneously sensitized mice. J Allergy Clin Immunol. (2019) 143:619–30.e7. doi: 10.1016/j.jaci.2018.05.032
144. Liang SC, Long AJ, Bennett F, Whitters MJ, Karim R, Collins M, et al. An IL-17F/A heterodimer protein is produced by mouse Th17 cells and induces airway neutrophil recruitment. J Immunol. (2007) 179:7791–9. doi: 10.4049/jimmunol.179.11.7791
145. Khan MA, Assiri AM, Broering DC. Complement mediators: key regulators of airway tissue remodeling in asthma. J Transl Med. (2015) 13:272. doi: 10.1186/s12967-015-0565-2
146. Rosethorne EM, Charlton SJ. Airway remodeling disease: primary human structural cells and phenotypic and pathway assays to identify targets with potential to prevent or reverse remodeling. J Exp Pharmacol. (2018) 10:75–85. doi: 10.2147/JEP.S159124
147. Holgate ST. A look at the pathogenesis of asthma: the need for a change in direction. Discov Med. (2010) 9:439–47.
148. Davies DE, Wicks J, Powell RM, Puddicombe SM, Holgate ST. Airway remodeling in asthma: new insights. J Allergy Clin Immunol. (2003) 111:215–25; quiz 26. doi: 10.1067/mai.2003.128
149. Prakash YS. Emerging concepts in smooth muscle contributions to airway structure and function: implications for health and disease. Am J Physiol Lung Cell Mol Physiol. (2016) 311:L1113–L40. doi: 10.1152/ajplung.00370.2016
150. Yap HM, Israf DA, Harith HH, Tham CL, Sulaiman MR. Crosstalk between signaling pathways involved in the regulation of airway smooth muscle cell hyperplasia. Front Pharmacol. (2019) 10:1148. doi: 10.3389/fphar.2019.01148
151. Tliba O, Panettieri RA Jr. Noncontractile functions of airway smooth muscle cells in asthma. Annu Rev Physiol. (2009) 71:509–35. doi: 10.1146/annurev.physiol.010908.163227
152. Van Eerdewegh P, Little RD, Dupuis J, Del Mastro RG, Falls K, Simon J, et al. Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature. (2002) 418:426–30. doi: 10.1038/nature00878
153. Haitchi HM, Powell RM, Shaw TJ, Howarth PH, Wilson SJ, Wilson DI, et al. ADAM33 expression in asthmatic airways and human embryonic lungs. Am J Respir Crit Care Med. (2005) 171:958–65. doi: 10.1164/rccm.200409-1251OC
154. Pan S, Shah SD, Panettieri RA Jr, Deshpande DA. Bnip3 regulates airway smooth muscle cell focal adhesion and proliferation. Am J Physiol Lung Cell Mol Physiol. (2019) 317:L758–L67. doi: 10.1152/ajplung.00224.2019
155. Wortley MA, Bonvini SJ. Transforming growth factor-β1: a novel cause of resistance to bronchodilators in asthma? Am J Respir Cell Mol Biol. (2019) 61:134–5. doi: 10.1165/rcmb.2019-0020ED
156. Ojiaku CA, Chung E, Parikh V, Williams JK, Schwab A, Fuentes AL, et al. Transforming growth factor-β1 decreases β2-agonist-induced relaxation in human airway smooth muscle. Am J Respir Cell Mol Biol. (2019) 61:209–18. doi: 10.1165/rcmb.2018-0301OC
157. Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, et al. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med. (2009) 15:1077–81. doi: 10.1038/nm.2005
158. Sutcliffe A, Hollins F, Gomez E, Saunders R, Doe C, Cooke M, et al. Increased nicotinamide adenine dinucleotide phosphate oxidase 4 expression mediates intrinsic airway smooth muscle hypercontractility in asthma. Am J Respir Crit Care Med. (2012) 185:267–74. doi: 10.1164/rccm.201107-1281OC
159. Sturrock A, Huecksteadt TP, Norman K, Sanders K, Murphy TM, Chitano P, et al. Nox4 mediates TGF-β1-induced retinoblastoma protein phosphorylation, proliferation, and hypertrophy in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. (2007) 292:L1543–L55. doi: 10.1152/ajplung.00430.2006
160. Shipe R, Burdick MD, Strieter BA, Liu L, Shim YM, Sung SS, et al. Number, activation, and differentiation of circulating fibrocytes correlate with asthma severity. J Allergy Clin Immunol. (2016) 137:750–7.e3. doi: 10.1016/j.jaci.2015.07.037
161. Zou Y, Song W, Zhou L, Mao Y, Hong W. House dust mite induces Sonic hedgehog signaling that mediates epithelialmesenchymal transition in human bronchial epithelial cells. Mol Med Rep. (2019) 20:4674–82. doi: 10.3892/mmr.2019.10707
162. Kuwabara Y, Kobayashi T, D'Alessandro-Gabazza CN, Toda M, Yasuma T, Nishihama K, et al. Role of matrix metalloproteinase-2 in eosinophil-mediated airway remodeling. Front Immunol. (2018) 9:2163. doi: 10.3389/fimmu.2018.02163
163. Zhai J, Insel M, Addison KJ, Stern DA, Pederson W, Dy A, et al. Club cell secretory protein deficiency leads to altered lung function. Am J Respir Crit Care Med. (2019) 199:302–12. doi: 10.1164/rccm.201807-1345OC
164. Chanda D, Otoupalova E, Hough KP, Locy ML, Bernard K, Deshane JS, et al. Fibronectin on the surface of extracellular vesicles mediates fibroblast invasion. Am J Respir Cell Mol Biol. (2019) 60:279–88. doi: 10.1165/rcmb.2018-0062OC
165. Hough KP, Wilson LS, Trevor JL, Strenkowski JG, Maina N, Kim YI, et al. Unique lipid signatures of extracellular vesicles from the airways of asthmatics. Sci Rep. (2018) 8:10340. doi: 10.1038/s41598-018-28655-9
166. Mainardi AS, Castro M, Chupp G. Bronchial thermoplasty. Clin Chest Med. (2019) 40:193–207. doi: 10.1016/j.ccm.2018.10.015
167. Thomson NC. Recent developments in bronchial thermoplasty for severe asthma. J Asthma Allergy. (2019) 12:375–87. doi: 10.2147/JAA.S200912
168. McKnight CG, Potter C, Finkelman FD. IL-4Rα expression by airway epithelium and smooth muscle accounts for nearly all airway hyperresponsiveness in murine allergic airway disease. Mucosal Immunol. (2019) 13:283–92. doi: 10.1038/s41385-019-0232-7
169. Ambhore NS, Kalidhindi RSR, Loganathan J, Sathish V. Role of differential estrogen receptor activation in airway hyperreactivity and remodeling in a murine model of asthma. Am J Respir Cell Mol Biol. (2019) 61:469–80. doi: 10.1165/rcmb.2018-0321OC
170. Talmon M, Rossi S, Lim D, Pollastro F, Palattella G, Ruffinatti FA, et al. Absinthin, an agonist of the bitter taste receptor hTAS2R46, uncovers an ER-to-mitochondria Ca2+-shuttling event. J Biol Chem. (2019) 294:12472–82. doi: 10.1074/jbc.RA119.007763
171. Blais-Lecours P, Laouafa S, Arias-Reyes C, Santos WL, Joseph V, Burgess JK, et al. Metabolic adaptation of airway smooth muscle cells to an SPHK2 substrate precedes cytostasis. Am J Respir Cell Mol Biol. (2020) 62:35–42. doi: 10.1165/rcmb.2018-0397OC
172. Nayak AP, Shah SD, Michael JV, Deshpande DA. Bitter taste receptors for asthma therapeutics. Front Physiol. (2019) 10:884. doi: 10.3389/fphys.2019.00884
173. Ingram JL. Give me some room to breathe! can targeting SPHK2 reduce airway smooth muscle thickening in asthma? Am J Respir Cell Mol Biol. (2020) 62:1–2. doi: 10.1165/rcmb.2019-0240ED
174. Saunders R, Kaul H, Berair R, Gonem S, Singapuri A, Sutcliffe AJ, et al. DP2 antagonism reduces airway smooth muscle mass in asthma by decreasing eosinophilia and myofibroblast recruitment. Sci Transl Med. (2019) 11:eaao6451. doi: 10.1126/scitranslmed.aao6451
175. Liu RM, Eldridge S, Watanabe N, Deshane J, Kuo HC, Jiang C, et al. Therapeutic potential of an orally effective small molecule inhibitor of plasminogen activator inhibitor for asthma. Am J Physiol Lung Cell Mol Physiol. (2016) 310:L328–L36. doi: 10.1152/ajplung.00217.2015
176. Pinti M, Appay V, Campisi J, Frasca D, Fulop T, Sauce D, et al. Aging of the immune system: focus on inflammation and vaccination. Eur J Immunol. (2016) 46:2286–301. doi: 10.1002/eji.201546178
177. Lu Y, Tan CT, Nyunt MS, Mok EW, Camous X, Kared H, et al. Inflammatory and immune markers associated with physical frailty syndrome: findings from Singapore longitudinal aging studies. Oncotarget. (2016) 7:28783–95. doi: 10.18632/oncotarget.8939
178. Fulop T, Herbein G, Cossarizza A, Witkowski JM, Frost E, Dupuis G, et al. Cellular senescence, immunosenescence and HIV. Interdiscip Top Gerontol Geriatr. (2017) 42:28–46. doi: 10.1159/000448542
179. Fulop T, Dupuis G, Witkowski JM, Larbi A. The role of immunosenescence in the development of age-related diseases. Rev Invest Clin. (2016) 68:84–91.
180. Fulop T, Dupuis G, Baehl S, Le Page A, Bourgade K, Frost E, et al. From inflamm-aging to immune-paralysis: a slippery slope during aging for immune-adaptation. Biogerontology. (2016) 17:147–57. doi: 10.1007/s10522-015-9615-7
181. Fulop T. Biological research into aging: from cells to clinic. Biogerontology. (2016) 17:1–6. doi: 10.1007/s10522-016-9633-0
182. Barth E, Srivastava A, Stojiljkovic M, Frahm C, Axer H, Witte OW, et al. Conserved aging-related signatures of senescence and inflammation in different tissues and species. Aging (Albany NY). (2019) 11:8556–72. doi: 10.18632/aging.102345
183. Meyer KC. Impact of aging on the lung. Semin Respir Crit Care Med. (2010) 31:519–20. doi: 10.1055/s-0030-1265892
184. Kling KM, Lopez-Rodriguez E, Pfarrer C, Muhlfeld C, Brandenberger C. Aging exacerbates acute lung injury-induced changes of the air-blood barrier, lung function, and inflammation in the mouse. Am J Physiol Lung Cell Mol Physiol. (2017) 312:L1–L12. doi: 10.1152/ajplung.00347.2016
185. Brandenberger C, Muhlfeld C. Mechanisms of lung aging. Cell Tissue Res. (2017) 367:469–80. doi: 10.1007/s00441-016-2511-x
186. Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. (2000) 908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x
187. Murray MA, Chotirmall SH. The impact of immunosenescence on pulmonary disease. Mediators Inflamm. (2015) 2015:692546. doi: 10.1155/2015/692546
188. Guibas GV, Mathioudakis AG, Tsoumani M, Tsabouri S. Relationship of allergy with asthma: there are more than the allergy “eggs” in the asthma “basket”. Front Pediatr. (2017) 5:92. doi: 10.3389/fped.2017.00092
189. Canan CH, Gokhale NS, Carruthers B, Lafuse WP, Schlesinger LS, Torrelles JB, et al. Characterization of lung inflammation and its impact on macrophage function in aging. J Leukoc Biol. (2014) 96:473–80. doi: 10.1189/jlb.4A0214-093RR
190. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. (2002) 23:549–55. doi: 10.1016/S1471-4906(02)02302-5
191. Marakalala MJ, Martinez FO, Pluddemann A, Gordon S. Macrophage heterogeneity in the immunopathogenesis of tuberculosis. Front Microbiol. (2018) 9:1028. doi: 10.3389/fmicb.2018.01028
192. Gordon S, Pluddemann A. Macrophage clearance of apoptotic cells: a critical assessment. Front Immunol. (2018) 9:127. doi: 10.3389/fimmu.2018.00127
193. Dranoff G, Mulligan RC. Activities of granulocyte-macrophage colony-stimulating factor revealed by gene transfer and gene knockout studies. Stem Cells. (1994) 12(Suppl 1):173–82; discussion 82–4.
194. Dranoff G, Crawford AD, Sadelain M, Ream B, Rashid A, Bronson RT, et al. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science. (1994) 264:713–6. doi: 10.1126/science.8171324
195. Sharma L, Wu W, Dholakiya SL, Gorasiya S, Wu J, Sitapara R, et al. Assessment of phagocytic activity of cultured macrophages using fluorescence microscopy and flow cytometry. Methods Mol Biol. (2014) 1172:137–45. doi: 10.1007/978-1-4939-0928-5_12
196. Takahashi R, Ishigami A, Kobayashi Y, Nagata K. Skewing of peritoneal resident macrophages toward M1-like is involved in enhancement of inflammatory responses induced by secondary necrotic neutrophils in aged mice. Cell Immunol. (2016) 304–305:44–8. doi: 10.1016/j.cellimm.2016.03.001
197. Boyd AR, Shivshankar P, Jiang S, Berton MT, Orihuela CJ. Age-related defects in TLR2 signaling diminish the cytokine response by alveolar macrophages during murine pneumococcal pneumonia. Exp Gerontol. (2012) 47:507–18. doi: 10.1016/j.exger.2012.04.004
198. Linehan E, Dombrowski Y, Snoddy R, Fallon PG, Kissenpfennig A, Fitzgerald DC. Aging impairs peritoneal but not bone marrow-derived macrophage phagocytosis. Aging Cell. (2014) 13:699–708. doi: 10.1111/acel.12223
199. Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-β induces development of the T(H)17 lineage. Nature. (2006) 441:231–4. doi: 10.1038/nature04754
200. Harrington LE, Mangan PR, Weaver CT. Expanding the effector CD4 T-cell repertoire: the Th17 lineage. Curr Opin Immunol. (2006) 18:349–56. doi: 10.1016/j.coi.2006.03.017
201. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. (2006) 441:235–8. doi: 10.1038/nature04753
202. Lasiglie D, Traggiai E, Federici S, Alessio M, Buoncompagni A, Accogli A, et al. Role of IL-1 β in the development of human T(H)17 cells: lesson from NLPR3 mutated patients. PLoS One. (2011) 6:e20014. doi: 10.1371/journal.pone.0020014
203. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. (2009) 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
204. Ivanov S, Bozinovski S, Bossios A, Valadi H, Vlahos R, Malmhall C, et al. Functional relevance of the IL-23-IL-17 axis in lungs in vivo. Am J Respir Cell Mol Biol. (2007) 36:442–51. doi: 10.1165/rcmb.2006-0020OC
205. Jagger A, Shimojima Y, Goronzy JJ, Weyand CM. Regulatory T cells and the immune aging process: a mini-review. Gerontology. (2014) 60:130–7. doi: 10.1159/000355303
206. Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. (1996) 183:2593–603. doi: 10.1084/jem.183.6.2593
207. Wang Y. H., Wills-Karp M. (2011). The potential role of interleukin-17 in severe asthma. Curr. Allergy Asthma Rep. 11, 388–394. doi: 10.1007/s11882-011-0210-y
208. Molet S, Hamid Q, Davoine F, Nutku E, Taha R, Page N, et al. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J Allergy Clin Immunol. (2001) 108:430–8. doi: 10.1067/mai.2001.117929
209. Salminen A, Kauppinen A, Kaarniranta K. AMPK activation inhibits the functions of myeloid-derived suppressor cells (MDSC): impact on cancer and aging. J Mol Med (Berl). (2019) 97:1049–64. doi: 10.1007/s00109-019-01795-9
210. Salminen A, Kaarniranta K, Kauppinen A. Immunosenescence: the potential role of myeloid-derived suppressor cells (MDSC) in age-related immune deficiency. Cell Mol Life Sci. (2019) 76:1901–18. doi: 10.1007/s00018-019-03048-x
211. Williams LM, Rudensky AY. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol. (2007) 8:277–84. doi: 10.1038/ni1437
212. Maynard CL, Harrington LE, Janowski KM, Oliver JR, Zindl CL, Rudensky AY, et al. Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3− precursor cells in the absence of interleukin 10. Nat Immunol. (2007) 8:931–41. doi: 10.1038/ni1504
213. Nagaraj S, Collazo M, Corzo CA, Youn JI, Ortiz M, Quiceno D, et al. Regulatory myeloid suppressor cells in health and disease. Cancer Res. (2009) 69:7503–6. doi: 10.1158/0008-5472.CAN-09-2152
214. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
215. Shevach EM. Mechanisms of Foxp3+ T regulatory cell-mediated suppression. Immunity. (2009) 30:636–45. doi: 10.1016/j.immuni.2009.04.010
216. McAnulty RJ. Fibroblasts and myofibroblasts: their source, function and role in disease. Int J Biochem Cell Biol. (2007) 39:666–71. doi: 10.1016/j.biocel.2006.11.005
217. Murtha LA, Morten M, Schuliga MJ, Mabotuwana NS, Hardy SA, Waters DW, et al. The role of pathological aging in cardiac and pulmonary fibrosis. Aging Dis. (2019) 10:419–28. doi: 10.14336/AD.2018.0601
218. Hardy SA, Mabotuwana NS, Murtha LA, Coulter B, Sanchez-Bezanilla S, Al-Omary MS, et al. Novel role of extracellular matrix protein 1 (ECM1) in cardiac aging and myocardial infarction. PLoS One. (2019) 14:e0212230. doi: 10.1371/journal.pone.0212230
219. Van Linthout S, Miteva K, Tschope C. Crosstalk between fibroblasts and inflammatory cells. Cardiovasc Res. (2014) 102:258–69. doi: 10.1093/cvr/cvu062
220. You K, Parikh P, Khandalavala K, Wicher SA, Manlove L, Yang B, et al. Moderate hyperoxia induces senescence in developing human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. (2019) 317:L525–L36. doi: 10.1152/ajplung.00067.2019
221. Parikh P, Britt RD Jr, Manlove LJ, Wicher SA, Roesler A, Ravix J, et al. Hyperoxia-induced cellular senescence in fetal airway smooth muscle cells. Am J Respir Cell Mol Biol. (2019) 61:51–60. doi: 10.1165/rcmb.2018-0176OC
222. Nardini C, Moreau JF, Gensous N, Ravaioli F, Garagnani P, Bacalini MG. The epigenetics of inflammaging: the contribution of age-related heterochromatin loss and locus-specific remodelling and the modulation by environmental stimuli. Semin Immunol. (2018) 40:49–60. doi: 10.1016/j.smim.2018.10.009
223. Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, et al. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med. (2014) 6:231ra47. doi: 10.1126/scitranslmed.3008182
224. Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. (2000) 279:L1005–28. doi: 10.1152/ajplung.2000.279.6.L1005
225. Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. (1956) 11:298–300. doi: 10.1093/geronj/11.3.298
226. Giuliani A, Prattichizzo F, Micolucci L, Ceriello A, Procopio AD, Rippo MR. Mitochondrial (Dys) function in inflammaging: do mitomirs influence the energetic, oxidative, and inflammatory status of senescent cells? Mediators Inflamm. (2017) 2017:2309034. doi: 10.1155/2017/2309034
227. Sauler M, Bazan IS, Lee PJ. Cell death in the lung: the apoptosis-necroptosis axis. Annu Rev Physiol. (2019) 81:375–402. doi: 10.1146/annurev-physiol-020518-114320
228. Srikrishna G, Freeze HH. Endogenous damage-associated molecular pattern molecules at the crossroads of inflammation and cancer. Neoplasia. (2009) 11:615–28. doi: 10.1593/neo.09284
229. Pinti M, Cevenini E, Nasi M, De Biasi S, Salvioli S, Monti D, et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur J Immunol. (2014) 44:1552–62. doi: 10.1002/eji.201343921
230. Handy DE, Castro R, Loscalzo J. Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation. (2011) 123:2145–56. doi: 10.1161/CIRCULATIONAHA.110.956839
231. Sidler C, Kovalchuk O, Kovalchuk I. Epigenetic regulation of cellular senescence and aging. Front Genet. (2017) 8:138. doi: 10.3389/fgene.2017.00138
232. Ishimi Y., Kojima M., Takeuchi F., Miyamoto T., Yamada M., Hanaoka F. (1987). Changes in chromatin structure during aging of human skin fibroblasts. Exp. Cell Res. 169, 458–67. doi: 10.1016/0014-4827(87)90206-0
233. Agarwal S, Banerjee S, Tuzcu EM, Kapadia SR. Influence of age on revascularization related costs of hospitalization among patients of stable coronary artery disease. Am J Cardiol. (2010) 105:1549–54. doi: 10.1016/j.amjcard.2010.01.012
234. Franceschi C, Garagnani P, Morsiani C, Conte M, Santoro A, Grignolio A, et al. The continuum of aging and age-related diseases: common mechanisms but different rates. Front Med (Lausanne). (2018) 5:61. doi: 10.3389/fmed.2018.00061
235. Grohmann U, Bronte V. Control of immune response by amino acid metabolism. Immunol Rev. (2010) 236:243–64. doi: 10.1111/j.1600-065X.2010.00915.x
236. King NE, Rothenberg ME, Zimmermann N. Arginine in asthma and lung inflammation. J Nutr. (2004) 134(10 Suppl):2830S–6S; discussion 53S. doi: 10.1093/jn/134.10.2830S
237. King ME, Mannino DM, Holguin F. Risk factors for asthma incidence. A review of recent prospective evidence. Panminerva Med. (2004) 46:97–110.
238. Hsu JY, King SL, Kuo BI, Chiang CD. Age of onset and the characteristics of asthma. Respirology. (2004) 9:369–72. doi: 10.1111/j.1440-1843.2004.00572.x
239. Deshane JS, Redden DT, Zeng M, Spell ML, Zmijewski JW, Anderson JT, et al. Subsets of airway myeloid-derived regulatory cells distinguish mild asthma from chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2015) 135:413–24.e15. doi: 10.1016/j.jaci.2014.08.040
240. Deshane J, Zmijewski JW, Luther R, Gaggar A, Deshane R, Lai JF, et al. Free radical-producing myeloid-derived regulatory cells: potent activators and suppressors of lung inflammation and airway hyperresponsiveness. Mucosal Immunol. (2011) 4:503–18. doi: 10.1038/mi.2011.16
241. Comhair SA, McDunn J, Bennett C, Fettig J, Erzurum SC, Kalhan SC. Metabolomic endotype of asthma. J Immunol. (2015) 195:643–50. doi: 10.4049/jimmunol.1500736
242. Papsdorf K, Brunet A. Linking lipid metabolism to chromatin regulation in aging. Trends Cell Biol. (2019) 29:97–116. doi: 10.1016/j.tcb.2018.09.004
243. Lawton KA, Berger A, Mitchell M, Milgram KE, Evans AM, Guo L, et al. Analysis of the adult human plasma metabolome. Pharmacogenomics. (2008) 9:383–97. doi: 10.2217/14622416.9.4.383
244. Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, et al. A PPAR γ-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. (2001) 7:161–71. doi: 10.1016/S1097-2765(01)00164-2
245. Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-γ dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. (2001) 7:48–52. doi: 10.1038/83336
246. Jiang C, Ting AT, Seed B. PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature. (1998) 391:82–6. doi: 10.1038/34184
247. Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature. (1998) 391:79–82. doi: 10.1038/34178
248. Ricote M, Huang J, Fajas L, Li A, Welch J, Najib J, et al. Expression of the peroxisome proliferator-activated receptor γ (PPARγ) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc Natl Acad Sci U S A. (1998) 95:7614–9. doi: 10.1073/pnas.95.13.7614
249. Ye C, Sutter BM, Wang Y, Kuang Z, Tu BP. A Metabolic function for phospholipid and histone methylation. Mol Cell. (2017) 66:180–93.e8. doi: 10.1016/j.molcel.2017.02.026
250. Barzilai N, Huffman DM, Muzumdar RH, Bartke A. The critical role of metabolic pathways in aging. Diabetes. (2012) 61:1315–22. doi: 10.2337/db11-1300
251. Klimentidis YC, Vazquez AI, de Los Campos G, Allison DB, Dransfield MT, Thannickal VJ. Heritability of pulmonary function estimated from pedigree and whole-genome markers. Front Genet. (2013) 4:174. doi: 10.3389/fgene.2013.00174
252. Moffatt MF, Kabesch M, Liang L, Dixon AL, Strachan D, Heath S, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. (2007) 448:470–3. doi: 10.1038/nature06014
Keywords: asthma, epithelium, airway remodeling, mesenchyme, immune cells
Citation: Hough KP, Curtiss ML, Blain TJ, Liu R-M, Trevor J, Deshane JS and Thannickal VJ (2020) Airway Remodeling in Asthma. Front. Med. 7:191. doi: 10.3389/fmed.2020.00191
Received: 17 February 2020; Accepted: 21 April 2020;
Published: 21 May 2020.
Edited by:
Hsiao-Chi Chuang, Taipei Medical University, TaiwanReviewed by:
Chih-Ming Weng, Taipei Medical University, TaiwanEleni Papakonstantinou, Aristotle University of Thessaloniki, Greece
Christina Pabelick, Mayo Clinic, United States
Brian Gregory George Oliver, University of Technology Sydney, Australia
Copyright © 2020 Hough, Curtiss, Blain, Liu, Trevor, Deshane and Thannickal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jessy S. Deshane, jessydeshane@uabmc.edu; Victor J. Thannickal, vthannickal@uabmc.edu