 Katja Bieber
Katja Bieber Khalaf Kridin
Khalaf Kridin Shirin Emtenani
Shirin Emtenani Katharina Boch
Katharina Boch Enno Schmidt
Enno Schmidt Ralf J. Ludwig
Ralf J. Ludwig- 1Lübeck Institute of Experimental Dermatology and Center for Research on Inflammation of the Skin, University of Lübeck, Lübeck, Germany
- 2Department of Dermatology and Center for Research on Inflammation of the Skin, University of Lübeck, Lübeck, Germany
Pemphigus and pemphigoid diseases are autoimmune bullous diseases characterized and caused by autoantibodies targeting adhesion molecules in the skin and/or mucous membranes. Personalized medicine is a new medical model that separates patients into different groups and aims to tailor medical decisions, practices, and interventions based on the individual patient`s predicted response or risk factors. An important milestone in personalized medicine in pemphigus and pemphigoid was achieved by verifying the autoimmune pathogenesis underlying these diseases, as well as by identifying and cloning several pemphigus/pemphigoid autoantigens. The latter has become the basis of the current, molecular-based diagnosis that allows the differentiation of about a dozen pemphigus and pemphigoid entities. The importance of autoantigen-identification in pemphigus/pemphigoid is further highlighted by the emergence of autoantigen-specific B cell depleting strategies. To achieve this goal, the chimeric antigen receptor (CAR) T cell technology, which is used for the treatment of certain hematological malignancies, was adopted, by generating chimeric autoantigen receptor (CAAR) T cells. In addition to these more basic science-driven milestones in personalized medicine in pemphigus and pemphigoid, careful clinical observation and epidemiology are again contributing to personalized medicine. The identification of clearly distinct clinical phenotypes in pemphigoid like the non-inflammatory and gliptin-associated bullous pemphigoid embodies a prominent instance of the latter. We here review these exciting developments in basic, translational, clinical, and epidemiological research in pemphigus and pemphigoid. Overall, we hereby aim to attract more researchers and clinicians to this highly interesting and dynamic field of research.
Pemphigus and Pemphigoid
Pemphigus and pemphigoid are autoimmune diseases of the skin and/or mucous membranes characterized and caused by autoantibodies targeting structural proteins (1). In individual patients, the specific pemphigus or pemphigoid disease is diagnosed based on the clinical presentation, the detection of tissue-bound autoantibodies and the autoantibody specificity (2–4). Albeit rare, pemphigus and pemphigoid diseases impose a major disease burden with a high unmet medical need (5, 6). The clinical hallmark of pemphigus and pemphigoid is (muco)-cutaneous blistering, which occurs intradermal in pemphigus and subepidermal in pemphigoid. In both diseases, autoantibodies are generated in a CD4-dependent fashion. As a general principle, blistering occurs directly as a result of autoantibody binding to the target antigens and via complement-independent mechanisms in pemphigus, whereas blistering in pemphigoid usually depends on the activation of innate immune responses through the Fc-portion of the autoantibodies (7, 8). Pemphigus is currently treated with high-dose corticosteroids and the anti-CD20 antibody rituximab, which achieves complete remissions in 80% of the patients (9). The most effective treatment of bullous pemphigoid (BP), by far the most common pemphigoid disease, is long-term application of superpotent topical or oral corticosteroids (10). Epidermolysis bullosa acquisita (EBA) is another pemphigoid disease characterized by a chronic course and is often more refractory to treatment as compared to BP. The main challenges in treating pemphigus are the relative long time needed to induce remissions, high rate of adverse events, and relapse after stopping treatment (11). In BP, relapses after cessation of treatment are the main challenge, as these leads to prolonged treatment with corticosteroids, which are partially responsible for the increased morbidity and mortality of the patients (12).
The current research on pemphigus and pemphigoid diseases is, in our opinion, based on the landmark discovery by Walter Lever in 1953, who, for the first time, clearly differentiated between pemphigus and pemphigoid diseases, mainly based on the histology of skin affected by either one of the diseases (13). This differentiation between pemphigus and pemphigoid based on lesional histopathology promoted tailoring specific treatments for patients with autoimmune bullous diseases (AIBDs), as patients with pemphigus necessitated more aggressive immunospuressive therapy as compared to their counterparts with BP. Subsequently, further milestones in personalized medicine in pemphigus and pemphigoid were made (Figure 1):
1. Identification of distinct patterns of autoantibody deposits in the skin
2. Discovery of unique autoantigens in distinct pemphigus and pemphigoid diseases
3. Defining pemphigus and pemphigoid as autoimmune diseases
4. Establishing the current, molecular-based diagnosis of pemphigus and pemphigoid
5. Exploring the functionally relevant molecules and pathways by the use of complex model systems
6. Defining unique pemphigus and pemphigoid variants based on epidemiology
7. Moving towards personalized treatment, selectively targeting specific, autoreactive B cells
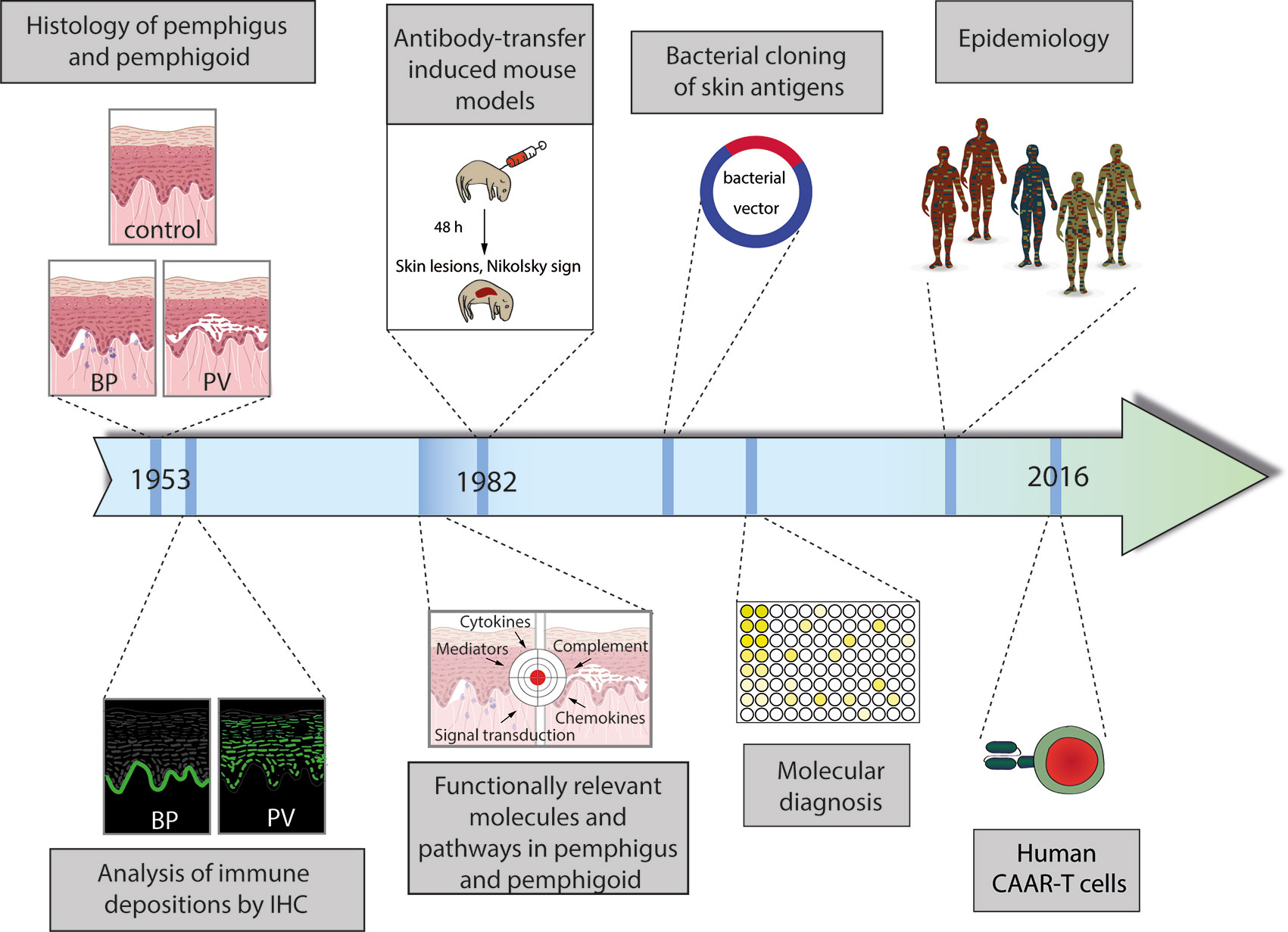
Figure 1 Schematic diagram of the milestones in personalized medicine in pemphigus and pemphigoid. (1) Distinction of pemphigus and pemphigoid based on clinical observation and histology of lesional skin. (2) Identification of distinct binding patterns of autoantibodies in pemphigus (exemplified by pemphigus vulgaris, PV) and pemphigoid (exemplified by bullous pemphigoid, BP). (3) Proof of the autoimmune pathogenesis of pemphigus and pemphigoid by transfer of patient IgG into mice with the subsequent development of pemphigus. (4) Identification and cloning of autoantigens. (5) Establishment of the current, molecular-based diagnostics of pemphigus and pemphigoid. (6) Identification of unique subgroups by careful clinical observation and epidemiology. (6) Individualized treatment by the use of chimeric autoantigen receptor (CAAR) T cells. More details are given in the text.
These milestones in personalized medicine in pemphigus and pemphigoid are presented in detail in the following sections.
Histological Differentiation Between Pemphigus and Pemphigoid
The term “pemphigus” was used as descriptive terms for skin diseases characterized by blisters since Hippocrates (460-370 B.C.) who described different types of fever associated with blistering as “pemphigus fever”. However, the term pemphigus in its present meaning was coined by Dr. Wichman in 1791 when describing a case of pemphigus (in today’s understanding). Thereafter, “pemphigus” was used as a synonym for any vesicular or bullous disease. This led to the emerge of several different types of “pemphigus” (14). In 1860, Dr. Hebra reinstated the concept established by Wichman, stating that “pemphigus” is always a chronic disease. Thus, all pemphigus and pemphigoid diseases were subsumed under the term “pemphigus” (15). Based on the prognosis, two different forms of “pemphigus” were differentiated: Malignant and benign pemphigus. In 1953, Walter Lever published his landmark histological observations where he distinguished between pemphigus, characterized by intraepidermal blistering, and BP, characterized by subepidermal blistering (13) (Figure 2). Taking the clinical presentation into account, he also coined the term mucous membrane pemphigoid (MMP) that is characterized with a histology similar to BP but with blistering at mucosal sites (13). Thus, Walter Levers’ careful clinical and histological observations still hold true and are a good example of how thorough clinical observations allow landmark discoveries.
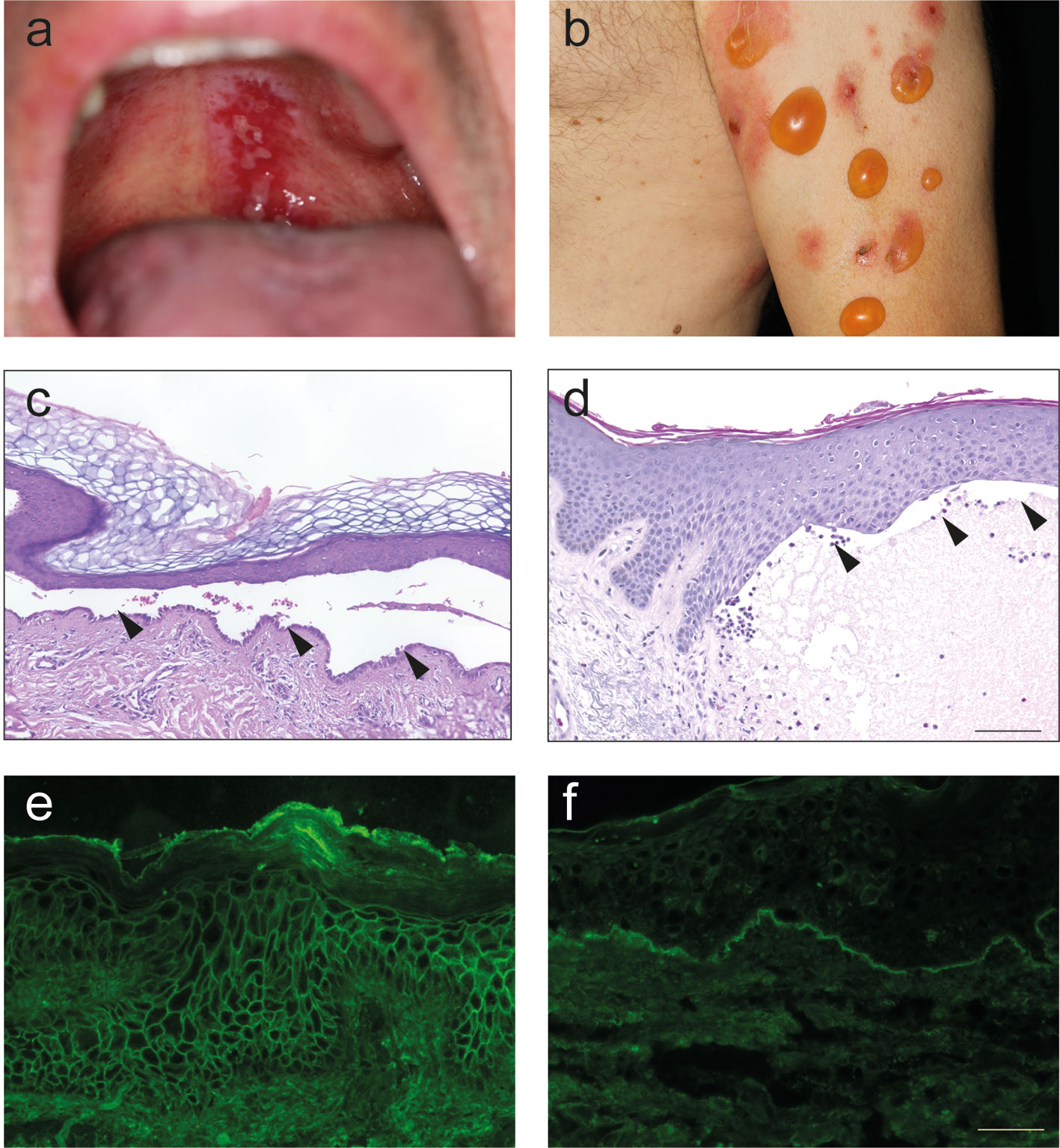
Figure 2 Clinical, histological and immunological features of pemphigus and pemphigoid. (A) Clinical presentation of pemphigus vulgaris with erosions at the upper palate. (B) Clinical presentation of bullous pemphigoid with blistering at erythematous or otherwise apparently healthy skin. (C) Lesional histopathology of a patient with pemphigus vulgaris showing suprabasal splitting with some acantholysis and the typical “row of tombstones” (arrows). (D) In a lesional biopsy of a patient with bullous pemphigoid, subepidermal splitting. In this case, interestingly, the dermal infiltrate is only marginally present. (E) Tissue-bound IgG in a perilesional skin biopsy of a pemphigus vulgaris patient, showing IgG deposits in a honeycomb-like pattern within the epidermis. (F) Tissue-bound IgG in a perilesional skin biopsy of a bullous pemphigoid patient, showing linear IgG deposits along the dermal-epidermal junction.
Identification of Unique Autoantibody Deposits in Pemphigus and Pemphigoid
About two decades after the discovery of Dr. Lever, IgG deposits were noted in patients with pemphigus (16) and pemphigoid (17). In pemphigus, immunoglobulin (Ig) deposits were found to be located at the surface of the keratinocytes. Thus, the term honeycomb-like fluorescence observed within the epidermis has been coined for this particular staining pattern (Figure 2). In pemphigoid diseases, linear Ig/complement (C) deposits along the dermal-epidermal junction are observed. For this, staining pattern the term “linear” Ig/C deposition has been established (Figure 2). More recently, unique patterns of IgG deposits have been observed in pemphigoid diseases. In contrast to all other pemphigoid diseases where the IgG deposits show a “n-serrated” pattern, the pemphigoid disease EBA displays an “u-serrated” pattern of both IgG and IgA (18–20). The identification of these patterns in direct immunofluorescence (IF) microscopy is a learnable skill (21), which is essential for the diagnosis of EBA because up to 60% of cases are seronegative (22). The observation of these unique staining patterns in pemphigus and pemphigoid (as well as in EBA) supported the histopathological observation in 1953 that pemphigus and pemphigoid are distinct diseases.
Defining Pemphigus and Pemphigoid as Autoimmune Diseases
The deposition of IgG in the skin suggested that pemphigus and pemphigoid may be caused by an immune response to self-antigens. Yet, direct proof of the autoimmune pathogenesis of pemphigus and pemphigoid were missing. According to the revised Witebsky’s postulates, such direct proof is the induction of the disease in experimental animals by transfer of patient serum or IgG (23). The autoimmune nature of pemphigus was demonstrated by Grant Anhalt and colleagues in 1982, when they induced skin blistering and erosions, accompanied by the histologic, ultrastructural, and immunological findings found in pemphigus patients, by transfer of pemphigus patient IgG into neonatal Balb/c mice (24). Attempts to reproduce the pemphigoid disease BP by transfer of patient IgG into mice failed because the transferred IgG did not bind to the skin of the mice (25). Several years later, in 1993, when the autoantigens in BP had been defined (BP180 and BP230), and the lack of cross-reactivity of human autoantibodies with murine BP180 had been demonstrated, Zhiou Liu and colleagues were able to induce experimental BP in mice by the transfer of rabbit anti-mouse BP180 IgG (26). Collectively, the defining of pemphigus and (later) pemphigoid as autoimmune diseases, and the demonstration of distinct clinical, histological and immunological features in experimental pemphigus and pemphigoid triggered the search for the autoantigens. Subsequently, the pathogenic relevance of several autoantigens and their corresponding antibodies has been demonstrated in different mouse models for pemphigus foliaceus (PF), paraneopalastic pemphigus (PNP), MMP, and EBA.
Identification of Distinct Autoantigens in Pemphigus and Pemphigoid
Identification of Ig deposits in the skin, as well as the proof of the autoimmune pathogenesis of pemphigus and pemphigoid initiated the search for putative autoantigens, which is still ongoing; i.e., in the case of anti-p200 pemphigoid (27). Using patient autoantibodies, and monoclonal antibodies against type VII collagen (COL7), COL7 was identified as the autoantigen in EBA, as early as in 1988 (28). In 1990, BP180 was cloned human from a keratinocyte library (29). One year later, both, desmoglein (Dsg) 1 and 3 were cloned and identified as the autoantigens in PF and pemphigus vulgaris (PV) (30–32). Subsequently, additional autoantigens in pemphigus and pemphigoid were cloned and/or identified (Table 1).
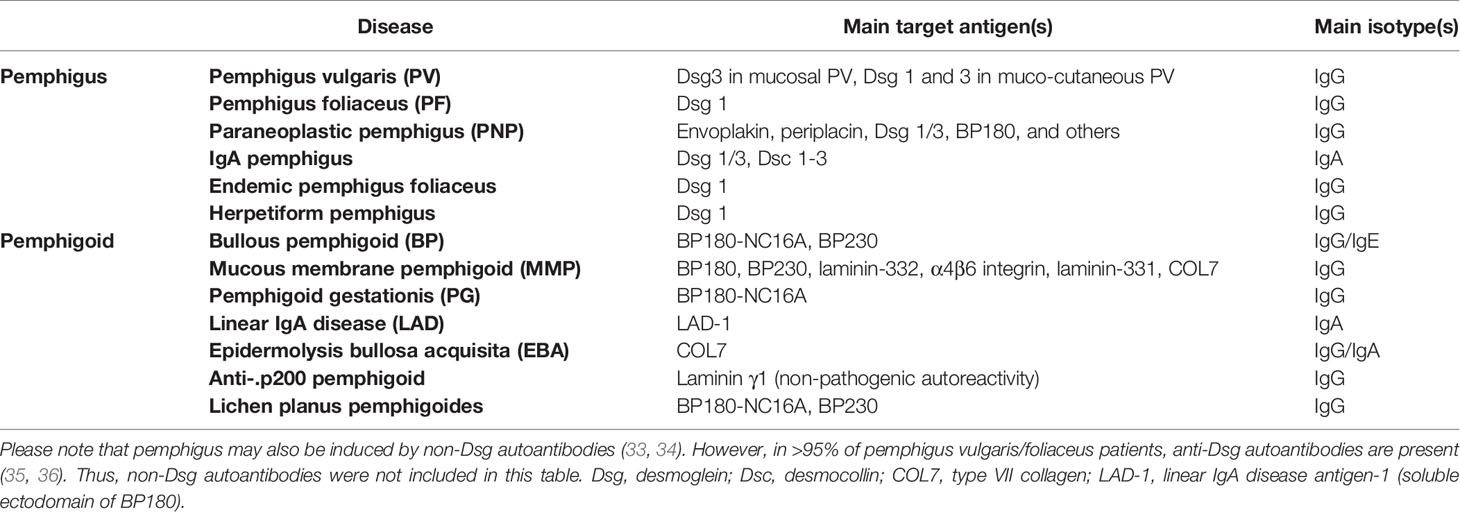
Table 1 Autoantigens in pemphigus and pemphigoid.
Molecular-Based Modern Diagnosis of Pemphigus and Pemphigoid
Definition of distinct autoantigens in pemphigus and pemphigoid diseases enabled the development of the currently used, molecular-based diagnosis of pemphigus and pemphigoid (37). If clinically suspected, the detection of tissue-bound autoantibodies (or C3) in a perilesional skin (or mucosal) biopsy is the gold standard for the diagnosis of pemphigus and pemphigoid. Depending on the location of the Ig or C3 deposits, pemphigus (deposits in the intercellular space, also termed honeycomb pattern), or pemphigoid (linear staining along the dermal-epidermal junction) are differentiated. EBA can be further differentiated based on the serration pattern (19). This pattern analysis is a learnable and important skill because EBA may be seronegative in 60% of the cases (21, 22).
If the diagnosis cannot be established based on the direct IF microscopy, indirect IF microscopy using different organ substrates, most frequently monkey esophagus and human salt-split skin, can further differentiate between the different pemphigus and pemphigoid diseases (37). Other less frequently utilized substrates are rat bladder for PNP and normal oral mucosa for MMP (38). While monkey esophagus is useful in the detection of circulating pemphigus-related autoantibodies, pemphigoid autoantibodies better bind to salt split skin. Of note, if a linear deposit of patient autoantibodies is observed at the roof of the blister of the artificially split skin, BP (or MMP in patients with predominant mucosal involvement) is diagnosed because its autoantigens (BP180 and BP230) are expressed at the blister roof. By contrast, laminin-332, COL7 and p200 are expressed at the blister floor. Hence, binding of patient autoantibodies to the blister floor in indirect IF microscopy on salt split skin requires further differentiation between MMP, EBA, and anti-p200 pemphigoid. The latter, as well as (semi)-quantitative determination of circulating autoantibody concentrations, can be achieved by detection of specific autoantibodies. This can be achieved using the recombinant immunodominant domains of the target antigens, i.e., Dsg1, Dsg3, envoplakin, BP180, BP230, laminin 332, and COL7 in commercial ELISA systems or biochip mosaics. In addition, specialized laboratories have established techniques (mainly Western blotting or immunoprecipitation) for the detection of autoantibodies against p200, selected chains of laminin-332, the ectodomain of BP180, or rare autoantigens. Thus, this molecular-based modern diagnosis of pemphigus and pemphigoid allows (in most cases) to diagnose individual pemphigus and pemphigoid diseases. As their treatment and prognosis greatly differs (2, 3), this allows to select the appropriate treatment for each patient. In addition to their use in diagnosis, longitudinal monitoring allows for early detection of relapses because circulating autoantibody concentrations correlate intraindividually with disease activity (39–41). Molecular characterization of some AIBD, however, is still possible only in highly specialized academic centers. A possible consequence is the delay experienced by patients in receiving the right diagnosis and optimal treatment.
A more recent development in the personalized management of pemphigoid is the identification of biomarkers other than the autoantibodies that allow to predict treatment response and/or relapse (42). In brief, persistence of elevated eosinophil cationic protein (ECP) in patients with BP was associated with relapse (43). In addition to persistence of elevated ECP levels, the presence of anti-COL7 autoantibodies (the autoantibody in EBA) (44), and increased CXCL10 serum levels (45) are also predictors of relapse in patients with BP. Relative to non-autoreactive B cells, autoreactive B cells of patients with PV showed overexpression of genes encoding for IL-1β, IL-23p19, and IL-12p35 pro-inflammatory cytokines and the IRF5 transcription factor. Relative to patients with active pemphigus, those experiencing complete remission following rituximab displayed under-expression of IL-1β and the CD27 memory marker genes (46).
Efforts were extensively made to establish a personalized approach to optimize management of patients with pemphigus. That is, to predict patients predisposed to early relapses under rituximab who may benefit from maintenance rituximab infusion at month 6. Increased severity score at baseline and increased levels of anti-Dsg1 and anti-Dsg3 three months following the first infusion were found to independently project post-rituximab early relapse, thus warranting to consider a maintenance dose of rituximab after 6 months (47).
Functionally Relevant Molecules and Pathways in Pemphigus and Pemphigoid
Employing keratinocyte cultures, ex vivo skin models, and the above-mentioned mouse models (48, 49), several disease pathways have been identified that provide the base for valuable novel therapeutic approaches. Dsgs are transmembrane desmosomal cadherin-like glycoproteins which function to maintain tissue integrity and facilitate cell-cell adhesion. IgG autoantibodies targeting Dsg3 and Dsg1 play the main etiopathogenetic role in the development of PV and PF, respectively. In pemphigus, monovalent fragments of anti-Dsg antibodies that lack the Fc portion are sufficient to cause acantholysis in vitro and in vivo (50). The exact sequence of events in anti-Dsg antibody-mediated acantholysis has not yet been fully understood. Three major events following the binding of anti-Dsg IgG have been described: (i) direct interference with Dsg transinteraction, a phenomenon termed steric hindrance, (ii) remodeling of Dsg expression on the cell surface leading to internalization and depletion of Dsg from the cell membrane, and (iii) signaling events that impair cytoskeletal architecture (3, 4). These mechanisms do not apply equally for Dsg1 IgG- and Dsg3 IgG-binding. Upon targeting of Dsg1, Ca2+ influx is induced and the ERK pathway is activated. In contrast, after binding of Dsg3-specific IgG, signaling via p38MAPK occurs in the epidermis but not in mucosal tissues, and SRC family of protein tyrosine kinases and EGFR pathways are activated (51, 52). Current data also strongly suggest that, in addition to Dsg1/3 autoantibodies, non-Dsg antibodies, as well as soluble Fas ligand contribute to the pemphigus phenotype (53–56).
In contrast to pemphigus disorders, in pemphigoid diseases, FcγR-mediated effects are pivotal for blister formation, and over the last decade several disease pathways and key molecules with functional relevance in these diseases have been described including several signaling molecules, leukotriene B4 (LTB4), and IL-17 (57–60). Furthermore, complement activation at the dermal-epidermal junction is generally accepted to be a cornerstone in recruiting neutrophils, eosinophils, and macrophages to this site (61). Of note, subtle differences in the impact of complement activation emerged between different pemphigoid diseases as well as between clinical variants of BP. In contrast, acantholysis in pemphigus appears to develop independently of complement activation although staining of C3 in the epithelium/epidermis is a diagnostic hall mark (3). Exploring these differences may uncover patient and diseases subgroups that can benefit from therapeutic interventions in targeting complement components. About 80% of BP patients reveal C3c deposition along the dermal-epidermal junction in perilesional biopsies. In patients with C3c deposition, anti-BP180 NC16A IgG serum levels were significantly higher and patients without blisters had significantly less C3c deposits along the dermal-epidermal junction (62). While no relation between the extent of skin lesions and C3c staining in the skin of patients was found, the complement activation capacity of autoantibodies in the ex vivo complement fixation assay correlated with diseases activity as measured by the Bullous Pemphigoid Disease Area Index (BPDAI) (63). Further support for the relevance of complement activation in BP comes from the finding of elevated levels of C3a in the serum of BP patients and the positive correlation of serum C3a levels with both anti-BP180 NC16A IgG and soluble CD46, a crucial complement regulatory protein in the complement activation (64). However, plasma concentrations of C3a, C4a, as well as C5a are not different between BP patients and age/sex matched controls. Furthermore, the plasma levels of these three complement components remain constant when evaluated in flares and in remission of BP (65). In the neonatal BP mouse model, where blisters typically develop 24–48 h after injection of rabbit IgG against the NC15A domain of BP180, the blistering phenotype was completely dependent on complement activation at the dermal-epidermal junction (66, 67). The same complement dependency was observed in a humanized mouse model of BP in which the human NC16A domain replaced the homologous murine NC15A region (68). In contrast, in another humanized mouse model of BP, in which the entire BP180 molecule had been replaced by the human protein, injection of polyclonal F(ab’)2 anti-NC16A IgG or non-complement-activating anti-NC16A IgG4 led to blister formation (69, 70). In line, Dainichi et al. reported on two BP patients without C3 deposits in the skin and IgG4 autoantibodies as main subclass that were unable to elicit complement activation ex vivo (71). Additional observations in this model with C3-deficient animals that were susceptible to the pathogenic effect of anti-BP180 IgG and recently, in a BP model in adult mice where transfer of anti-BP180 NC15A IgG in C5aR1-deficient mice led to a reduction of skin lesions by 50% (70, 72, 73) pointed to complement-independent mechanisms in BP pathophysiology. Interestingly, similar to the BP neonatal mouse model, in adult mouse models of EBA and anti-laminin 332 MMP, complement activation appeared as prerequisite for a blistering phenotype (74–76). Unravelling the complex scenario of complement activation in pemphigoid disorders will certainly help to identify patient subpopulations and to tailor more specific and safe treatments for these diseases. The dose-dependent inhibition of the BP IgG-induced C3 deposition at the dermal-epidermal junction in the in vitro complement fixation on cryosections of human skin by (i) the anti-C1s antibody TNT003, (ii) the low-molecular-weight heparin tinzaparin sodium, and (iii) the dual C5/LTB4 antagonist coversin, all of the which disturbing the normal activity of complement pathway (65, 77, 78) directs us to further explore complement inhibition as valuable therapeutic target in BP.
Epidemiology Defines Unique Variations Within Several Pemphigus and Pemphigoid Diseases
As its name indicates, PV is the most prevalent subtype of pemphigus, comprising up to 70% of all cases (79). PV is typified by a variable geographic and ethnic distribution, with annual incidence rates ranging between 0.8 and 16.1 cases per million population in different regions (80). Congruently, a predisposition for developing pemphigus was reported in some ethnic groups, namely, Ashkenazi Jews and individuals of Mediterranean origin (80). In a recent population-based study, the incidence of PV was 3.6-fold increased among Jews as compared to Arabs in Israel (81). In a retrospective study conducted in Connecticut, the US, the annual incidence of PV was almost eight-fold higher among people of Jewish ancestry than among those belonging to other ethnic groups (82). These epidemiological observations have been strongly substantiated by genetic studies disclosing an association of several HLA-class II genes, HLA-DRB1*04, and HLA-A*10, with the occurrence of PV among Ashkenazi Jews (83–85). Subsequently, a polymorphic variant in ST18 gene was associated with PV in Jewish and Egyptian but not in German patients (86). Hence, despite an underlying genetic pre-disposition in PV, the disease may manifest also in the absence of certain genetic predisposing factors. Collectively, this points towards a polygenic genetic risk to develop PV, as well as points towards the environment as a potential driver of disease pathogenesis, as reported for other inflammatory diseases (87).
Sporadic PF is an uncommon disease constituting 20%–30% of all pemphigus cases. Its estimated annual incidence in Europe and the United States is less than one case per million population (80). HLA-DRB1*04 was associated with increased risk for sporadic PF in Brazilian, Italian, French, and Dutch populations (88, 89). An association with HLA-DRB1*0101 was found among patients with sporadic PF originating from Mexico (90). Nevertheless, no ethnic predilection in the occurrence of PF was noted in Israel, as the adjusted incidence rate of the disease was comparable between Jews and Arabs (81). Apart from sporadic cases, endemic subtypes of PF have been described in Brazil (folgo selvagem), Colombia, and Tunisia (80). Although patients with endemic PF are clinically, histologically, and immunologically indistinguishable from those with sporadic PF (80, 91), the former is characterized by a patterned geographic distribution, familial predisposition, and younger age of presentation (92, 93).
BP is the most common subepidermal AIBDs worldwide. BP characteristically affects the elderly and is seen mainly among patients older than 75 years. While the general annual incidence of BP has been reported to range between 2.4 and 23.0 cases per million population, it rises exponentially to 312 cases per million population in individuals older than 80 years (94). Several lines of evidence accumulated to suggest a notable increasing incidence of BP by 1.9 to 4.3 folds over the past two decades (95). Several putative interpretations were postulated to account for this observation, the most plausible of which is the growing exposure to certain medications implicated with the induction of BP (95). Dipeptidyl-peptidase IV inhibitors (DPP4i), also known as gliptins, a recently introduced anti-diabetic class of oral medications, emerged as a potential trigger of BP (96–98). It is yet to be decisively determined whether patients with DPP4i-associated BP follow a distinct clinical and immunological profile. While European studies did not depict distinct features for these patients distinguishing them from typical BP, DPP4i-associated BP patients originating from Japan were more likely to present with non-inflammatory BP and to target the immunodominant domain of BP180 antigen (NC16A) less frequently (99–101). Thus, careful clinical observation has led to the identification of a unique clinical presentation of BP. Given that the initial observations of unique immunological and genetic features of DPP4i-asscociated BP can be confirmed, this would allow to treat the patients according to the underlying, disease-promoting pathways. In the case of DPP4i-associated BP, this is relatively easy because BP usually clears after stopping DPP4i treatment.
CAAR T Cells as a Potential and Personalized Cure of Pemphigus and Pemphigoid
The most recent advance towards a personalized treatment approach is the development of chimeric autoantigen receptor (CAAR) T cells for the treatment of pemphigus (102). Based on the breakthrough discovery of chimeric antigen receptor (CAR) T cells for the treatment of hematologic malignancies (103), Aimee Payne and colleagues developed a recombinant T cell receptor by fusing the autoantigen in PV (Dsg3) to CD137-CD3ζ. These Dsg3 CAAR T cells exhibited specific cytotoxicity against B/plasma cells expressing the B cell receptor specific for Dsg3. Next, experimental pemphigus was induced in mice by transfer of Dsg3-hybridoma cell lines. When mice were additionally injected with Dsg3 CAAR T cells, they were protected from induction of experimental pemphigus (102). Subsequent work from the same group expanded the work to additional autoantigens in pemphigus, namely, Dsg1 (104). Currently, a phase I clinical trial is conducted on autologous Dsg3 CAAR T cells in mucosal PV (https://cabalettabio.com/clinical-trials/, accessed May 31, 2020). By selectively targeting autoreactive B cells, using the CARR T cell technology, a highly personalized treatment approach for PV is currently under development (105). Overall, given successful completion of this (and subsequent) clinical trials, a new era of managing B cell-driven autoimmune diseases (106) will emerge.
Future Perspectives
The emergence of treatments selectively targeting autoreactive B cells, e.g., by CAAR T cells is expected to significantly change the treatment of pemphigus. It is highly intriguing to estimate whether CAAR T cells the same potential additionally in pemphigoid diseases. Alternatively, in another approach, immunization of PV patients with Dsg3-coated nanoparticles may specifically suppress autoimmunity against Dsg3 and is currently performed in a phase I clinical trial in PV with Dsg3. Targeting IL-17 and eotaxin is currently being assessed as a potential therapeutic approach in BP (107). The success of these treatments will, however, depend on an expansion of the molecular diagnosis that will allow to precisely define the autoimmune response in individual patients. In parallel, molecular diagnosis is also expected to help define distinct (sub)-groups of pemphigus and pemphigoid diseases that most likely will be more and more based on molecular signatures. In the long term, one may envision that curative and safe treatments for pemphigus and pemphigoid will be available.
Author Contributions
KBi, KK, SE, KBo, ES, and RL wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Excellence Cluster “Precision Medicine in Chronic Inflammation” (EXC 2167) from the Deutsche Forschungsgemeinschaft.
Conflict of Interest
During the last 3 years, ES has received research funding from Novartis, UCB, Incyte, Biotest, Euroimmun, Dompe, Admirx, Synthon, TxCell, ArgenX, and Fresenius Medical Care and fees for consulting or speaking and/or travel expenses from Novartis, UCB, Biotest, TxCell, ArgenX, Roche, Genentech, Imevax, Amryth, Thermo Fisher, AstraZeneca, True North Therapeutics, Bristol-Myers Squibb, and Fresenius Medical Care. RL has received research funding from Almirall, True North Therapeutics, UCB Pharma, ArgenX, TxCell, Topadur, Incyte, and Admirx and fees for consulting or speaking from ArgenX, Immunogenetics, Novartis, and Lilly.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Cianfarani F, Zambruno G, Castiglia D, Odorisio T. Pathomechanisms of Altered Wound Healing in Recessive Dystrophic Epidermolysis Bullosa. Am J Pathol (2017) 187:1445–53. doi: 10.1016/j.ajpath.2017.03.003
2. Schmidt E, Zillikens D. Pemphigoid diseases. Lancet (2013) 381:320–32. doi: 10.1016/S0140-6736(12)61140-4
3. Schmidt E, Kasperkiewicz M, Joly J. Pemphigus. Lancet (2019) 394:882–94. doi: 10.1016/S0140-6736(19)31778-7
4. Kasperkiewicz M, Ellebrecht CT, Takahashi H, Yamagami J, Zillikens D, Payne AS, et al. Pemphigus. Nat Rev Dis Primers (2017) 3:17026. doi: 10.1038/nrdp.2017.26
5. Lamberts A, Yale M, Grando SA, Horváth B, Zillikens D, Jonkman MF. Unmet Needs in Pemphigoid Diseases: An International Survey Amongst Patients, Clinicians and Researchers. Acta Derm Venereol (2019) 99:224–5. doi: 10.2340/00015555-3052
6. Ständer S, Färber B, Radeke S, Schmidt E, Zillikens D, Ludwig RJ. Assessment of the healthcare costs for pemphigus and bullous pemphigoid patients in an academic center in Germany.[letter]. Br J Dermatol (2020) 182:1296–7. doi: 10.1111/bjd.18731
7. Hammers CM, Stanley JR. Recent Advances in Understanding Pemphigus and Bullous Pemphigoid. J Invest Dermatol (2020) 140:733–41. doi: 10.1016/j.jid.2019.11.005
8. Egami S, Yamagami J, Amagai M. Autoimmune bullous skin diseases, pemphigus and pemphigoid. J Allergy Clin Immunol (2020) 145:1031–47. doi: 10.1016/j.jaci.2020.02.013
9. Joly P, Maho-Vaillant M, Prost-Squarcioni C, Hebert V, Houivet E, Calbo S, et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet (2017) 389(10083):2031–40. doi: 10.1016/S0140-6736
10. Joly P, Roujeau JC, Benichou J, Picard C, Dreno B, Delaporte E, et al. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. N Engl J Med (2002) 346:321–7. doi: 10.1056/NEJMoa011592
11. Schmidt E, Spindler V, Eming R, Amagai M, Antonicelli F, Baines JF, et al. Meeting Report of the Pathogenesis of Pemphigus and Pemphigoid Meeting in Munich, September 2016. J Invest Dermatol (2017) 137:1199–203. doi: 10.1016/j.jid.2017.01.028
12. Kirtschig G, Middleton P, Bennett C, Murrell DF, Wojnarowska F, Khumalo NP. Interventions for bullous pemphigoid. Cochrane Database Syst Rev (2010) 2010(10):CD002292. doi: 10.1002/14651858.CD002292.pub3
15. Hebra F. Acute Exantheme und Hautkrankheiten. Handbuch der speciellen Pathologie und Therapie, dritter Band. Verlag Ferdinand Enke (1860) p. 572–601.
16. Meurer M, Millns JL, Rogers RS, Jordon RE. Oral pemphigus vulgaris. A report of ten cases. Arch Dermatol (1977) 113:1520–4. doi: 10.1001/archderm.113.11.1520
17. Provost TT, Tomasi TB. Immunopathology of bullous pemphigoid. Basement membrane deposition of IgE, alternate pathway components and fibrin. Clin Exp Immunol (1974) 18:193–200.
18. Vodegel RM, Jonkman MF, Pas HH, de Jong MC. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol (2004) 151:112–8. doi: 10.1111/j.1365-2133.2004.06006.x
19. Meijer JM, Atefi I, Diercks GFH, Vorobyev A, Zuiderveen J, Meijer HJ, et al. Serration pattern analysis for differentiating epidermolysis bullosa acquisita from other pemphigoid diseases. J Am Acad Dermatol (2018) 78:754–759.e6. doi: 10.1016/j.jaad.2017.11.029
20. Senatore S, Maglie R, Salemme A, Caproni M, Di Zenzo G, Antiga E. Serration pattern analysis as a tool for the diagnosis of immunoglobulin A-mediated epidermolysis bullosa acquisita.[letter]. J Dermatol (2020) 47(5):e198–9. doi: 10.1111/1346-8138.15161
21. Terra JB, Meijer JM, Jonkman MF, Diercks GF. The n- versus u-serration is a learnable criterion to differentiate pemphigoid from epidermolysis bullosa acquisita in direct immunofluorescence serration pattern analysis. Br J Dermatol (2013) 169:100–5. doi: 10.1111/bjd.12308
22. Buijsrogge JJ, Diercks GF, Pas HH, Jonkman MF. The many faces of epidermolysis bullosa acquisita after serration pattern analysis by direct immunofluorescence microscopy. Br J Dermatol (2011) 165:92–8. doi: 10.1111/j.1365-2133.2011.10346.x
23. Rose NR, Bona C. Defining criteria for autoimmune diseases (Witebsky’s postulates revisited). Immunol Today (1993) 14:426–30. doi: 10.1016/0167-5699(93)90244-F
24. Anhalt GJ, Labib RS, Voorhees JJ, Beals TF, Diaz LA. Induction of pemphigus in neonatal mice by passive transfer of IgG from patients with the disease. N Engl J Med (1982) 306:1189–96. doi: 10.1056/NEJM198205203062001
25. Anhalt GJ, Diaz LA. Animal models for bullous pemphigoid. Clin Dermatol (1987) 5:117–25. doi: 10.1016/0738-081X(87)90056-3
26. Liu Z, Diaz LA, Troy JL, Taylor AF, Emery DJ, Fairley JA, et al. A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest (1993) 92:2480–8. doi: 10.1172/JCI116856
27. Lau I, Goletz S, Holtsche MM, Zillikens D, Fechner K, Schmidt E. Anti-p200 pemphigoid is the most common pemphigoid disease with serum antibodies against the dermal side by indirect immunofluorescence microscopy on human salt-split skin.[letter]. J Am Acad Dermatol (2019) 81(5):1195–7. doi: 10.1016/j.jaad.2019.03.077
28. Woodley DT, Burgeson RE, Lunstrum G, Bruckner-Tuderman L, Reese MJ, Briggaman RA. Epidermolysis bullosa acquisita antigen is the globular carboxyl terminus of type VII procollagen. J Clin Invest (1988) 81:683–7. doi: 10.1172/JCI113373
29. Diaz LA, Ratrie H, Saunders WS, Futamura S, Squiquera HL, Anhalt GJ, et al. Isolation of a human epidermal cDNA corresponding to the 180-kD autoantigen recognized by bullous pemphigoid and herpes gestationis sera. Immunolocalization of this protein to the hemidesmosome. J Clin Invest (1990) 86:1088–94. doi: 10.1172/JCI114812
30. Nilles LA, Parry DA, Powers EE, Angst BD, Wagner RM, Green KJ. Structural analysis and expression of human desmoglein: a cadherin-like component of the desmosome. J Cell Sci (1991) 99:809–21.
31. Amagai M, Klaus-Kovtun V, Stanley JR. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell (1991) 67:869–77. doi: 10.1016/0092-8674(91)90360-B
32. Koulu L, Kusumi A, Steinberg MS, Klaus-Kovtun V, Stanley JR. Human autoantibodies against a desmosomal core protein in pemphigus foliaceus. J Exp Med (1984) 160:1509–18. doi: 10.1084/jem.160.5.1509
33. Kalantari-Dehaghi M, Anhalt GJ, Camilleri MJ, Chernyavsky AI, Chun S, Felgner PL, et al. Pemphigus vulgaris autoantibody profiling by proteomic technique. PLoS One (2013) 8:e57587. doi: 10.1371/journal.pone.0057587
34. Sinha AA, Sajda T. The evolving story of autoantibodies in Pemphigus vulgaris: development of the “super compensation hypothesis”. Front Med (2018) 5:218. doi: 10.3389/fmed.2018.00218
35. Mindorf S, Dettmann IM, Krüger S, Fuhrmann T, Rentzsch K, Karl I, et al. International Autoimmune Bullous Diseases Study Group. Routine detection of serum antidesmocollin autoantibodies is only useful in patients with atypical pemphigus. Exp Dermatol (2017) 26(12):1267–70. doi: 10.1111/exd.13409
36. van Beek N, Krüger S, Fuhrmann T, Lemcke S, Goletz S, Probst C, et al. Multicenter prospective study on multivariant diagnostics of autoimmune bullous dermatoses using the BIOCHIP technology. J Am Acad Dermatol (2020) 83(5):1315–22. doi: 10.1016/j.jaad.2020.01.049
37. Witte M, Zillikens D, Schmidt E. Diagnosis of Autoimmune Blistering Diseases. Front Med (2018) 5:296. doi: 10.3389/fmed.2018.00296
38. Maglie R, Borgi A, Caproni M, Antiga E. Indirect immunofluorescence in mucous membrane pemphigoid: which substrate should be used[letter]. Br J Dermatol (2019) 180(5):1266–7. doi: 10.1111/bjd.17694
39. Sitaru C, Dahnrich C, Probst C, Komorowski L, Blocker I, Schmidt E, et al. Enzyme-linked immunosorbent assay using multimers of the 16th non-collagenous domain of the BP180 antigen for sensitive and specific detection of pemphigoid autoantibodies. Exp Dermatol (2007) 16:770–7. doi: 10.1111/j.1600-0625.2007.00592.x
40. Schmidt E, Dahnrich C, Rosemann A, Probst C, Komorowski L, Saschenbrecker S, et al. Novel ELISA systems for antibodies to desmoglein 1 and 3: correlation of disease activity with serum autoantibody levels in individual pemphigus patients. Exp Dermatol (2010) 19:458–63. doi: 10.1111/j.1600-0625.2010.01069.x
41. Di Zenzo G, Thoma-Uszynski S, Fontao L, Calabresi V, Hofmann SC, Hellmark T, et al. Multicenter prospective study of the humoral autoimmune response in bullous pemphigoid. Clin Immunol (2008) 128:415–26. doi: 10.1016/j.clim.2008.04.012
42. Giusti D, Le Jan S, Gatouillat G, Bernard P, Pham BN, Antonicelli F. Biomarkers related to bullous pemphigoid activity and outcome. Exp Dermatol (2017) 26:1240–7. doi: 10.1111/exd.13459
43. Giusti D, Gatouillat G, Le Jan S, Plée J, Bernard P, Antonicelli F, et al. Eosinophil Cationic Protein (ECP), a predictive marker of bullous pemphigoid severity and outcome. Sci Rep (2017) 7:4833. doi: 10.1038/s41598-017-04687-5
44. Giusti D, Gatouillat G, Le Jan S, Plée J, Bernard P, Antonicelli F, et al. Anti-Type VII Collagen Antibodies Are Identified in a Subpopulation of Bullous Pemphigoid Patients With Relapse. Front Immunol (2018) 9:570. doi: 10.3389/fimmu.2018.00570
45. Riani M, Le Jan S, Plée J, Durlach A, Le Naour R, Haegeman G, et al. Bullous pemphigoid outcome is associated with CXCL10-induced matrix metalloproteinase 9 secretion from monocytes and neutrophils but not lymphocytes. J Allergy Clin Immunol (2016) 139(3):863–72. doi: 10.1016/j.jaci.2016.08.012
46. Hébert V, Petit M, Maho-Vaillant M, Golinski ML, Riou G, Derambure C, et al. Modifications of the Transcriptomic Profile of Autoreactive B Cells From Pemphigus Patients After Treatment With Rituximab or a Standard Corticosteroid Regimen. Front Immunol (2019) 10:1794. doi: 10.3389/fimmu.2019.01794
47. Mignard C, Maho-Vaillant M, Golinski ML, Balayé P, Prost-Squarcioni C, Houivet E, et al. Factors Associated With Short-term Relapse in Patients With Pemphigus Who Receive Rituximab as First-line Therapy: A Post Hoc Analysis of a Randomized Clinical Trial. JAMA Dermatol (2020) 156:545–52.
48. Burmester IAK, Emtenani S, Johns JG, Ludwig RJ, Hammers CM, Hundt JE. Translational use of a standardized full human skin organ culture model in autoimmune blistering diseases. Curr Protoc Pharmacol (2019) 85(1):e56. doi: 10.1002/cpph.56
49. Kasprick A, Bieber K, Ludwig RJ. Drug discovery for pemphigoid diseases. Curr Protoc Pharmacol (2019) 84:e55. doi: 10.1002/cpph.55
50. Payne AS, Ishii K, Kacir S, Lin C, Li H, Hanakawa Y, et al. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest (2005) 115:888–99. doi: 10.1172/JCI24185
51. Egu DT, Sigmund AM, Schmidt E, Spindler V, Walter E, Waschke J. A new ex vivo human oral mucosa model reveals that p38MAPK inhibition is not effective to prevent autoantibody-induced mucosal blistering in pemphigus. Br J Dermatol (2020) 182:987–94. doi: 10.1111/bjd.18237
52. Walter E, Vielmuth F, Wanuske MT, Seifert M, Pollmann R, Eming R, et al. Role of Dsg1- and Dsg3-Mediated Signaling in Pemphigus Autoantibody-Induced Loss of Keratinocyte Cohesion. Front Immunol (2019) 10:1128. doi: 10.3389/fimmu.2019.01128
53. Lotti R, Shu E, Petrachi T, Marconi A, Palazzo E, Quadri M, et al. Soluble Fas Ligand Is Essential for Blister Formation in Pemphigus. Front Immunol (2018) 9:370. doi: 10.3389/fimmu.2018.00370
54. Sajda T, Sinha AA. Autoantibody Signaling in Pemphigus Vulgaris: Development of an Integrated Model. Front Immunol (2018) 9:692. doi: 10.3389/fimmu.2018.00692
55. Chernyavsky A, Amber KT, Agnoletti AF, Wang C, Grando SA. Synergy among non-desmoglein antibodies contributes to the immunopathology of desmoglein antibody-negative pemphigus vulgaris. J Biol Chem (2019) 294:4520–8. doi: 10.1074/jbc.RA118.006743
56. Spindler V, Waschke J. Pemphigus-A Disease of Desmosome Dysfunction Caused by Multiple Mechanisms. Front Immunol (2018) 9:136. doi: 10.3389/fimmu.2018.00136
57. Samavedam UK, Mitschker N, Kasprick A, Bieber K, Schmidt E, Laskay T, et al. Whole-Genome Expression Profiling in Skin Reveals SYK As a Key Regulator of Inflammation in Experimental Epidermolysis Bullosa Acquisita. Front Immunol (2018) 9:249. doi: 10.3389/fimmu.2018.00249
58. Koga H, Kasprick A, Lopez R, Auli M, Pont M, Godessart N, et al. Therapeutic effect of a novel PI3Kδ inhibitor in experimental epidermolysis bullosa acquisita. Front Immunol (2018) 9:1558. doi: 10.3389/fimmu.2018.01558
59. Chakievska L, Holtsche MM, Künstner A, Goletz S, Petersen BS, Thaci D, et al. IL-17A is functionally relevant and a potential therapeutic target in bullous pemphigoid. J Autoimmun (2019) 96:104–12. doi: 10.1016/j.jaut.2018.09.003
60. Sezin T, Krajewski M, Wutkowski A, Mousavi S, Chakievska L, Bieber K, et al. The Leukotriene B4 and its Receptor BLT1 Act as Critical Drivers of Neutrophil Recruitment in Murine Bullous Pemphigoid-Like Epidermolysis Bullosa Acquisita. J Invest Dermatol (2017) 137:1104–13. doi: 10.1016/j.jid.2016.12.021
61. Edwards G, Diercks GFH, Seelen MAJ, Horvath B, van Doorn MBA, Damman J. Complement Activation in Autoimmune Bullous Dermatoses: A Comprehensive Review. Front Immunol (2019) 10:1477. doi: 10.3389/fimmu.2019.01477
62. Romeijn TR, Jonkman MF, Knoppers C, Pas HH, Diercks GF. Complement in bullous pemphigoid: results from a large observational study.[letter]. Br J Dermatol (2017) 176(2):517–9. doi: 10.1111/bjd.14822
63. Chiorean RM, Baican A, Mustafa MB, Lischka A, Leucuta DC, Feldrihan V, et al. Complement-Activating Capacity of Autoantibodies Correlates With Disease Activity in Bullous Pemphigoid Patients. Front Immunol (2018) 9:2687. doi: 10.3389/fimmu.2018.02687
64. Qiao P, Dang E, Cao T, Fang H, Zhang J, Qiao H, et al. Dysregulation of mCD46 and sCD46 contribute to the pathogenesis of bullous pemphigoid. Sci Rep (2017) 7:145. doi: 10.1038/s41598-017-00235-3
65. Kasprick A, Holtsche MM, Rose EL, Hussain S, Schmidt E, Petersen F, et al. The anti-C1s antibody TNT003 prevents complement activation in the skin induced by bullous pemphigoid autoantibodies. J Invest Derm (2018) 138:458–61. doi: 10.1016/j.jid.2017.08.030
66. Liu Z, Giudice GJ, Swartz SJ, Fairley JA, Till GO, Troy JL, et al. The role of complement in experimental bullous pemphigoid. J Clin Invest (1995) 95:1539–44. doi: 10.1172/JCI117826
67. Nelson KC, Zhao M, Schroeder PR, Li N, Wetsel RA, Diaz LA, et al. Role of different pathways of the complement cascade in experimental bullous pemphigoid. J Clin Invest (2006) 116:2892–900. doi: 10.1172/JCI17891
68. Liu Z, Sui W, Zhao M, Li Z, Li N, Thresher R, et al. Subepidermal blistering induced by human autoantibodies to BP180 requires innate immune players in a humanized bullous pemphigoid mouse model. J Autoimmun (2008) 31:331–8. doi: 10.1016/j.jaut.2008.08.009
69. Natsuga K, Nishie W, Shinkuma S, Ujiie H, Nishimura M, Sawamura D, et al. Antibodies to Pathogenic Epitopes on Type XVII Collagen Cause Skin Fragility in a Complement-Dependent and -Independent Manner. J Immunol (2012) 188(11):5792-9. doi: 10.4049/jimmunol.1003402
70. Ujiie H, Sasaoka T, Izumi K, Nishie W, Shinkuma S, Natsuga K, et al. Bullous Pemphigoid Autoantibodies Directly Induce Blister Formation without Complement Activation. J Immunol (2014) 193:4415–28. doi: 10.4049/jimmunol.1400095
71. Dainichi T, Nishie W, Yamagami Y, Sonobe H, Ujiie H, Kaku Y, et al. Bullous pemphigoid suggestive of complement-independent blister formation with anti-BP180 IgG4 autoantibodies. Br J Dermatol (2016) 175:187–90. doi: 10.1111/bjd.14411
72. Karsten CM, Beckmann T, Holtsche MM, Tillmann J, Tofern S, Schulze FS, et al. Tissue Destruction in Bullous Pemphigoid Can Be Complement Independent and May Be Mitigated by C5aR2. Front Immunol (2018) 9:488. doi: 10.3389/fimmu.2018.00488
73. Schulze FS, Beckmann T, Nimmerjahn F, Ishiko A, Collin M, Kohl J, et al. Fc gamma receptors III and IV mediate tissue destruction in a novel adult mouse model of bullous pemphigoid. Am J Pathol (2014) 184:2185–96. doi: 10.1016/j.ajpath.2014.05.007
74. Heppe EN, Tofern S, Schulze FS, Ishiko A, Shimizu A, Sina C, et al. Experimental laminin 332 mucous membrane pemphigoid critically involves C5aR1 and reflects clinical and immunopathological characteristics of the human disease. J Invest Dermatol (2017) 137:1709–18. doi: 10.1016/j.jid.2017.03.037
75. Karsten CM, Pandey MK, Figge J, Kilchenstein R, Taylor PR, Rosas M, et al. Anti-inflammatory activity of IgG1 mediated by Fc galactosylation and association of FcgammaRIIB and dectin-1. Nat Med (2012) 18:1401–6. doi: 10.1038/nm.2862
76. Iwata H, Witte M, Samavedam UK, Gupta Y, Shimizu A, Ishiko A, et al. Radiosensitive Hematopoietic Cells Determine the Extent of Skin Inflammation in Experimental Epidermolysis Bullosa Acquisita. J Immunol (2015) 195:1945–54. doi: 10.4049/jimmunol.1501003
77. Gutjahr A, Heck F, Emtenani S, Hammers AK, Hundt JE, Muck P, et al. Bullous pemphigoid autoantibody-mediated complement fixation is abolished by the low-molecular-weight heparin tinzaparin sodium.[letter]. Br J Dermatol (2019) 181(3):593–4. doi: 10.1111/bjd.18156
78. Sezin T, Murthy S, Attah C, Seutter M, Holtsche MM, Hammers CM, et al. Dual inhibition of complement factor 5 and leukotriene B4 synergistically suppresses murine pemphigoid disease. JCI Insight (2019) 4(15):e128239. doi: 10.1172/jci.insight.128239
79. Joly P, Litrowski N. Pemphigus group (vulgaris, vegetans, foliaceus, herpetiformis, brasiliensis). Clin Dermatol (2011) 29:432–6. doi: 10.1016/j.clindermatol.2011.01.013
80. Kridin K. Pemphigus group: overview, epidemiology, mortality, and comorbidities. Immunol Res (2018) 66:255–70. doi: 10.1007/s12026-018-8986-7
81. Kridin K, Zelber-Sagi S, Bergman R. Pemphigus Vulgaris and Pemphigus Foliaceus: Differences in Epidemiology and Mortality. Acta Derm Venereol (2017) 97:1095–9. doi: 10.2340/00015555-2706
82. Simon DG, Krutchkoff D, Kaslow RA, Zarbo R. Pemphigus in Hartford County, Connecticut, from 1972 to 1977. Arch Dermatol (1980) 116:1035–7. doi: 10.1001/archderm.116.9.1035
83. Ahmed AR, Wagner R, Khatri K, Notani G, Awdeh Z, Alper CA, et al. Major histocompatibility complex haplotypes and class II genes in non-Jewish patients with pemphigus vulgaris. Proc Natl Acad Sci U S A (1991) 88:5056–60. doi: 10.1073/pnas.88.11.5056
84. Krain LS, Terasaki PI, Newcomer VD, Mickey MR. Increased frequency of HL-A10 in pemphigus vulgaris. Arch Dermatol (1973) 108:803–5. doi: 10.1001/archderm.108.6.803
85. Mobini N, Yunis EJ, Alper CA, Yunis JJ, Delgado JC, Yunis DE, et al. Identical MHC markers in non-Jewish Iranian and Ashkenazi Jewish patients with pemphigus vulgaris: possible common central Asian ancestral origin. Hum Immunol (1997) 57:62–7. doi: 10.1016/S0198-8859(97)00182-1
86. Sarig O, Bercovici S, Zoller L, Goldberg I, Indelman M, Nahum S, et al. Population-specific association between a polymorphic variant in ST18, encoding a pro-apoptotic molecule, and pemphigus vulgaris. J Invest Dermatol (2012) 132:1798–805. doi: 10.1038/jid.2012.46
87. Vorobyev A, Gupta Y, Sezin T, Koga H, Bartsch YC, Belheouane M, et al. Gene-diet interactions associated with complex trait variation in an advanced intercross outbred mouse line. Nat Commun (2019) 10:4097. doi: 10.1038/s41467-019-11952-w
88. de Sena Nogueira Maehara L, De-Souza-Santana FC, Porro AM, Marcos EVC, Ura S, Nolte IM, et al. HLA class II alleles of susceptibility and protection in Brazilian and Dutch pemphigus foliaceus.[letter]. Br J Dermatol (2018) 178(3):e212–4. doi: 10.1111/bjd.16022
89. Lombardi ML, Mercuro O, Ruocco V, Lo Schiavo A, Lombari V, Guerrera V, et al. Common human leukocyte antigen alleles in pemphigus vulgaris and pemphigus foliaceus Italian patients. J Invest Dermatol (1999) 113:107–10. doi: 10.1046/j.1523-1747.1999.00626.x
90. del Mar Sáez-de-Ocariz M, Vega-Memije ME, Zúñiga J, Salgado N, Ruíz J, Balbuena A, et al. HLA-DRB1*0101 is associated with foliaceous pemphigus in Mexicans.[letter]. Int J Dermatol (2005) 44(4):350. doi: 10.1111/j.1365-4632.2005.02038.x
91. Korman NJ. New immunomodulating drugs in autoimmune blistering diseases. Dermatol Clin (2001) 19:637–48, viii. doi: 10.1016/S0733-8635(05)70305-3
92. Abrèu-Velez AM, Hashimoto T, Bollag WB, Tobón Arroyave S, Abrèu-Velez CE, Londoño ML, et al. A unique form of endemic pemphigus in northern Colombia. J Am Acad Dermatol (2003) 49:599–608. doi: 10.1067/S0190-9622(03)00851-X
93. Khachemoune A, Guldbakke KK, Ehrsam E. Pemphigus foliaceus: a case report and short review. Cutis (2006) 78:105–10.
94. Kridin K. Subepidermal autoimmune bullous diseases: overview, epidemiology, and associations. Immunol Res (2018) 66(1):6-17. doi: 10.1007/s12026-017-8975-2
95. Kridin K, Ludwig RJ. The Growing Incidence of Bullous Pemphigoid: Overview and Potential Explanations. Front Med (Lausanne) (2018) 5:220. doi: 10.3389/fmed.2018.00220
96. Skandalis K, Spirova M, Gaitanis G, Tsartsarakis A, Bassukas ID. Drug-induced bullous pemphigoid in diabetes mellitus patients receiving dipeptidyl peptidase-IV inhibitors plus metformin. J Eur Acad Dermatol Venereol (2012) 26:249–53. doi: 10.1111/j.1468-3083.2011.04062.x
97. Pasmatzi E, Monastirli A, Habeos J, Georgiou S, Tsambaos D. Dipeptidyl peptidase-4 inhibitors cause bullous pemphigoid in diabetic patients: report of two cases. Diabetes Care (2011) 34:e133. doi: 10.2337/dc11-0804
98. Aouidad I, Fite C, Marinho E, Deschamps L, Crickx B, Descamps V. A case report of bullous pemphigoid induced by dipeptidyl peptidase-4 inhibitors. JAMA Dermatol (2013) 149:243–5. doi: 10.1001/jamadermatol.2013.1073
99. Izumi K, Nishie W, Mai Y, Wada M, Natsuga K, Ujiie H, et al. Autoantibody Profile Differentiates between Inflammatory and Noninflammatory Bullous Pemphigoid. J Invest Dermatol (2016) 136(11):2201–10 doi: 10.1016/j.jid.2016.06.622
100. Horikawa H, Kurihara Y, Funakoshi T, Umegaki-Arao N, Takahashi H, Kubo A, et al. Unique clinical and serological features of bullous pemphigoid associated with dipeptidyl peptidase-4 inhibitors.[letter]. Br J Dermatol (2018) 178(6):1462–3. doi: 10.1111/bjd.16479
101. Ujiie H, Muramatsu K, Mushiroda T, Ozeki T, Miyoshi H, Iwata H, et al. HLA-DQB1*03:01 as a Biomarker for Genetic Susceptibility to Bullous Pemphigoid Induced by DPP-4 Inhibitors. J Invest Dermatol (2018) 138:1201–4. doi: 10.1016/j.jid.2017.11.023
102. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science (2016) 353:179–84. doi: 10.1126/science.aaf6756
103. Mardiana S, Gill S. CAR T Cells for Acute Myeloid Leukemia: State of the Art and Future Directions. Front Oncol (2020) 10:697. doi: 10.3389/fonc.2020.00697
104. Lee J, Werth VP, Hall RP, Eming R, Fairley JA, Fajgenbaum DC, et al. Perspective from the 5th International Pemphigus and Pemphigoid Foundation scientific conference. Front Med (Lausanne) (2018) 5:306. doi: 10.3389/fmed.2018.00306
105. Ellebrecht CT, Lundgren DK, Payne AS. On the mark: genetically engineered immunotherapies for autoimmunity. Curr Opin Immunol (2019) 61:69–73. doi: 10.1016/j.coi.2019.08.005
106. Ludwig RJ, Vanhoorelbeke K, Leypoldt F, Kaya Z, Bieber KM, McLachlan SM, et al. Mechanisms of autoantibody-induced pathology. Front Immunol (2017) 8:603. doi: 10.3389/fimmu.2017.00603
Keywords: precision medicine, pemphigus, pemphigoid, diagnosis, treatment
Citation: Bieber K, Kridin K, Emtenani S, Boch K, Schmidt E and Ludwig RJ (2021) Milestones in Personalized Medicine in Pemphigus and Pemphigoid. Front. Immunol. 11:591971. doi: 10.3389/fimmu.2020.591971
Received: 05 August 2020; Accepted: 16 November 2020;
Published: 11 January 2021.
Edited by:
Heiko Mühl, Goethe University Frankfurt, GermanyReviewed by:
Volker Spindler, University of Basel, SwitzerlandMiriam Wittmann, University of Leeds, United Kingdom
Roberto Maglie, University of Florence, Italy
Copyright © 2021 Bieber, Kridin, Emtenani, Boch, Schmidt and Ludwig. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ralf J. Ludwig, ralf.ludwig@uksh.de