 Alessandra Colamatteo1Teresa Micillo2Sara Bruzzaniti3Clorinda Fusco1Silvia Garavelli3
Alessandra Colamatteo1Teresa Micillo2Sara Bruzzaniti3Clorinda Fusco1Silvia Garavelli3 Veronica De Rosa3,4
Veronica De Rosa3,4 Mario Galgani3Maria Immacolata Spagnuolo5Francesca Di Rella6Annibale A. Puca7,8
Mario Galgani3Maria Immacolata Spagnuolo5Francesca Di Rella6Annibale A. Puca7,8 Paola de Candia7
Paola de Candia7 Giuseppe Matarese1,3*
Giuseppe Matarese1,3*- 1Treg Cell Lab, Dipartimento di Medicina Molecolare e Biotecnologie Mediche, Università degli Studi di Napoli “Federi II”, Naples, Italy
- 2Dipartimento di Biologia, Università degli Studi di Napoli “Federico II”, Naples, Italy
- 3Laboratorio di Immunologia, Istituto di Endocrinologia e Oncologia Sperimentale, Consiglio Nazionale delle Ricerche (IEOS-CNR), Naples, Italy
- 4Fondazione Santa Lucia, Unità di Neuroimmunologia, Rome, Italy
- 5Dipartimento di Scienze Mediche Traslazionali, Università degli Studi di Napoli “Federico II”, Naples, Italy
- 6Dipartimento di Senologia, Oncologia Medica, IRCCS–Fondazione G. Pascale, Naples, Italy
- 7Department of Cardiovascular Diseases, IRCCS MultiMedica, Milan, Italy
- 8Department of Medicine and Surgery, University of Salerno, Baronissi, Italy
Distinct metabolic pathways are known to regulate growth, differentiation, survival, and activation of immune cells by providing energy and specific biosynthetic precursors. Compelling experimental evidence demonstrates that effector T cell functions are coupled with profound changes in cellular metabolism. Importantly, the effector T cell-dependent “anti-self” response characterizing the autoimmune diseases is accompanied by significant metabolic alterations. MicroRNAs (miRNAs), evolutionary conserved small non-coding RNA molecules that affect gene expression by binding to target messenger RNAs, are now known to regulate multiple functions of effector T cells, including the strength of their activation, thus contributing to immune homeostasis. In this review, we will examine the most recent studies that describe miRNA direct involvement in the metabolic reprogramming that marks effector T cell functions. In particular, we will focus on the work showing a connection between miRNA regulatory function and the molecular network dysregulation that leads to metabolic pathway derangement in autoimmunity. Finally, we will also speculate on the possibility that the interplay between miRNAs and metabolism in T cells may help identify novel miRNA-based therapeutic strategies to treat effector T cell immunometabolic alterations in pathological conditions such as autoimmunity and chronic inflammation.
Introduction
The immune system encompasses a variety of cellular subsets that are highly dynamic and specialized in several activities essential for host defense and tissue homeostasis (1–4). T cells are a crucial component of the adaptive immune system, with a unique nature that makes them able to respond rapidly to environmental changes (5–7). T cell activation and function are deeply related to specific metabolic programs necessary to regulate T cell signaling and support their growth, differentiation, and effector function. Indeed, the ability of intracellular metabolism to integrate signals and nutrients to produce energy is fundamental to determine a specific T cell fate (8–10). Over the past decade, increasing studies have highlighted the role of microRNAs (miRNAs) in the metabolic control of immune cells. miRNAs are a class of small non-coding RNAs involved in the fine-tune regulation of gene expression. Several mechanisms have been uncovered by which miRNAs control T cell function, including regulation of intracellular metabolic pathways. Under specific conditions, such as inflammation, the biogenesis of miRNAs may be modified. Aberrant expression of miRNAs may influence T cell metabolic reprogramming, leading to pathological phenomena, including chronic inflammatory disorders and autoimmune conditions.
In this review, we discuss recent literature reporting the involvement of miRNAs in the metabolic control of immune responses, with particular emphasis on those impacting on T lymphocyte differentiation and function, in health and autoimmunity.
Metabolic Regulation of T Cell Response
T lymphocytes play a crucial role in host defense and coordination of immune response (11). Upon antigen recognition via the T cell receptor (TCR) in the presence of co-stimulatory signals, T lymphocytes clonally expand and produce cytokines to eliminate infected or transformed cells (12, 13). In particular, during an acute infection, antigen-specific T cells proliferate and differentiate into effector T (Teff) cells: CD8+ cytotoxic T lymphocytes (CTLs) that rapidly mediate the clearance of infected cells and CD4+ Teff cells that became functional specialized in distinct T helper (Th) cell subset [Th1, Th2, Th17, follicular Th (Tfh), and regulatory T (Treg) cells] (14, 15). Their dysregulated activation leads to a wide spectrum of autoimmune and inflammatory conditions (16–19).
Compelling evidence indicate that intracellular metabolic programs adopted by T cells finely regulate immune response (20, 21). T cell activation determines an increased biosynthetic demand, which requires rapid changes to generate metabolic intermediates for T cell growth, proliferation, and function (20–22). Both quiescent naïve and memory T cells are characterized by metabolic pathways that supply energy for survival and migration, including oxidative phosphorylation (OXPHOS), fatty acid oxidation (FAO), and amino acid oxidation [Figure 1; (10, 23)]. Conversely, activation of T cells drives transcriptional changes, causing downregulation of oxidative metabolism and upregulation of biosynthetic pathways, such as aerobic glycolysis that promotes an increase in biochemical intermediates, necessary for nucleotide, amino acid, and fatty acid synthesis (24). This anabolic program increases nutrient uptake at the expense of ATP production (25, 26). Teff cells require high levels of glucose to proliferate and differentiate in distinct T cell subsets (27). The increased glucose metabolism is controlled by glucose transporters, glycolytic enzymes, multi-protein complexes, and transcriptional factors that coordinate glucose utilization to generate pyruvate (28, 29). Under hypoxic conditions, pyruvate can be converted to lactate by the lactate dehydrogenase (LDH) (anaerobic glycolysis); on the other hand, higher oxygen levels promote pyruvate transfer into the mitochondria to supply intermediates of the tricarboxylic (TCA) cycle (30). TCA cycle starts from acetyl-CoA to generate citrate, which is consumed and regenerated throughout multiple biochemical reactions; nicotinamide adenine dinucleotide (NADH), flavin adenine dinucleotide (FADH2), and one GTP or ATP molecule are produced in each cycle [Figure 1; (31)]. Subsequently, NADH and FADH2 provide electrons to generate ATP via OXPHOS [Figure 1; (31)]. Alternatively, glucose-6-phosphate (G6P), derived from the first enzymatic step of glycolysis, can be directed into the pentose phosphate pathway (PPP) that provides ribose for the synthesis of nucleotides and reducing equivalents, such as nicotinamide adenine dinucleotide phosphate (NADPH), for lipid and cholesterol biogenesis [Figure 1; (32)].
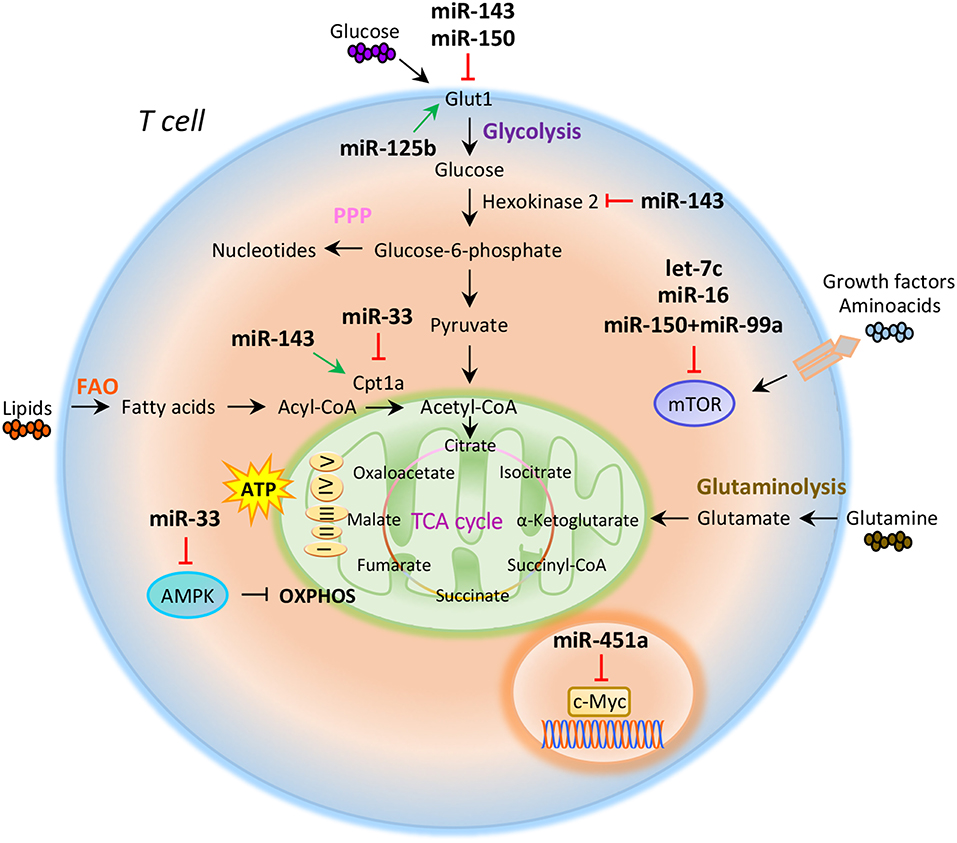
Figure 1. miRNAs orchestrate T cell metabolic reprogramming. Schematic representation of the main metabolic programs controlled by miRNAs in T cells: Glycolysis, pentose phosphate pathway (PPP), fatty acid oxidation (FAO), tricarboxylic acid (TCA) cycle, glutaminolysis, and oxidative phosphorylation (OXPHOS).
The increased glucose flow into the pentose phosphate pathway is also accompanied by an increase in glutamine metabolism (glutaminolysis), an energy-producing process required for fast biosynthesis of macromolecules necessary for lipid production (33, 34). Lipids play an important role in the regulation and maintenance of membrane properties, bioenergetic demands, and cell signaling (35). Upon TCR activation, T cells must increase their lipid content at each cell division for new plasma membrane generation; moreover, lipids, especially long fatty acids, are necessary to generate energy through the FAO [Figure 1; (36)]. Long-lived memory T cells preferentially use FAO to fuel the TCA cycle and OXPHOS, and maintain ATP production (37). Distinct metabolic and nutrient sensors, including the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway, hypoxia-inducible factor 1α (HIF1α), c-Myc, and AMP-activated kinase (AMPK), integrate external stimuli and nutrient availability with intracellular metabolic processes (38–42). mTOR is a serine/threonine kinase with a key role in the regulation of T cell metabolism; it integrates multiple signals in response to nutrients, growth factors, energy, and stress to coordinate immune response upon TCR activation (43, 44). mTOR acts through two main signaling complexes, mTORC1 and mTORC2, which differ for their structure and sensitivity to the inhibitor rapamycin (43–46). PI3K/AKT/mTOR signaling pathway activation induced by TCR engagement, CD28 co-stimulation, or IL-2 receptor leads to an increase of glucose uptake via upregulation of Glucose transporter 1 (Glut1) levels, mediated by mTORC1 complex (45, 47). Instead, mTORC2, which mainly responds to growth signals, controls T cell proliferation and survival by AKT phosphorylation that affects glycogen synthase kinase-3 β (GSK-3β) (48, 49). Moreover, mTORC1 activates glycolytic program in T cells also thanks to the downstream transcription factors HIF1α and its target genes (50). HIF1α is an oxygen sensor rapidly activated under hypoxic conditions, which determines the transcription of target genes including erythropoietin (EPO), vascular endothelial growth factor (VEGF), and glycolytic enzymes (51, 52). HIF1α in turn induces the expression of intermediates required for glycolysis, such as Glut1, in the presence of low oxygen availability (53). HIF1α can also increase glycolysis by inducing the expression of pyruvate dehydrogenase kinase 1 (PDK1), a key metabolic enzyme that favors the conversion of pyruvate to lactate (53). mTORC1 is also responsible for the activation of c-Myc, another transcription factor with pleiotropic effects involved in cell proliferation, growth, and metabolism (38, 54). As a metabolic checkpoint, c-Myc is rapidly activated after TCR engagement and induces the transcription of enzymatic mediators of glycolysis, such as lactate dehydrogenase A (LDHA), pyruvate kinase isoenzyme type M2 (PKM2), hexokinase 2 (HK2), and Glut1 (55). c-Myc also induces the expression of transcription factor activating enhancer binding protein 4 (AP-4) which in turn upregulates the abovementioned glycolytic enzymes (56). Conversely, AMPK drives activated T cells to engage OXPHOS and maintain ATP levels under low glucose concentration (57). AMPK is activated in response to energy deprivation and under stress conditions, such as infections, inflammation, and DNA damage (58). AMPK is also a sensor of energy homeostasis and inhibits energy-consuming metabolism by increasing cellular AMP levels during energy deprivation, favoring ATP production (57, 59). Furthermore, AMPK controls catabolic metabolism through the inhibition of acetyl-CoA carboxylase (ACC), a crucial metabolic enzyme of fatty acid biosynthesis, promoting FAO (59). Under nutrient deprivation, AMPK also inhibits mTORC1 complex activity in T cells (57). Indeed, it has been observed that AMPK loss in naïve T cells upregulates mTOR activity and induces glycolysis (57). In all, these findings suggest that metabolism represents a critical checkpoint for T cell activation and function. Distinct levels of regulation (epigenetic, transcriptional, and translational) control and coordinate this cross-talk to ensure the appropriate energetic status underlying the specific immune cell function.
miRNA Biogenesis and Biological Function
miRNAs are a class of small, non-coding RNAs of 21–25 nucleotides involved in post-transcriptional control of gene expression, through base pairing with complementary sequences in the 3′ untranslated regions (3′UTR) (58). miRNAs are transcribed by RNA polymerase II or III as long variable-length transcripts named primary miRNA transcripts (pri-miRNAs), with a 5′cap and a 3′poly (A) tail [Figure 2; (60)]. Pri-miRNAs are processed by Drosha ribonuclease III and its partner named Di George syndrome Critical Region 8 (DGCR8), to produce a hairpin RNA of about 65 nucleotides known as pre-miRNA molecule [Figure 2; (61)]. After nuclear processing, the pre-miRNAs are exported from the nucleus to the cytoplasm by the GTP-binding nuclear protein Ran (RanGTP)/exportin-5 (XPO5) complex and cleaved into a miRNA duplex of about 21 nucleotides (ds-miRNAs) by a second endoribonucleolytic Dicer and its cofactor TRBP (trans-activator RNA binding protein) [Figure 2; (62)]. ds-miRNAs are loaded into the Argonaute protein (Ago2), which facilitates incorporation of the guide strand into the RNA-induced silencing complex (RISC), while the passenger strand is degraded [Figure 2; (63)]. The guide strand binds target messenger RNA (mRNA) based on sequence complementarity; base pairing match induces degradation of target mRNA, while the imperfect complementarity results in suppression of translation [Figure 2; (64)].
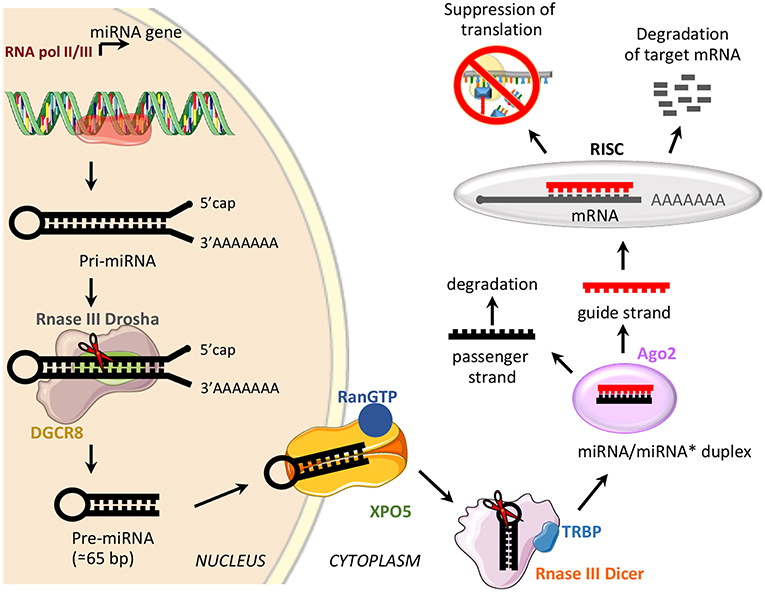
Figure 2. miRNA biogenesis and function. Schematic representation of miRNA biogenesis pathway and biological function. Di George syndrome Critical Region 8 (DGCR8), exportin-5 (XPO5), GTP-binding nuclear protein Ran (RanGTP), trans-activator RNA binding protein (TRBP), Argonaute protein 2 (Ago2), and RNA-induced silencing complex (RISC).
The biological relevance of miRNAs is highlighted by the discovery that a single miRNA could interact with hundreds of target mRNAs, and each gene transcript may have several sites of miRNA recognition (65, 66). The finely tuned control of gene expression requires the production of the appropriate level of specific miRNAs in a well-defined time frame (65, 66). Since miRNAs are involved in post-transcriptional regulation of several cellular processes, dysregulation or dysfunction of their biogenesis leads to a wide range of human diseases, ranging from cancer to autoimmune disorders (67–69). Specific miRNAs have been described to regulate the function and homeostasis of several immune cell populations (70). Compelling evidence has demonstrated that deletion of Drosha or Dicer within T cell compartment affects T cell development, differentiation and function (71, 72). In this context, it has been shown that Dicer deletion at an early stage of T cell differentiation compromises the survival of TCR alphabeta (α/β) chain cells while it is dispensable for CD4 or CD8 T cell lineage commitment (73, 74). Furthermore, Chong et al. highlighted the essential role of Drosha and Dicer in Treg cells, as specific deletion of one or both of them results in impaired expression of the transcription factor forkhead box P3 (FoxP3), the master gene of Treg cell development and function (71). Among all the miRNAs involved in the control of T cell fate, miR-125b has been shown to restrain the expression of genes encoding molecules important for differentiation of naïve into effector and memory T cells, targeting interferon-γ (IFN-γ), interleukin 2 receptor β (IL-2Rβ), interleukin 10 receptor α (IL-10RA), and Blimp-1 (PRDM1) genes (75).
miR-214 and miR-182 act through different mechanisms to control T lymphocyte activation (76). Specifically, up-regulation of miR-124 in T cells, after TCR stimulation, promotes T cell activation through the inhibition of phosphatase and tensin homolog (PTEN), a negative regulator of T cell activation. Mechanistically, activation via CD28 induces upregulation of miR-214, which targets the 3′UTR of Pten, causing a reduction in PTEN levels promoting T cell activation (76). Constitutive overexpression of miR-182 by IL-2 supports clonal expansion of Th cells, by reducing the transcription factor Foxo1, a suppressor of resting Th cell proliferation (77).
Compelling experimental evidence reports that several miRNAs can also influence T cell differentiation (78–80). In more detail, miR-155, expressed at high levels in Treg cells, is involved in the modulation of the suppressor of cytokine signaling 1 (SOCS1) and the signal transducers and activators of transcription 5 (STAT5) activity, key molecules for Treg cell differentiation and function (81). Data from animal models revealed that mice lacking miR-146a are more susceptible to chronic inflammation and autoimmune disorders secondarily to the loss of peripheral T cell tolerance. These findings suggest that miR-146 may be considered a key regulator of T cell response (82). In all, miRNA network can be considered as a crucial regulator of cell biology and, although studies focusing on miRNAs are progressively increasing, their impact on T cell function is still only partially explored.
Role of miRNAs in T Cell Metabolic Reprogramming
The role of miRNAs in the regulation of metabolic reprogramming in cancer cells has been largely investigated, thus opening the study of their potential role in the modulation of T cell metabolism (83–85). Several studies have suggested that miRNAs mainly act on key metabolic enzymes or transporters of energetic nutrients, thus impacting on T cell proliferation and differentiation (86, 87). In this context, Zhang et al. identified miR-143 as a regulator of T cell metabolism that reduces glucose uptake through the inhibition of glucose receptor Glut1. The authors found that, by specifically reducing glycolysis during TCR-dependent activation, miR-143 promotes memory T cell differentiation and metabolic reprogramming (88). In addition, miR-143 stimulates T cell memory differentiation also through the control of two enzymes, hexokinase II and carnitine palmitoyltransferase 1a (Cpt1a), which regulate glucose oxidation and oxidative phosphorylation, respectively [Figure 1; (88)]. miR-150 is an additional miRNA that finely tunes T cell glycolytic reprogramming, secondarily to co-stimulatory signals. Recent findings suggest that CD46 signaling, which is activated downstream of CD28 co-stimulation during T cell activation, leads to a rapid downregulation of miR-150 expression, which targets solute carrier family 2 member 1 (SLC2A1) gene, encoding the glucose transporter Glut1 [Figure 1; (89)]. Through a direct interference with Glut1-dependent glycolysis, miR-150 profoundly impacts on T cell activation in the absence of a proper co-stimulatory signal (89). On the contrary, metabolic reprogramming of activated T cells seems to be associated with the upregulation of miRNAs promoting glucose uptake and downregulation of those stimulating catabolic pathways. In this context, Liu et al. have studied the role of miR-125b, overexpressed in T cell acute lymphoblastic leukemia (T-ALL) (90). The authors revealed that miR-125b regulates glucose uptake in T cell via Glut1 by reducing the expression of TNF-α-induced protein 3 (TNFAIP3), which inhibits the activation of nuclear factor k B (NF-kB) [Figure 1; (90)]. These findings suggest that, through the enhancement of glucose metabolism and oxygen consumption, deregulation of miR-125b contributes to abnormal differentiation of T cell in T cell leukemia (90).
It has been reported that CD28 engagement during T cell activation promotes the expression of Cpt1a, a key enzyme for mitochondrial FAO. This process is finely regulated by miR-33 that, in the absence of CD28 signal, attenuates Cpt1a expression, interfering with the metabolic demand central for future recall of memory T cells [Figure 1; (91)]. Several studies have also reported that miRNAs control T cell metabolic reprogramming by targeting important metabolic checkpoints, such as AMPK, mTOR, and c-Myc (92–94). Ouimet et al. unveiled that miR-33 targets AMPK, inducing an unbalance between aerobic glycolysis and mitochondrial OXPHOS (92). miR-33 affects oxidative phosphorylation and induces macrophages M2 polarization through direct targeting of AMPK [Figure 1; (92)]. The same authors revealed that miR-33 inhibition increased macrophage expression of the retinoic acid (RA)-producing enzyme aldehyde dehydrogenase family 1, subfamily A2 (ALDH1A2), and of retinal dehydrogenase enzyme, with subsequent production of RA, which favors differentiation of FoxP3+ CD4+ T cells (92).
Several other miRNAs are involved in the control of mTOR pathway; in particular, an increased activation of mTOR, leading to S6 and AKT phosphorylation, has been observed in the absence of co-stimulatory signals in CD4+ T cells deficient for RNaseIII enzyme Dicer, a key component of miRNAs biogenesis (93). Two miRNAs are involved in the fine regulation of mTOR and Rictor mRNA expression. Specifically, the downregulation of let-7c miRNA and miR-16 in Dicer-deficient CD4+ T cells causes TCR signaling amplification and increased IL-2 production, due to overexpression of mTOR and Rictor mRNAs [Figure 1; (93)]. Subsequently, restoration of mTOR and Rictor expression by genetic manipulation determines reduction of IL-2 production at levels sufficient to avoid anergy in response to TCR engagement (93, 94). Thus, let-7c miRNA and miR-16 control the balance between activation and anergy through post-transcriptional control of mTOR components in T cells [Figure 1; (93, 94)].
mTOR pathway is also regulated by miR-150 expressed at high levels during differentiation of naive CD4+ T cells into Treg cells; miR-150 efficiently represses mTOR in cooperation with miR-99a, and this promotes Treg cell differentiation [Figure 1; (87)]. In addition, a recent report showed that miR-451a directly targets c-Myc in Jurkat T cells; indeed, miR-451a negatively correlated with c-Myc expression in CD4+ T cells from dilated cardiomyopathy subjects. These results suggest a role of the miR-451/c-Myc pathway in CD4+ T cell proliferation and activation [Figure 1; (95)].
These data support the idea that, through the modulation of intracellular metabolic programs, miRNAs are able to influence T cell fate and differentiation (96). Understanding the mechanism by which miRNAs target metabolism in T cells may lead to therapeutic strategies for immune-related diseases.
T Cell Metabolic Alteration in Autoimmunity: the miRNA Connection
Given the impact of miRNAs in the regulation of T cell metabolic programs, aberrant miRNA expression could interfere with T cell fate with consequent loss of immune homeostasis and autoimmunity (97–100). Autoimmune diseases afflict about 7–9% of the worldwide population; in particular, type 1 diabetes (T1D), systemic lupus erythematosus (SLE), and multiple sclerosis (MS) have been increasing in the last few years (101–104). The role of T cell metabolism in the control of immune function and how its alteration could influence immune response during autoimmunity have been well-characterized (24, 105–111). Naïve T lymphocytes are metabolically inactive, and their differentiation is controlled by metabolic reprogramming, involving glycolysis, FAO, and OXPHOS (10, 112–115). It is well-known that activated T cells require high levels of metabolic compounds to maintain their viability and function (57). In particular, it has been shown that glycolysis sustains cytokine production in Teff cells (57); moreover, FAO and OXPHOS play an important role to increase the inflammatory capacity of memory T cells, and this allows a rapid response upon antigen recall (116, 117). Thus, an aberrant metabolic environment could influence the development of inflammation and autoimmune disorder by fueling the differentiation and activation of pathogenic T cells.
miRNAs were shown to regulate metabolism-related genes in T1D, an autoimmune disease characterized by persistent hyperglycemia secondarily to pancreatic β-cell destruction and insulin deficiency (118, 119). Increased glucose levels in the extracellular microenvironment determine a metabolic reprogramming that fuels autoreactive Teff cell activation and IFN-γ secretion (57, 114, 116). Glucose-activated Teff cells upregulate Glut4 and insulin receptor substrate (IRS)-1 on their surface to sustain glycolytic rate and produce pro-inflammatory cytokines and reactive oxygen species (ROS) that promote the autoimmune response (120). In this context, let-7 family of miRNAs, which control several genes involved in glucose homeostasis, insulin resistance, and cell differentiation, has been reported to control important reguatory mechanisms in T1D subjects [Table 1; (121–123)]. Let-7 miRNAs are one of the largest and highly conserved family of miRNAs expressed in T cells, present in multiple copies in the genome; the number of let-7 miRNAs differs between species; for example, in humans, there are 10 mature let-7 miRNAs (124). It has been shown that let-7 miRNAs target multiple genes related to glucose response and the insulin-PI3K-mTOR pathway, such as insulin-like growth factor 1 receptor (IGF1R), insulin receptor (INSR), and IRS-2 but also regulate genes involved in the effector functions of CTLs (i.e., granzyme A, granzyme B, perforin 1, and eomesodermin) (121, 122, 125). In particular, it has been shown that four members of the let-7 miRNAs family (let-7a, let-7e, let-7f, and let-7g) were higher in PBMCs from T1D subjects (126); however, how these miRNAs are involved in T1D pathogenesis is poorly understood. Together, these results suggest that an altered insulin–PI3K–mTOR pathway in T cells of T1D individuals may determine an aberrant glucose uptake as a consequence of a defective regulation of insulin receptor genes. This impaired metabolism could favor the differentiation and activation of pathological Teff cells in T1D patients. Furthermore, serum miR-378 negatively correlates with insulinoma-associated protein 2 (IA2A) and the Zinc transporter 8 (Znt8) autoantibodies in T1D subjects (127). This miRNA is involved in the control of several metabolic processes, such as glycolysis, mitochondrial oxidation, and fatty acid metabolism, through the interference with the expression of mannose-1-phosphate guanylyltransferase (GDP), dimethylarginine dimethylaminohydrolase 1 (DDAH1), LDHA, and carnitine O-acetyltransferase (CRAT) enzymes [Table 1; (127–129)]. One hypothesis is that serum levels of miR-378 could reflect metabolic alteration that promotes differentiation of pathogenic T cells in T1D subjects (Table 1). In this context, Zhou et al. have investigated by in silico analysis that 27 miRNAs out of 530 are located in nine human insulin-dependent diabetes mellitus (IDDM) loci associated with T1D susceptibility (130). Among them, miR-16-2, miR-551b, and miR-877 target specific genes involved in the activation of Teff cells, such as CD28, Fas ligand (FasL), and the autoimmune regulator (AIRE), respectively [Table 1; (130)].
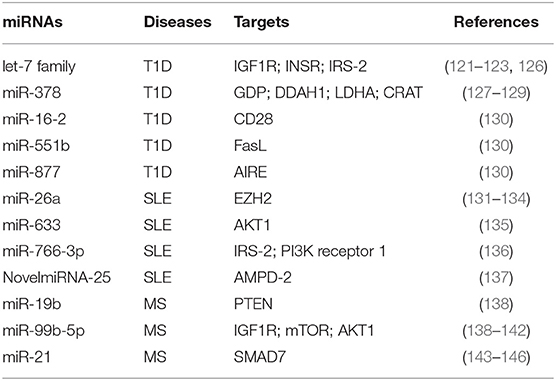
Table 1. miRNAs involved in T cell metabolic reprogramming, during autoimmune diseases: type 1 diabetes (T1D), systemic lupus erythematosus (SLE), and multiple sclerosis (MS).
miR-26a is an additional miRNA associated with T cell dysfunction, glucose metabolism, and autoimmune disease development (131, 132). Its expression in T cells is regulated by glucose availability, and it is able to target the epigenetic regulator enhancer of zeste homolog 2 (EZH2), a histone-lysine-N-methyltransferase, well-known to improve effector T cell function by inhibiting Notch signaling repressors [Table 1; (133, 147)]. In SLE subjects, the levels of miR-26a in CD4+ T cells negatively correlate with disease severity; this suggests that the reduced miRNA regulation of EZH2, secondarily to an increased glycolytic activity in CD4+ T cells, sustains their activation (134, 148). Several defects in metabolic pathways of T cells from SLE subjects have been described, especially those related to mitochondrial dysfunction (149–151). SLE is a complex multifactorial autoimmune disease where loss of tolerance determines the generation of antinuclear antibodies produced by B lymphocytes and tissue damage by autoreactive Teff cells (150, 152). Mitochondrial membrane hyperpolarization occurs in CD4+ T cells from SLE subjects, leading to increase of ROS and depletion of ATP and glutathione, determining an impaired T cell activation and cell death (149, 153, 154). Of note, mounting evidence show a role for mTOR as a sensor of mitochondrial dysfunction in Teff cell differentiation during SLE (155, 156). It has been recently reported that miR-633 that targets the AKT/mTOR pathway is significantly reduced in CD4+ T cells from SLE-affected subjects, and its expression negatively correlates with disease activity [Table 1; (135)]. In more detail, miR-633 inhibits the AKT/mTOR signaling and increases the induction of several cytokines, such as IL-4, IL-17, and IFN-γ, thus contributing to disease pathogenesis [Table 1; (135)]. Together with the abovementioned study, others reported an increase of miR-766-3p in CD4+ T cells from SLE subjects. Potential target genes of this miRNA are IRS-2 and PI3K receptor 1, both involved in the PI3K/AKT/mTOR pathway [Table 1; (136)]. These data suggest that CD4+ T cells of SLE subjects have an impaired PI3K signaling, which could affect their differentiation and function, leading to the development of autoimmunity. Recently, Guo et al. reported the upregulation of several miRNAs associated to metabolic pathways in PBMCs from SLE patients, such as NovelmiRNA-25 and miR-1273h-5p (137). NovelmiRNA-25 targets the enzyme adenosine monophosphate deaminase (AMPD)-2 involved in purine nucleobase or nucleotide metabolism by converting AMP to inosine monophosphate (IMP) [Table 1; (137)]. The overexpression of NovelmiRNA-25 associates with a downregulation of AMPD-2 protein in PBMCs from SLE subjects, contributing to AMP accumulation that improved the activation of pro-inflammatory pathways [Table 1; (137, 157)]. Moreover, increased levels of AMP contribute to activate AMPK, a key enzyme for cellular metabolic reprogramming (42). Of note, NovelmiRNA-25 positively correlates with disease activity, suggesting an important role for this miRNA as a biomarker to predict the activation of pathogenic T cells.
The role of miRNAs in the regulation of autoreactive T cell function has been reported also in MS (158–160). MS is the most common chronic inflammatory demyelinating disease of the central nervous system (CNS), characterized by autoreactive T cells able to target myelin-based antigens, leading to demyelinating lesions and neuronal degeneration (161). During MS, the demyelinating process associates with metabolic reprogramming in neuronal cell bodies sustaining chronic inflammation through the release of pro-inflammatory cytokines (162, 163). These events promote the activation of CD4+ T cells, which further increase neuronal damage (162). Mounting evidence highlights the close relationship between T cell metabolic alterations and neurodegeneration in MS (105, 164, 165). Impaired glycolytic engagement has been described in Tconv cells from naïve-to-treatment relapsing remitting (RR)-MS subjects during the generation of induced (i) Treg cells, which contributes to loss of immune tolerance (105). In more detail, the glycolytic enzyme enolase-1 accumulates in the nuclei—secondarily to the reduced engagement in the glycolytic cascade—and constrains the induction of FoxP3 expression during the generation of Treg cells (105). Moreover, other key enzymes involved in glycolysis and mitochondrial respiration are reduced in CD4+ T cells from RR-MS subjects, such as aldolase, hexokinase 1, Glut1, dihydrolipoamide S-acetyltransferase (DLAT), and dihydrolipoamide S-succinyltransferase (DLST); interestingly, restoration of these enzymes after IFN-β-1a treatment correlates with disease amelioration (165). Several studies also reported that alterations in the PI3K/AKT/mTOR pathway, which controls T cell activation and metabolism, ameliorate the clinical course of MS (166–169). Also, rapamycin—an immunosuppressant drug that inhibits mTOR by destabilizing the mTOR–Raptor complex—controls disease progression in experimental autoimmune encephalomyelitis (EAE) mice by suppressing Teff cell functions (170, 171). In this context, miR-19b and the miR-99 family are associated with the mTOR pathway, affecting effector T cell activation during MS [Table 1; (138)]. By targeting PTEN, the negative regulator of the PI3K/AKT/mTOR signaling pathway, miR-19b enhances mTOR activity sustaining pathogenic Th17 cell development [Table 1; (138)]. Other reports revealed also that the miR-99 family modulates the PI3K/AKT/mTOR signaling pathways. In particular, the miR-99b-5p, a member of this miRNA family, is significantly higher in splenocytes of EAE mice; in silico analysis confirmed that it may target multiple genes, such as IGF1R, mTOR, and AKT1 [Table 1; (139–141)]. Combined miRNA and mRNA expression analysis confirmed these data also in human disease; indeed, miR-99b-5p levels are upregulated in PBMCs from pediatric MS subjects (142). These results suggest an important role for miR-99 family, in particular the miR-99b-5p, in T cell activation during MS through a hyper-activation of the mTOR pathway in pathogenic lymphocytes.
Furthermore, recent reports suggest that fumaric acid ester (FAE)—a Krebs cycle intermediate used for MS therapy—induces hypermethylation of the miR-21 locus in CD4+ T cells, and this constrains Th17 cell differentiation and function [Table 1; (143)]. In more detail, FAE treatment reduces Th17 cells, by direct hypermethylation of CpG sites spanning the MIR-21 promoter. Several studies have shown that miR-21 is upregulated in PBMCs from MS subjects and also in CNS-infiltrating T cells of EAE mice (144, 145). As a therapeutic tool in MS subjects, FAE selectively reduces miR-21 transcripts in Th17 cells and indirectly increases its target—the small mothers against decapentaplegic homolog 7 (SMAD7)—an inhibitor of their differentiation [Table 1; (144, 146)].
Taken together, these findings support the existence of a cross-talk between metabolic programs and miRNA network in T cells. Through a strong impact on the intracellular molecular pathways that control T cell differentiation and function, miRNA dysregulation leads to an imbalance between autoreactive T cell activation and regulatory function with consequent loss of immunological tolerance.
Concluding Remarks
Our understanding of the link between T cell metabolism and miRNA expression has substantially increased in the past decade. The ability of several miRNAs to regulate and reprogram metabolic pathways that drive T cell function and differentiation may represent a hallmark to improve the comprehension of the molecular processes underlying the pathogenesis of autoimmune disorders. However, further studies are required to better understand the connection among miRNAs, T cell metabolism, and loss of immunological tolerance. The precise mechanisms by which miRNAs target the genes encoding for enzymes, multi-protein complex, and transcription factors related to T cell metabolism and how their alteration associates with the development of autoimmune disorders remain to be dissected. Considering the increasing important role of miRNAs in the immune homeostasis, therapeutic approaches could represent an innovative way to control the aberrant metabolism sustaining autoreactive T cell clones.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer SB and handling editor declared their shared affiliation.
Acknowledgments
This paper was supported by grants from the Juvenile Diabetes Research Foundation (JDRF n. 2-SRA-2018-479-S-B to MG and n. 1-SRA-2018-477-S-B to PdC) and the National Multiple Sclerosis Society (NMSS n. PP-1804-30725 to MG and PP-1606-24687 to PdC), Fondazione Italiana Sclerosi Multipla (FISM n. 2018/R/4 to VD, n. 2016/R/18 to GM, and n. 2016/R/10 to PdC), European Research Council Grant (ERC menTORingTregs n. 310496) and Telethon (n. GGP 17086) to GM, the Università degli Studi di Napoli Federico II (STAR Program Linea 1−2018), and Ministero della Salute (n. GR-2016-02363725) to VD.
Part of images used in the figure preparation were from the Motifolio drawing toolkits (www.motifolio.com) and from Servier Medical Art (http://www.servier.fr/servier-medical-art).
References
1. Mak TW, Saunders ME, Jett BD. Primer to the immune response. In: Components of the Immune System. Academic Cell Press; Elsevier (2014). p. 21–54.
2. Kumar BV, Kratchmarov R, Miron M, Carpenter DJ, Senda T, Lerner H, et al. Functional heterogeneity of human tissue-resident memory T cells based on dye efflux capacities. JCI Insight. (2018) 3:123568. doi: 10.1172/jci.insight.123568
3. Koenderman L, Buurman W, Daha MR. The innate immune response. Immunol Lett. (2014) 162:95–102. doi: 10.1016/j.imlet.2014.10.010
4. Chaplin DD. Overview of the immune response. J Allergy Clin Immunol. (2010) 125:S3–23. doi: 10.1016/j.jaci.2009.12.980
6. Mak TW, Saunders ME, Jett BD. Primer to the immune response. In: T Cell Development, Activation and Effector Functions. Academic Cell Press; Elsevier (2014). p. 197–226.
7. Mak TW, Saunders ME, Jett BD. Primer to the immune response. In: Regulation of Immune Responses in the Periphery. Academic Cell Press; Elsevier (2014). p. 227–46.
8. Norata GD, Caligiuri G, Chavakis T, Matarese G, Netea MG, Nicoletti A, et al. The cellular and molecular basis of translational immunometabolism. Immunity. (2015) 43:421–34. doi: 10.1016/j.immuni.2015.08.023
9. Gerriets VA, Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol. (2012) 33:168–73. doi: 10.1016/j.it.2012.01.010
10. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. (2013) 31:259–83. doi: 10.1146/annurev-immunol-032712-095956
11. Jiang H, Chess L. Regulation of immune responses by T cells. N Engl J Med. (2006) 354:1166–76. doi: 10.1056/NEJMra055446
12. Brownlie RJ, Zamoyska R. T cell receptor signalling networks: branched, diversified and bounded. Nat Rev Immunol. (2013) 13:257–69. doi: 10.1038/nri3403
13. Chambers CA, Allison JP. Co-stimulation in T cell responses. Curr Opin Immunol. (1997) 9:396–404.
14. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. (2012) 12:749–61. doi: 10.1038/nri3307
15. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. (2010) 28:445–89. doi: 10.1146/annurev-immunol-030409-101212
16. Suárez-Fueyo A, Bradley SJ, Tsokos GC. T cells in systemic lupus erythematosus. Curr Opin Immunol. (2016) 43:32–8. doi: 10.1016/j.coi.2016.09.001
17. Speiser DE, Ho PC, Verdeil G. Regulatory circuits of T cell function in cancer. Nat Rev Immunol. (2016) 16:599–611. doi: 10.1038/nri.2016.80
18. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
19. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
20. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. (2013) 38:633–43. doi: 10.1016/j.immuni.2013.04.005
21. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic instruction of immunity. Cell. (2017) 169:570–86. doi: 10.1016/j.cell.2017.04.004
22. Wang R, Green DR. Metabolic checkpoints in activated T cells. Nat Immunol. (2012) 13:907–15. doi: 10.1038/ni.2386
23. Dumitru C, Kabat AM, Maloy KJ. Metabolic adaptations of CD4(+) T cells in inflammatory disease. Front Immunol. (2018) 9:540. doi: 10.3389/fimmu.2018.00540
24. Bantug GR, Galluzzi L, Kroemer G, Hess C. The spectrum of T cell metabolism in health and disease. Nat Rev Immunol. (2018) 18:19–34. doi: 10.1038/nri.2017.99
25. Jones RG, Thompson CB. Revving the engine: signal transduction fuels T cell activation. Immunity. (2007) 27:173–8.
26. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
27. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. (2011) 186:3299–303. doi: 10.4049/jimmunol.1003613
28. Maciver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J Leukoc Biol. (2008) 84:949–57. doi: 10.1189/jlb.0108024
29. Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell. (2007) 18:1437–46. doi: 10.1091/mbc.e06-07-0593
30. Olson KA, Schell JC, Rutter J. Pyruvate and metabolic flexibility: Illuminating a path toward selective cancer therapies. Trends Biochem Sci. (2016) 41:219–30. doi: 10.1016/j.tibs.2016.01.002
31. Park BV, Pan F. Metabolic regulation of T cell differentiation and function. Mol Immunol. (2015) 68:497–506. doi: 10.1016/j.molimm.2015.07.027
32. Cho ES, Cha YH, Kim HS, Kim NH, Yook JI. The pentose phosphate pathway as a potential target for cancer therapy. Biomol Ther. (2018) 26:29–38. doi: 10.4062/biomolther.2017.179
33. Newsholme EA, Crabtree B, Ardawi MS. Glutamine metabolism in lymphocytes: its biochemical, physiological and clinical importance. Q J Exp Physiol. (1985) 70:473–89.
34. Newsholme EA, Crabtree B, Ardawi MS. The role of high rates of glycolysis and glutamine utilization in rapidly dividing cells. Biosci Rep. (1985) 5:393–400.
35. Ingólfsson HI, Melo MN, van Eerden FJ, Arnarez C, Lopez CA, Wassenaar TA, et al. Lipid organization of the plasma membrane. J Am Chem Soc. (2014) 136:14554–9. doi: 10.1021/ja507832e
36. Lochner M, Berod L, Sparwasser T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol. (2015) 36:81–91. doi: 10.1016/j.it.2014.12.005
37. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. (2009) 460:103–7. doi: 10.1038/nature08097
38. Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. (2010) 39:171–83. doi: 10.1016/j.molcel.2010.06.022
39. Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev Immunol. (2012) 30:39–68. doi: 10.1146/annurev-immunol-020711-075024
40. Corcoran SE, O'Neill LA. HIF1α and metabolic reprogramming in inflammation. J Clin Invest. (2016) 126:3699–707. doi: 10.1172/JCI84431
41. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
42. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. (2011) 13:1016–23. doi: 10.1038/ncb2329
43. Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev. (2012) 249:43–58. doi: 10.1111/j.1600-065X.2012.01152.x
44. Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. (2009) 122:3589–94. doi: 10.1242/jcs.051011
45. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
46. Liu C, Chapman NM, Karmaus PW, Zeng H, Chi H. mTOR and metabolic regulation of conventional and regulatory T cells. J Leukoc Biol. (2015) 97:837–47. doi: 10.1189/jlb.2RI0814-408R
47. Siska PJ, van der Windt GJ, Kishton RJ, Cohen S, Eisner W, MacIver NJ, et al. Suppression of Glut1 and glucose metabolism by decreased Akt/mTORC1 signaling drives T cell impairment in B cell leukemia. J Immunol. (2016) 97:2532–40. doi: 10.4049/jimmunol.1502464
48. Koromilas AE. Negative regulation of mTORC2 by glycogen synthase kinase-3β: an adaptive process to stress with an anticancer therapeutic potential? Fut Oncol. (2011) 7:845–8. doi: 10.2217/fon.11.59
49. Chen CH, Shaikenov T, Peterson TR, Aimbetov R, Bissenbaev AK, Lee SW, et al. ER stress inhibits mTORC2 and Akt signaling through GSK-3β-mediated phosphorylation of rictor. Sci Signal. (2001) 4:ra10. doi: 10.1126/scisignal.2001731
50. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. (2011) 208:1367–76. doi: 10.1084/jem.20110278
51. Semenza GL. Oxygen sensing, homeostasis, and disease. N Engl J Med. (2011) 365:537–47. doi: 10.1056/NEJMra1011165
52. Semenza GL. Oxygen homeostasis. Wiley Interdiscip Rev Syst Biol Med. (2010) 2:336–61. doi: 10.1002/wsbm.69
53. Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. (1998) 12:149–62.
54. Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. (2008) 8:976–90. doi: 10.1038/nrc2231
55. Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. (2009) 15:6479–83. doi: 10.1158/1078-0432.CCR-09-0889
56. Chou C, Pinto AK, Curtis JD, Persaud SP, Cella M, Lin CC, et al. c-Myc-induced transcription factor AP4 is required for host protection mediated by CD8+ T cells. Nat Immunol. (2014) 15:884–93. doi: 10.1038/ni.2943
57. Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, Yurchenko E, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity. (2015) 42:41–54. doi: 10.1016/j.immuni.2014.12.030
58. Ebert MS, Sharp PA. Roles for microRNAs in conferring robustness to biological processes. Cell. (2012) 149:515–24. doi: 10.1016/j.cell.2012.04.005
59. Ma EH, Poffenberger MC, Wong AH, Jones RG. The role of AMPK in T cell metabolism and function. Curr Opin Immunol. (2017) 46:45–52. doi: 10.1016/j.coi.2017.04.004
60. Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. (2004) 23:4051–60. doi: 10.1038/sj.emboj.7600385
61. Han. J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. The Drosha–DGCR8 complex in primary microRNA processing. Genes Dev. (2004) 18:3016–27. doi: 10.1101/gad.1262504
62. Wu. K, He J, Pu W, Peng Y. The role of Exportin-5 in MicroRNA biogenesis and cancer. Genom Proteom Bioinform. (2018) 16:120–6. doi: 10.1016/j.gpb.2017.09.004
63. Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. (2005) 436:740–4. doi: 10.1038/nature03868
64. Denzler R, McGeary SE, Title AC, Agarwal V, Bartel DP, Stoffel M. Impact of microRNA levels, target-site complementarity, and cooperativity on competing endogenous RNA-regulated gene expression. Mol Cell. (2016) 64:565–79. doi: 10.1016/j.molcel.2016.09.027
65. Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. (2008) 455:64–71. doi: 10.1038/nature07242
66. Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. (2008) 455:58–63. doi: 10.1038/nature07228
67. Pauley KM, Cha S, Chan EK. MicroRNA in autoimmunity and autoimmune diseases. J Autoimmun. (2009) 32:189–94. doi: 10.1016/j.jaut.2009.02.012
68. Esquela-Kerscher A, Slack FJ. Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer. (2006) 6:259–69. doi: 10.1038/nrc1840
69. Mehta A, Baltimore D. MicroRNAs as regulatory elements in immune system logic. Nat Rev Immunol. (2016) 16:279–94. doi: 10.1038/nri.2016.40
70. Jeker LT, Bluestone JA. Small RNA regulators of T cell-mediated autoimmunity. J Clin Immunol. (2010) 30:347–57. doi: 10.1007/s10875-010-9392-7
71. Chong MM, Rasmussen JP, Rudensky AY, Littman DR. The RNAseIII enzyme Drosha is critical in T cells for preventing lethal inflammatory disease. J Exp Med. (2008) 205:2005–17. doi: 10.1084/jem.20071219090508c
72. Kwon SC, Baek SC, Choi YG, Yang J, Lee YS, Woo JS, et al. Molecular basis for the single-nucleotide precision of primary microRNA processing. Mol Cell. (2019) 73:505–18.e5. doi: 10.1016/j.molcel.2018.11.005
73. Cobb BS, Nesterova TB, Thompson E, Hertweck A, O'Connor E, Godwin J, et al. T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J Exp Med. (2005) 201:1367–73. doi: 10.1084/jem.20050572
74. Muljo SA, Ansel KM, Kanellopoulou C, Livingston DM, Rao A, Rajewsky K. Aberrant T cell differentiation in the absence of Dicer. J Exp Med. (2005) 202:261–9. doi: 10.1084/jem.20050678
75. Rossi RL, Rossetti G, Wenandy L, Curti S, Ripamonti A, Bonnal RJ, et al. Distinct microRNA signatures in human lymphocyte subsets and enforcement of the naive state in CD4+ T cells by the microRNA miR-125b. Nat Immunol. (2011) 12:796–803. doi: 10.1038/ni.2057
76. Jindra PT, Bagley J, Godwin JG, Iacomini J. Costimulation-dependent expression of microRNA-214 increases the ability of T cells to proliferate by targeting Pten. J Immunol. (2010) 185:990–7. doi: 10.4049/jimmunol.1000793
77. Stittrich AB, Haftmann C, Sgouroudis E, Kühl AA, Hegazy AN, Panse I, et al. The microRNA miR-182 is induced by IL-2 and promotes clonal expansion of activated helper T lymphocytes. Nat Immunol. (2010) 11:1057–62. doi: 10.1038/ni.1945
78. Kroesen BJ, Teteloshvili N, Smigielska-Czepiel K, Brouwer E, Boots AM, et al. Immuno-miRs: critical regulators of T-cell development, function and ageing. Immunology. (2015) 144:1–10. doi: 10.1111/imm.12367
79. Tsitsiou E, Lindsay MA. microRNAs and the immune response. Curr Opin Pharmacol. (2009) 9:514–20. doi: 10.1016/j.coph.2009.05.003
80. Ha TY. The role of microRNAs in regulatory T cells and in the immune response. Immune Netw. (2011) 11:11–41. doi: 10.4110/in.2011.11.1.11
81. Lu LF, Thai TH, Calado DP, Chaudhry A, Kubo M, Tanaka K, et al. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. (2009) 30:80–91. doi: 10.1016/j.immuni.2008.11.010
82. Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med. (2011) 208:1189–201. doi: 10.1084/jem.20101823
83. Pinweha P, Rattanapornsompong K, Charoensawan V, Jitrapakdee S. MicroRNAs and oncogenic transcriptional regulatory networks controlling metabolic reprogramming in cancers. Comput Struct Biotechnol J. (2016) 14:223–33. doi: 10.1016/j.csbj.2016.05.005
84. Tomasetti M, Amati M, Santarelli L, Neuzil J. MicroRNA in metabolic re-programming and their role in tumorigenesis. Int J Mol Sci. (2016) 17:E754. doi: 10.3390/ijms17050754
85. Frediani JN, Fabbri M. Essential role of miRNAs in orchestrating the biology of the tumor microenvironment. Mol. Cancer. (2016) 15:42. doi: 10.1186/s12943-016-0525-3
86. Henao-Mejia J, Williams A, Goff LA, Staron M, Licona-Limón P, Kaech SM, et al. The microRNA miR-181 is a critical cellular metabolic rheostat essential for NKT cell ontogenesis and lymphocyte development and homeostasis. Immunity. (2013) 38:984–97. doi: 10.1016/j.immuni.2013.02.021
87. Warth SC, Hoefig KP, Hiekel A, Schallenberg S, Jovanovic K, Klein L, et al. Induced miR-99a expression represses Mtor cooperatively with miR-150 to promote regulatory T-cell differentiation. EMBO J. (2015) 34:1195–213. doi: 10.15252/embj.201489589
88. Zhang T, Zhang Z, Li F, Ping Y, Qin G, Zhang C, et al. miR-143 regulates memory T cell differentiation by reprogramming T cell metabolism. J Immunol. (2018) 201:2165–75. doi: 10.4049/jimmunol.1800230
89. King BC, Esguerra JL, Golec E, Eliasson L, Kemper C, Blom AM. CD46 activation regulates miR-150-mediated control of GLUT1 expression and cytokine secretion in human CD4+ T cells. J Immunol. (2016) 196:1636–45. doi: 10.4049/jimmunol.1500516
90. Liu Z, Smith KR, Khong HT, Huang J, Ahn EE, Zhou M, et al. miR-125b regulates differentiation and metabolic reprogramming of T cell acute lymphoblastic leukemia by directly targeting A20. Oncotarget. (2016) 7:78667–79. doi: 10.18632/oncotarget.12018
91. Klein Geltink RI, O'Sullivan D, Corrado M, Bremser A, Buck MD, Buescher JM, et al. Mitochondrial priming by CD28. Cell. (2017) 171:385–97.e11. doi: 10.1016/j.cell.2017.08.018
92. Ouimet M, Ediriweera HN, Gundra UM, Sheedy FJ, Ramkhelawon B, Hutchison SB, et al. MicroRNA-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J Clin Invest. (2015) 125:4334–48. doi: 10.1172/JCI81676
93. Marcais A, Blevins R, Graumann J, Feytout A, Dharmalingam G, Carroll T, et al. microRNA-mediated regulation of mTOR complex components facilitates discrimination between activation and anergy in CD4 T cells. J Exp Med. (2014) 211:2281–95. doi: 10.1084/jem.20132059
94. Merkenschlager M, Marcais A. microRNAs calibrate T cell responses by regulating mTOR. Oncotarget. (2015) 6:34059–60. doi: 10.18632/oncotarget.6107
95. Zeng Z, Wang K, Li Y, Xia N, Nie S, Lv B, et al. Down-regulation of microRNA-451a facilitates the activation and proliferation of CD4(+) T cells by targeting Myc in patients with dilated cardiomyopathy. J Biol Chem. (2017) 292:6004–13. doi: 10.1074/jbc.M116.765107
96. Ji Y, Hocker JD, Gattinoni L. Enhancing adoptive T cell immunotherapy with microRNA therapeutics. Semin Immunol. (2016) 28:45–53. doi: 10.1016/j.smim.2015.11.006
97. Rodríguez-Galán A, Fernández-Messina L, Sánchez-Madrid F. Control of immunoregulatory molecules by miRNAs in T cell activation. Front Immunol. (2018) 9:2148. doi: 10.3389/fimmu.2018.02148
98. Yao Q, Song Z, Wang B, Zhang JA. Emerging roles of microRNAs in the metabolic control of immune cells. Cancer Lett. (2018) 433:10–7. doi: 10.1016/j.canlet.2018.06.024
99. O'Connell RM, Rao DS, Baltimore D. microRNA regulation of inflammatory responses. Annu Rev Immunol. (2012) 30:295–312. doi: 10.1146/annurev-immunol-020711-075013
100. Long H, Wang X, Chen Y, Wang L, Zhao M, Lu Q. Dysregulation of microRNAs in autoimmune diseases: Pathogenesis, biomarkers and potential therapeutic targets. Cancer Lett. (2018) 428:90–103. doi: 10.1016/j.canlet.2018.04.016
101. Theofilopoulos AN, Kono DH, Baccala R. The multiple pathways to autoimmunity. Nat Immunol. (2017) 18:716–24. doi: 10.1038/ni.3731
102. Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet. (2014) 383:69–82. doi: 10.1016/S0140-6736(13)60591-7
103. Furst DE, Clarke AE, Fernandes AW, Bancroft T, Greth W, Iorga SR. Incidence and prevalence of adult systemic lupus erythematosus in a large US managed-care population. Lupus. (2013) 22:99–105. doi: 10.1177/0961203312463110
104. Koch-Henriksen N, Sørensen PS. The changing demographic pattern of multiple sclerosis epidemiology. Lancet Neurol. (2010) 9:520–32. doi: 10.1016/S1474-4422(10)70064-8
105. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol. (2015) 16:1174–84. doi: 10.1038/ni.3269
106. Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, et al. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. (2015) 7:274ra18. doi: 10.1126/scitranslmed.aaa0835
107. Yin Y, Choi SC, Xu Z, Zeumer L, Kanda N, Croker BP, et al. Glucose oxidation is critical for CD4+ T cell activation in a mouse model of systemic lupus erythematosus. J Immunol. (2016) 196:80–90. doi: 10.4049/jimmunol.1501537
108. Nath N, Giri S, Prasad R, Salem ML, Singh AK, Singh I. 5-Aminoimidazole-4-carboxamide ribonucleoside: a novel immunomodulator with therapeutic efficacy in experimental autoimmune encephalomyelitis. J Immunol. (2005) 175:566–74. doi: 10.4049/jimmunol.175.1.566
109. Carbone F, De Rosa V, Carrieri PB, Montella S, Bruzzese D, Porcellini A, et al. Regulatory T cell proliferative potential is impaired in human autoimmune disease. Nat Med. (2014) 20:69–74. doi: 10.1038/nm.3411
110. Procaccini C, De Rosa V, Galgani M, Abanni L, Calì G, Porcellini A, et al. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity. (2010) 33:929–41. doi: 10.1016/j.immuni.2010.11.024
111. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature. (2013) 499:485–90. doi: 10.1038/nature12297
113. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol. (2010) 185:1037–44. doi: 10.4049/jimmunol.0903586
114. Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. (2013) 153:1239–51. doi: 10.1016/j.cell.2013.05.016
115. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. (2013) 38:225–36. doi: 10.1016/j.immuni.2012.10.020
116. Amersfoort J, Kuiper J. T cell metabolism in metabolic disease-associated autoimmunity. Immunobiology. (2017) 222:925–36. doi: 10.1016/j.imbio.2017.03.001
117. van der Windt GJ, O'Sullivan D, Everts B, Huang SC, Buck MD, Curtis JD, et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci USA. (2013) 110:14336–41. doi: 10.1073/pnas.1221740110
118. Katsarou A, Gudbjörnsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, et al. Type 1 diabetes mellitus. Nat Rev Dis Primers. (2017) 3:17016. doi: 10.1038/nrdp.2017.16
119. Atkinson MA. Losing a grip on the notion of β-cell specificity for immune responses in type 1 diabetes: can we handle the truth? Diabetes. (2014) 63:3572–4. doi: 10.2337/db14-1069
120. Stentz FB, Kitabchi AE. Hyperglycemia-induced activation of human T-lymphocytes with de novo emergence of insulin receptors and generation of reactive oxygen species. Biochem Biophys Res Commun. (2005) 335:491–5. doi: 10.1016/j.bbrc.2005.07.109
121. Frost RJ, Olson EN. Control of glucose homeostasis and insulin sensitivity by the Let-7 family of microRNAs. Proc Natl Acad Sci USA. (2011) 108:21075–80. doi: 10.1073/pnas.1118922109
122. Zhu H, Shyh-Chang N, Segrè AV, Shinoda G, Shah SP, Einhorn WS, et al. The Lin28/let-7 axis regulates glucose metabolism. Cell. (2011) 147:81–94. doi: 10.1016/j.cell.2011.08.033
123. Erener S, Marwaha A, Tan R, Panagiotopoulos C, Kieffer TJ. Profiling of circulating microRNAs in children with recent onset of type 1 diabetes JCI. Insight. (2017) 2:e89656. doi: 10.1172/jci.insight.89656
124. Roush S, Slack FJ. The let-7 family of microRNAs. Trends Cell Biol. (2008) 18:505–16. doi: 10.1016/j.tcb.2008.07.007
125. Wells AC, Daniels KA, Angelou CC, Fagerberg E, Burnside AS, Markstein M, et al. Modulation of let-7 miRNAs controls the differentiation of effector CD8 T cells. Elife. (2017) 6:e26398. doi: 10.7554/eLife.26398
126. Collares CV, Evangelista AF, Xavier DJ, Rassi DM, Arns T, Foss-Freitas MC, et al. Identifying common and specific microRNAs expressed in peripheral blood mononuclear cell of type 1, type 2, and gestational diabetes mellitus patients. BMC Res Notes. (2013) 6:491. doi: 10.1186/1756-0500-6-491
127. Åkerman L, Casas R, Ludvigsson J, Tavira B, Skoglund C. Serum miRNA levels are related to glucose homeostasis and islet autoantibodies in children with high risk for type 1 diabetes. PLoS ONE. (2018) 13:e0191067. doi: 10.1371/journal.pone.0191067
128. Krist B, Florczyk U, Pietraszek-Gremplewicz K, Józkowicz A, Dulak J. The role of miR-378a in metabolism, angiogenesis, and muscle biology. Int J Endocrinol. (2015) 2015:281756. doi: 10.1155/2015/281756
129. Carrer M, Liu N, Grueter CE, Williams AH, Frisard MI, Hulver MW, et al. Control of mitochondrial metabolism and systemic energy homeostasis by microRNAs 378 and 378*. Proc Natl Acad Sci USA. (2012) 109:15330–5. doi: 10.1073/pnas.1207605109
130. Zhou L, He H, Mi JX, Li C, Lee B, Mi QS. MicroRNA genes. Ann N Y Acad Sci. (2008) 1150:72–5. doi: 10.1196/annals.1447.026
131. Ma H, Zhang S, Shi D, Mao Y, Cui J. MicroRNA-26a promotes regulatory T cells and suppresses autoimmune diabetes in mice. Inflammation. (2016) 39:1–9. doi: 10.1007/s10753-015-0215-0
132. Honardoost MA, Kiani-Esfahani A, Ghaedi K, Etemadifar M, Salehi M. miR-326 and miR-26a, two potential markers for diagnosis of relapse and remission phases in patient with relapsing-remitting multiple sclerosis. Gene. (2014) 544:128–33. doi: 10.1016/j.gene.2014.04.069
133. Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol. (2016) 17:95–103. doi: 10.1038/ni.3313
134. Coit P, Dozmorov MG, Merrill JT, McCune WJ, Maksimowicz-McKinnon K, Wren JD, et al. Epigenetic reprogramming in naive CD4+ T cells favoring T cell activation and non-Th1 effector T cell immune response as an early event in lupus flares. Arthritis Rheum. (2016) 68:2200–9. doi: 10.1002/art.39720
135. Chen S, Wang Y, Qin H, Lin J, Xie L, Chen S, et al. Downregulation of miR-633 activated AKT/mTOR pathway by targeting AKT1 in lupus CD4(+) T cells. Lupus. (2019) 28:510–9. doi: 10.1177/0961203319829853
136. Jafari Ghods F, Topal Sarikaya A, Arda N, Hamuryudan V. MiRNA and mRNA profiling in systemic lupus reveals a novel set of cytokine-related miRNAs and their target genes in cases with and without renal involvement. Kidney Blood Press Res. (2017) 42:1322–37. doi: 10.1159/000485987
137. Guo G, Wang H, Shi X, Ye L, Wu K, Lin K, et al. Novel miRNA-25 inhibits AMPD2 in peripheral blood mononuclear cells of patients with systemic lupus erythematosus and represents a promising novel biomarker. J Transl Med. (2018) 16:370. doi: 10.1186/s12967-018-1739-5
138. Liu SQ, Jiang S, Li C, Zhang B, Li QJ. miR-17-92 cluster targets phosphatase and tensin homology and Ikaros Family Zinc Finger 4 to promote TH17-mediated inflammation. J Biol Chem. (2014) 289:12446–56. doi: 10.1074/jbc.M114.550723
139. Venkatesha SH, Dudics S, Song Y, Mahurkar A, Moudgil KD. The miRNA expression profile of experimental autoimmune encephalomyelitis reveals novel potential disease biomarkers. Int J Mol Sci. (2018) 19:E3990. doi: 10.3390/ijms19123990
140. Jin Y, Tymen SD, Chen D, Fang ZJ, Zhao Y, Dragas D, et al. MicroRNA-99 family targets AKT/mTOR signaling pathway in dermal wound healing. PLoS ONE. (2013) 8:e64434. doi: 10.1371/journal.pone.0064434
141. Cao F, Liu T, Sun S, Feng S. The role of the miR-99b-5p/mTOR signaling pathway in neuroregeneration in mice following spinal cord injury. Mol Med Rep. (2017) 16:9355–60. doi: 10.3892/mmr.2017.7816
142. Liguori M, Nuzziello N, Licciulli F, Consiglio A, Simone M, Viterbo RG, et al. Combined microRNA and mRNA expression analysis in pediatric multiple sclerosis: an integrated approach to uncover novel pathogenic mechanisms of the disease. Hum Mol Genet. (2018) 27:66–79. doi: 10.1093/hmg/ddx385
143. Ntranos A, Ntranos V, Bonnefil V, Liu J, Kim-Schulze S, He Y, et al. Fumarates target the metabolic-epigenetic interplay of brain-homing T cells in multiple sclerosis. Brain. (2019) 142:647–61. doi: 10.1093/brain/awy344
144. Fenoglio C, Cantoni C, De Riz M, Ridolfi E, Cortini F, Serpente M, et al. Expression and genetic analysis of miRNAs involved in CD4+ cell activation in patients with multiple sclerosis. Neurosci Lett. (2011) 504:9–12. doi: 10.1016/j.neulet.2011.08.021
145. Mycko MP, Cichalewska M, Machlanska A, Cwiklinska H, Mariasiewicz M, Selmaj KW. MicroRNA-301a regulation of a T-helper 17 immune response controls autoimmune demyelination. Proc Natl Acad Sci USA. (2012) 109:E1248–57. doi: 10.1073/pnas.1114325109
146. Murugaiyan G, da Cunha AP, Ajay AK, Joller N, Garo LP, Kumaradevan S, et al. MicroRNA-21 promotes Th17 differentiation and mediates experimental autoimmune encephalomyelitis. J Clin Invest. (2015) 125:1069–80. doi: 10.1172/JCI74347
147. Yang XP, Jiang K, Hirahara K, Vahedi G, Afzali B, Sciume G, et al. EZH2 is crucial for both differentiation of regulatory T cells and T effector cell expansion. Sci Rep. (2015) 5:10643. doi: 10.1038/srep10643
148. Yin Y, Qiu S, Peng Y. Functional roles of enhancer of zeste homolog 2 in gliomas. Gene. (2016) 576:189–94. doi: 10.1016/j.gene.2015.09.080
149. Perl A, Gergely P Jr, Banki K. Mitochondrial dysfunction in T cells of patients with systemic lupus erythematosus. Int Rev Immunol. (2004) 23:293–313. doi: 10.1080/08830180490452576
150. Perl A, Hanczko R, Doherty E. Assessment of mitochondrial dysfunction in lymphocytes of patients with systemic lupus erythematosus. Methods Mol Biol. (2012) 900:61–89. doi: 10.1007/978-1-60761-720-4_4
151. Morel L. Immunometabolism in systemic lupus erythematosus. Nat Rev Rheumatol. (2017) 13:280–90. doi: 10.1038/nrrheum.2017.43
152. Katsuyama T, Tsokos GC, Moulton VR. Aberrant T cell signaling and subsets in systemic lupus erythematosus. Front Immunol. (2018) 9:1088. doi: 10.3389/fimmu.2018.01088
153. Gergely P, Jr, Grossman C, Niland B, Puskas F, Neupane H, Allam F, et al. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. (2002) 46:175–90. doi: 10.1002/1529-0131(200201)46:1<175::AID-ART10015>3.0.CO;2-H
154. Perl A, Hanczko R, Telarico T, Oaks Z, Landas S. Oxidative stress, inflammation and carcinogenesis are controlled through the pentose phosphate pathway by transaldolase. Trends Mol Med. (2011) 17:395–403. doi: 10.1016/j.molmed.2011.01.014
155. Fernandez D, Perl A. mTOR signaling: a central pathway to pathogenesis in systemic lupus erythematosus? Discov Med. (2010) 9:173–8.
156. Lai ZW, Borsuk R, Shadakshari A, Yu J, Dawood M, Garcia R, et al. Mechanistic target of rapamycin activation triggers IL-4 production and necrotic death of double-negative T cells in patients with systemic lupus erythematosus. J Immunol. (2013) 191:2236–46. doi: 10.4049/jimmunol.1301005
157. Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5'-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther. (2006) 112:358–404. doi: 10.1016/j.pharmthera.2005.04.013
158. Junker A, Hohlfeld R, Meinl E. The emerging role of microRNAs in multiple sclerosis. Nat Rev Neurol. (2011) 7:56–9. doi: 10.1038/nrneurol.2010.179
159. Chen C, Zhou Y, Wang J, Yan Y, Peng L, Qiu W. Dysregulated microRNA involvement in multiple sclerosis by induction of T helper 17 cell differentiation. Front Immunol. (2018) 9:1256. doi: 10.3389/fimmu.2018.01256
160. Liu C, Yang H, Shi W, Wang T, Ruan Q. MicroRNA-mediated regulation of T helper type 17/regulatory T-cell balance in autoimmune disease. Immunology. (2018) 155:427–34. doi: 10.1111/imm.12994
161. Compston A. The pathogenesis and basis for treatment in multiple sclerosis. Clin Neurol Neurosurg. (2004) 106:246–8. doi: 10.1016/j.clineuro.2004.02.007
162. Vallée A, Lecarpentier Y, Guillevin R, Vallée JN. Demyelination in multiple sclerosis: Reprogramming energy metabolism and potential PPARγ agonist treatment approaches. Int J Mol Sci. (2018) 19:E1212. doi: 10.3390/ijms19041212
163. Yuan S, Shi Y, Tang SJ. Wnt signaling in the pathogenesis of multiple sclerosis-associated chronic pain. J Neuroimmune Pharmacol. (2012) 7:904–13. doi: 10.1007/s11481-012-9370-3
164. Procaccini C, Santopaolo M, Faicchia D, Colamatteo A, Formisano L, de Candia P, et al. Role of metabolism in neurodegenerative disorders. Metabolism. (2016) 65:1376–90. doi: 10.1016/j.metabol.2016.05.018
165. La Rocca C, Carbone F, De Rosa V, Colamatteo A, Galgani M, Perna F, et al. Immunometabolic profiling of T cells from patients with relapsing-remitting multiple sclerosis reveals an impairment in glycolysis and mitochondrial respiration. Metabolism. (2017) 77:39–46. doi: 10.1016/j.metabol.2017.08.011
166. Liu Y, Zhang DT, Liu XG. mTOR signaling in T cell immunity and autoimmunity. Int Rev Immunol. (2015) 34:50–66. doi: 10.3109/08830185.2014.933957
167. Giacoppo S, Pollastro F, Grassi G, Bramanti P, Mazzon E. Target regulation of PI3K/Akt/mTOR pathway by cannabidiol in treatment of experimental multiple sclerosis. Fitoterapia. (2017) 116:77–84. doi: 10.1016/j.fitote.2016.11.010
168. Mammana S, Bramanti P, Mazzon E, Cavalli E, Basile MS, Fagone P, et al. Preclinical evaluation of the PI3K/Akt/mTOR pathway in animal models of multiple sclerosis. Oncotarget. (2018) 9:8263–77. doi: 10.18632/oncotarget.23862
169. Salmond RJ. mTOR regulation of glycolytic metabolism in T cells. Front Cell Dev Biol. (2018) 6:122. doi: 10.3389/fcell.2018.00122
170. Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. (2002) 110:163–75. doi: 10.1016/s0092-8674(02)00808-5
Keywords: T cells, metabolic regulation, immunometabolism, miRNAs, autoimmune diseases
Citation: Colamatteo A, Micillo T, Bruzzaniti S, Fusco C, Garavelli S, De Rosa V, Galgani M, Spagnuolo MI, Di Rella F, Puca AA, de Candia P and Matarese G (2019) Metabolism and Autoimmune Responses: The microRNA Connection. Front. Immunol. 10:1969. doi: 10.3389/fimmu.2019.01969
Received: 14 June 2019; Accepted: 05 August 2019;
Published: 28 August 2019.
Edited by:
Anna Ohradanova-Repic, Medical University of Vienna, AustriaReviewed by:
Stephan Blüml, Medical University of Vienna, AustriaFederica Marelli-Berg, Queen Mary University of London, United Kingdom
Copyright © 2019 Colamatteo, Micillo, Bruzzaniti, Fusco, Garavelli, De Rosa, Galgani, Spagnuolo, Di Rella, Puca, de Candia and Matarese. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppe Matarese, giuseppe.matarese@unina.it