 Vilma Mantovani1,2†
Vilma Mantovani1,2† Sofia Bin2,3†
Sofia Bin2,3† Claudio Graziano1
Claudio Graziano1 Irene Capelli3
Irene Capelli3 Raffaella Minardi2,3
Raffaella Minardi2,3 Valeria Aiello3
Valeria Aiello3 Enrico Ambrosini1
Enrico Ambrosini1 Carlotta Pia Cristalli2,3
Carlotta Pia Cristalli2,3 Alessandro Mattiaccio2
Alessandro Mattiaccio2 Milena Pariali2
Milena Pariali2 Sara De Fanti4
Sara De Fanti4 Flavio Faletra5
Flavio Faletra5 Enrico Grosso6
Enrico Grosso6 Rachele Cantone6
Rachele Cantone6 Elena Mancini7
Elena Mancini7 Francesca Mencarelli8
Francesca Mencarelli8 Andrea Pasini8
Andrea Pasini8 Anita Wischmeijer9
Anita Wischmeijer9 Nicola Sciascia10
Nicola Sciascia10 Marco Seri2
Marco Seri2 Gaetano La Manna3*
Gaetano La Manna3*- 1Medical Genetics Unit, S. Orsola-Malpighi University Hospital, Bologna, Italy
- 2Center for Applied Biomedical Research (CRBA), University of Bologna, Bologna, Italy
- 3Nephrology, Dialysis and Transplantation Unit, Department of Experimental, Diagnostic and Specialty Medicine (DIMES), S. Orsola-Malpighi University Hospital, Bologna, Italy
- 4Department of Biological, Geological and Environmental Sciences, University of Bologna, Bologna, Italy
- 5Medical Genetics Unit, Institute for Maternal and Child Health - IRCCS “Burlo Garofolo”, Trieste, Italy
- 6Medical Genetics Unit, AOU Città della Salute e della Scienza, Turin, Italy
- 7Nephrology, Dialysis and Hypertension Unit, S. Orsola-Malpighi University Hospital, Bologna, Italy
- 8Pediatrics Unit, S. Orsola-Malpighi University Hospital, Bologna, Italy
- 9Clinical Genetics Service and South Tyrol Coordination Center for Rare Diseases, Department of Pediatrics, Regional Hospital of Bolzano, Bolzano, Italy
- 10Radiology Unit, S. Orsola-Malpighi University Hospital, Bologna, Italy
Introduction: Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common inherited disorders in humans and the majority of patients carry a variant in either PKD1 or PKD2. Genetic testing is increasingly required for diagnosis, prognosis, and treatment decision, but it is challenging due to segmental duplications of PKD1, genetic and allelic heterogeneity, and the presence of many variants hypomorphic or of uncertain significance. We propose an NGS-based testing strategy for molecular analysis of ADPKD and its phenocopies, validated in a diagnostic setting.
Materials and Methods: Our protocol is based on high-throughput simultaneous sequencing of PKD1 and PKD2 after long range PCR of coding regions, followed by a masked reference genome alignment, and MLPA analysis. A further screening of additional 14 cystogenes was performed in negative cases. We applied this strategy to analyze 212 patients with a clinical suspicion of ADPKD.
Results and Discussion: We detected causative variants (interpreted as pathogenic/likely pathogenic) in 61.3% of our index patients, and variants of uncertain clinical significance in 12.5%. The majority (88%) of genetic variants was identified in PKD1, 12% in PKD2. Among 158 distinct variants, 80 (50.6%) were previously unreported, confirming broad allelic heterogeneity. Eleven patients showed more than one variant. Segregation analysis indicated biallelic disease in five patients, digenic in one, de novo variant with unknown phase in two. Furthermore, our NGS protocol allowed the identification of two patients with somatic mosaicism, which was undetectable with Sanger sequencing.
Among patients without PKD1/PKD2 variants, we identified three with possible alternative diagnosis: a patient with biallelic mutations in PKHD1, confirming the overlap between recessive and dominant PKD, and two patients with variants in ALG8 and PRKCSH, respectively.
Genotype-phenotype correlations showed that patients with PKD1 variants predicted to truncate (T) the protein experienced end-stage renal disease 9 years earlier than patients with PKD1 non-truncating (NT) mutations and >13 years earlier than patients with PKD2 mutations. ADPKD-PKD1T cases showed a disease onset significantly earlier than ADPKD-PKD1NT and ADPK-PKD2, as well as a significant earlier diagnosis. These data emphasize the need to combine clinical information with genetic data to achieve useful prognostic predictions.
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common inherited disorders in humans, with an estimated prevalence between 1/400 and 1/1000 in the general population (Torres et al., 2007). It is characterized by the development and progressive enlargement of renal cysts, which often leads to end-stage renal disease (ESRD) (Gabow, 1993; Grantham et al., 2011). ADPKD is mainly caused by mutation in either PKD1 (74–85% of patients) or PKD2 (15–26%) (Rossetti et al., 2007; Barua et al., 2009; Carrera et al., 2016), with PKD2 variants being relatively more frequent in Japan (Kurashige et al., 2015). Furthermore, mutations in GANAB and DNAJB11 were recently shown to cause polycystic kidney in a small percentage (0.3%) of affected subjects (Porath et al., 2016; Cornec-Le Gall et al., 2018). Patients with PKD1 mutations usually develop a more severe disease than patients with PKD2, with more cysts at an earlier age (Hateboer et al., 1999; Torres et al., 2007), and truncating mutations in PKD1 lead to earlier ESRD than non-truncating ones (Cornec-Le Gall et al., 2013). Intra and inter familial disease severity is also reported, suggesting a role for environmental factors and the genetic background. Additional genetic defects are sometime identified in rare cases with early onset disease, i.e., mutations in the second PKD allele, or in other genes such as PKHD1 or HNF1B (Rossetti et al., 2009; Bergmann et al., 2011; Audrézet et al., 2016). Somatic mosaicism and hypomorphic alleles contribute to further increase the genetic complexity of this condition (Connor et al., 2008; Rossetti et al., 2009; Vujic et al., 2010).
Although the diagnosis of ADPKD is usually based on imaging studies, a genetic test is currently indicated in patients without a positive family history, with atypical clinical or imaging features, and in helping the identification of a familial donor for renal transplantation. In addition, a genetic diagnosis may guide clinical management and treatments, and it may improve the accuracy of genetic counseling. Genetic analysis is not uniformly required by nephrologists in standard clinical practice. However, information on specific mutations may help to predict disease progression using the PROPKD score, which can guide the selection of patients who will best benefit from the use of Tolvaptan, a vasopressin 2 receptor antagonist (Cornec-Le Gall et al., 2016, 2019).
The cost of analysis and technical challenges prevented a widespread application of PKD1 sequencing. This gene has six pseudogenes with high sequence homology, in a duplicated region adjacent to the original PKD1 locus. In addition, its large size and high GC content, the extensive allelic heterogeneity of PKD1 (and PKD2 as well) and genetic heterogeneity further complicate the molecular screening. Next-generation sequencing (NGS) technologies revolutionized the approach to molecular diagnostics and have recently been applied to ADPKD as well, with different strategies. Targeted re-sequencing was performed by generating long-range (LR) locus-specific PCR amplicons and multiplexing them from individuals samples as bar-coded libraries for NGS (Rossetti et al., 2012; Tan et al., 2014; Kinoshita et al., 2016). An alternative approach was a sequence capture-based NGS, with improved algorithms in order to overcome unspecific capture of pseudogenes (Trujillano et al., 2014; Eisenberger et al., 2015). A long-read single-molecule sequencing was recently suggested as a strategy to overcome PKD1 complexities (Borràs et al., 2017). Finally, a comprehensive approach with 140 candidate gene-panel for cystic and glomerular nephropathies has been proposed (Bullich et al., 2018). All these methods have strengths and pitfalls, and should be further validated.
Here, we propose an NGS-based testing strategy for molecular diagnosis of ADPKD and its phenocopies, validated in a diagnostic setting. Our workflow included LR-PCR targeted re-sequencing for both PKD1 and PKD2, combined with a specific alignment pipeline for PKD1. Large rearrangements were assessed by multiplex-ligation probe amplification (MLPA). Additional cystogenes, namely GANAB, DNAJB11, LRP5, PMM2, PRKCSH, SEC63, SEC61B, ALG8, PKHD1, DZIP1L, HNF1B, UMOD, INF2, and REN (Porath et al., 2016; Cornec-Le Gall et al., 2017; Bullich et al., 2018), were also tested by NGS targeted re-sequencing in PKD1/2 negative patients, after clinical revision.
Materials and Methods
Patient Recruitment
Most patients were recruited from the Units of Nephrology and Medical Genetics of Bologna University Hospital, a few from partner centers. Written informed consents for genetic analysis and publication of results were obtained from the patients or their parents/guardians in compliance with national ethics regulation. The families received pre- and post-test genetic counseling.
The 212 patients enrolled in the study belonged to the following three groups: (1) a validation cohort included 21 ADPKD patients that had previously undergone genetic diagnosis by Sanger sequencing of PKD1 and PKD2; (2) a group of 36 patients with a clinical diagnosis of severe ADPKD, who had already received a kidney transplantation, was included as a confirmation cohort; (3) a third group was a discovery cohort, including 155 unrelated patients who had been consecutively referred for ADPKD genetic testing from 2014 to 2018. In addition, 143 family members were tested for the possible pathogenic variants detected in the probands.
Main clinical features of patients submitted to the study are summarized in Table 1, while the most significant clinical histories are extrapolated and described in the Results section.
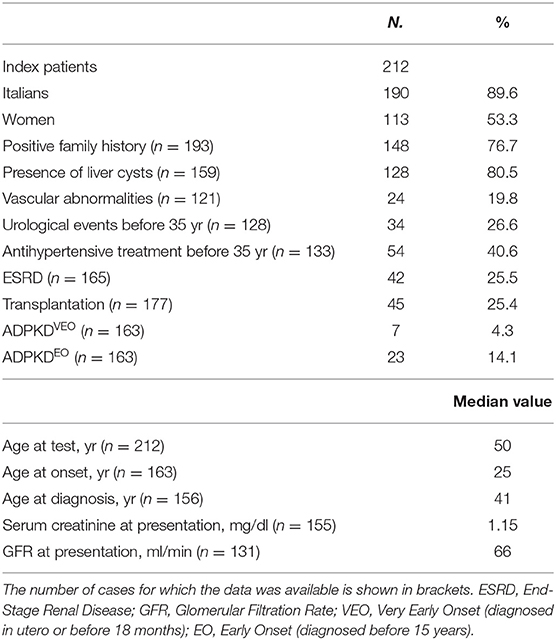
Table 1. Clinical characteristics of study patients.
The diagnosis of ADPKD relies primarily on imaging, although some cases are diagnosed by genetic testing. Typical ultrasonography imaging reveals large kidneys with multiple bilateral cysts. Imaging is also the first choice for pre-symptomatic diagnosis of patients with a positive family history. Specifically, Pei's criteria suggest that the presence of a total of three or more kidney cysts for at-risk individuals aged 15–39 years and 2 or more cysts in each kidney for at-risk individuals aged 40–59 years are sufficient for a clinical diagnosis. The absence of kidney cysts on ultrasound excludes ADPKD in patients older than 40 years. If ultrasonography images are equivocal, magnetic resonance imaging (MRI) or computed tomography (CT) may clarify the diagnosis (Pei et al., 2009).
Library Preparation
Genomic DNA was isolated from EDTA peripheral blood using the semi-automatic Maxwell 16 instrument (Promega Corporation, Madison, WI USA).
The PKD1 (MIM#601313 HGNC:9008, RefSeq NM _001009944.2) entire coding region, including exon-intron boundaries (at least 50 bp) and most of the 5′ and 3′ untranslated regions were amplified in eight long-range (LR) PCR using specific primers anchored on the rare mismatch sequencing between PKD1 and pseudogenes. We combined previously described primers (Audrézet et al., 2012; Tan et al., 2014; Kinoshita et al., 2016) with primers designed by us and we optimized the PCR protocols. A LR-PCR method was validated also for PKD2 (MIM#173910 HGNC:9009, RefSeq NM_000297.3), using previously described primers (Tan et al., 2014). Primers and PCR conditions are reported in Supplementary Table 1.
For each patient, all 13 LR-PCR products (37.8 kb for PKD1 and 35.9 kb for PKD2) were pooled together in equimolar manner (50 pM each), the pools were then subjected to enzymatic digestion using Ion Xpress Plus Fragment Library Kit, in order to obtain about 200 bp fragments. Fragmented LR-PCRs were used to construct barcoded libraries using Ion Xpress Barcode Adapters following the manufacturer's protocol. A single barcode was used for simultaneous analysis of both genes for each patient. Size selection of fragments was performed by E-Gel Size Select II 2% agarose gel (Thermo Fisher Scientific Inc.).
Patients without PKD1/PKD2 genetic defects were revised by clinicians and, if appropriate, were tested for mutations in additional genes. To this aim, we developed a multiplexed PCR NGS for a panel including the novel described ADPKD genes (GANAB, MIM#600666 HGNC:4138 RefSeq NM_198335.3; DNAJB11, MIM#611341 HGNC:14889 RefSeq NM_016306.5), the autosomal dominant polycystic liver disease (ADPLD) genes (LRP5, MIM#603506 HGNC:6697 RefSeq NM_002335.3; PMM2 MIM#601785 HGNC:9115 RefSeq NM_000303.2; PRKCSH, MIM#177060 HGNC:9411 RefSeq NM_002743.3; SEC63, MIM#608648 HGNC:21082 RefSeq NM_007214.4; SEC61B, MIM#609214 HGNC:16993 RefSeq NM_006808.2; ALG8, MIM#608103 HGNC:23161 RefSeq NM_024079.4), the two genes for ARPKD (PKHD1, MIM#606702 HGNC:9016 RefSeq NM_138694.3 and DZIP1L, MIM#617570 HGNC:26551 RefSeq NM_173543.2), and additional cystogenes (HNF1B, MIM#189907 HGNC:11630 RefSeq NM_000458.3; UMOD, MIM#191845 HGNC:12559 RefSeq NM_001008389.2; INF2, MIM#610982 HGNC:23791 RefSeq NM_022489.3; REN, MIM#179820 HGNC:9958 RefSeq NM_000537.3). Amplicon-based libraries were generated from 10 ng of DNA per sample using Ion AmpliSeq Library Kit Plus according to the manufacturer's instructions (Thermo Fisher Scientific Inc.).
Template Generation and Sequencing
The barcoded libraries were quantified using Ion Library TaqMan Quantitation Kit and pooled in equimolar manner (9 pM), then the emulsion PCR was performed using the Ion OneTouch 2 System (Thermo Fisher Scientific Inc.). The enriched emulsion-PCR was then prepared for sequencing protocol using Ion PGM Hi-Q Sequencing Kit, loaded onto an Ion 318v2 chip and sequenced with the Ion Personal Genome Machine (PGM). For PKD1 and PKD2 genes, 12 patients were simultaneously analyzed in each chip. Recently, we introduced the HID Ion Chef instrument for emulsion PCR and enrichment, united with Ion Gene Studio System S5 (Thermo Fisher Scientific Inc). We simultaneously analyzed 16 patients on an Ion 520 chip. For the other genes, the number of patients per chip has been calculated to obtain an average cover of at least 500x.
Data Analysis
The raw sequencing data were transferred to the Torrent Server where the Torrent SuiteTM performed the alignment to a reference genome in order to generate Fastq files, Binary Alignment Map (BAM) in conjunction with Binary Alignment Index (BAI) and Variant Call Format (VCF) files. PKD1 reads were aligned against a modified reference genome based on chromosome 16 of Human Genome 38 (Grch38), where all the nucleotides outside the PKD1 locus were masked and replaced with “Ns.” All the VCF files were uploaded into Ion Reporter software (Thermo Fisher Scientific Inc.) selecting the Annotation Variant workflow in order to associate to each variant the nucleotide change in mRNA transcript, the aminoacidic change, the exons or IVSs and the function.
The BAM/BAI files, generated following the alignment, were visualized using Integrative Genome Viewer (IGV) software1. IGV was used to assess the depth of coverage of the sequencing reads, zygosity, quality of the sequencing reads and the mapping quality.
Filtering and Classification of the Gene Variants
Variant filtering based on population frequency was performed using population databases ExAC, gnomAD (Lek et al., 2016), 1000Genomes (1000 Genomes Project Consortium et al., 2015) and dbSNP database to include only rare alleles (minor allele frequency ≤1%).
The variants were then annotated according to the guidelines published by the Human Genome Variation Society2 (Den Dunnen and Antonarakis, 2000) and classified in five categories, according to the American College of Medical Genetics and Genomics (ACMG) standards (Richards et al., 2015). Segregation analysis was performed in additional affected family members whenever possible. The protein truncating (PT) mutations (frameshift, non-sense, canonical splice-site and large-gene rearrangements) were considered pathogenic (P). The non-truncating (NT) variants (in-frame insertion/deletion, non-synonymous and synonymous variants) were attributed to one of the other four categories, by using the VarSomeClinical platform3. We also checked variants in mutation database PKDB4, in Leiden Open Variations PKD Database5, in Human Gene Mutation Database (Stenson et al., 2017), in ClinVar archive6 and by segregation analysis, and we re-classified them when appropriate.
In addition to the five categories defined by ACMG standards, we classified some PKD variants as hypomorphic (H), according to the literature or databases. We also re-classified H some non-truncating PKD1 alleles showing incomplete penetrance through family study.
Missense variants predicted to be neutral, rare synonymous and intronic variants predicted as not affecting function, deep intronic and frequent synonymous variants in non-conserved sequence domains were classified likely benign (LB) or benign (B) and are not reported.
We performed an additional bioinformatics analysis for missense variants of uncertain significance (VUS) and for H variants by using PyMOL, an open source molecular graphics system 2.0 Schrodinger, LLC7. Five variants in PKD1 and two variants in PKD2, lying in the crystalized portion of the proteins (PDB number: 6A70), were submitted to the tool.
Sanger Sequencing and MLPA
All the variants considered as pathogenic, likely pathogenic and VUS were confirmed by Sanger sequencing on 3730 DNA Analyzer (Applied Biosystem, Foster City, CA, USA). Coding and splicing regions with low coverage (<20x) have also been sequenced by the Sanger method. For the exons 1-33 of PKD1, we performed a nested PCR of the interested region from LR amplicons before proceeding to Sanger sequencing.
Samples without variants classified as P or LP in PKD1/PKD2, including those presenting a VUS, were analyzed by MLPA for detection of large gene rearrangements, using commercially kits SALSA MLPA P351 PKD1 and SALSA MLPA P352 PKD1-PKD2 probemix (MRC-Holland, Amsterdam, NL). Fragment analysis of the PCR product was carried out on 3730 DNA Analyzer and the electropherograms were analyzed by Coffalyzer software (MRC Holland, Amsterdam, NL).
Statistical Analyses
Continuous variables were reported as mean if normally distributed and median if not, and discrete variables were reported as percentages. The statistical significance has been tested by Pearson's Goodness of Fit Chi-square for categorical variables. Fisher Exact test has been applied in the case of a value less than five in one of the cells. One-way analysis of variance was performed for continuous traits measurement of central tendency. We performed the analysis using the Statistix 8 software (Analytical Software, FL, USA).
Results
Identification of PKD1 or PKD2 Genetic Defects in Probands
The NGS performed on LR-PCR libraries for PKD1 and PKD2 showed an excellent read depth, providing an average base coverage of 1,156x without gaps. On average, 92% of target sequences were covered >20x, but the regions with lower coverage were mainly in deep introns. The exon 1 of PKD1, usually difficult to sequence due to its high GC content, resulted entirely sequenced, with an average coverage at 100x.
We adopted the recently suggested nosology, describing the disease and the genetic etiology of ADPKD, classifying the patients as ADPKD-PKD1T/NT or ADPKD-PKD2, as shown in Table 2 (Eckardt et al., 2015; Cornec-Le Gall et al., 2017). Patients harboring more than one variant were classified ADPKD-PKD1/2T/NT based on the main variant.
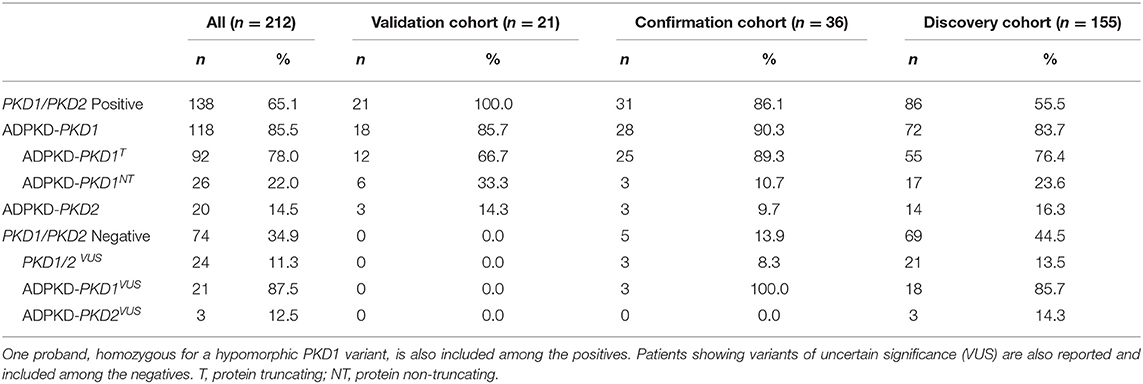
Table 2. Patient groups and positives for pathogenic/likely pathogenic variants in PKD1/PKD2.
To validate our method, LR-PCR re-sequencing was performed on DNA from 18 ADPKD-PKD1 and 3 ADPKD-PKD2 patients who had previously undergone genetic diagnosis by Sanger sequencing. All the 21 potentially causative variants known to be present in PKD1 (9 missense, 4 non-sense, 4 splicing and 4 frameshift), as well as the three in PKD2 (2 frameshift and 1 missense), were correctly detected.
In order to confirm the accuracy of the method, we tested 36 additional patients with a clinical diagnosis of ADPKD, who had already undergone kidney transplantation. In 28 individuals we identified possible pathogenic variants (P or LP) in PKD1, 25 of which were T and three NT. In three cases, protein truncating variants in PKD2 were detected. Three patients showed a variant of uncertain significance in PKD1. The low prevalence of PKD2 mutations (9.7%) compared to PKD1 (90.3%) in this cohort was consistent with the disease severity in these patients.
We then applied the method to a group of 155 newly recruited patients (discovery cohort). Among these cases, composed of patients with varying degrees of disease severity, 72 patients resulted positive for potentially causative variants in the PKD1 gene, 55 T and 17 NT, whereas 14 patients showed genetic defects in PKD2 gene. In 21 cases, we identified a variant classified as VUS, 18 in PKD1 and three in PKD2. In this discovery cohort, the prevalence of PKD2 mutations (16.3%) is higher when compared to the confirmation cohort.
Overall, in confirmation and discovery cohorts, we detected causative variants in 117 out of 191 patients, obtaining a diagnostic rate of 61.3%; 85.5% of positives showed variants in PKD1 gene, whereas 14.5% showed variants in PKD2. Diagnostic rates are heterogeneous among different studies and can vary in relation to the criteria for variant classification and the proportion of typical/familial cases. A diagnostic rate of 80% was reported in the only large Italian cohort of ADPKD patients, including “highly likely” and “likely” pathogenic variants (Carrera et al., 2016), but studies in other European populations report lower diagnostic rates (Hoefele et al., 2011; Obeidova et al., 2014).
Accuracy of the Method and Mutation Spectrum
Since we re-sequenced all the possible pathogenic variants by Sanger method, we have been able to calculate sensitivity, specificity and accuracy of NGS, using the Sanger as gold standard. Our NGS on LR-PCR for PKD1 and PKD2 genes provided 100% sensitivity, 63% specificity and 92% accuracy. Although we cannot rule out that some variants have not been called by NGS, we have not recorded false negative. On the other hand, detection of false positive variants was not so rare (22/221, 10%) and usually involved GC reach regions and/or homopolymers. However, the frequencies of the two called alleles in these cases was usually unbalanced (Supplementary Table 2).
Two cases of allele-drop out of the wild-type allele were detected, using LR-PCR amplification of exons 13–21 of PKD1. In both cases, replacing the LR-PCR primers with those previously described by Rossetti et al. (2012) for exons 15–21, the heterozygous state of the mutations was called correctly. For this reason, we decided to implement our analysis with this last LR-PCR, to avoid possible pitfalls (Supplementary Table 1).
Types and numbers of the variants classified in the P, LP, VUS, and H categories are listed in Table 3. Among the 158 different genetic alterations detected in our study, 80 (50.6%) had not been previously described, 73 of which were found in PKD1 and 7 in PKD2. Missense and non-sense were the most frequent variants in both genes, followed by frameshift in PKD1 and by splicing and frameshift in PKD2. Six variants in PKD1 occurred de-novo, three of which were not previously described: c.3236del p.(Asp1079Alafs*25), c.8860G>T p.(Glu2954*), and c.9201+1G>A. One de-novo and novel variant was also detected in PKD2: c.992G>A p.(Cys331Tyr).
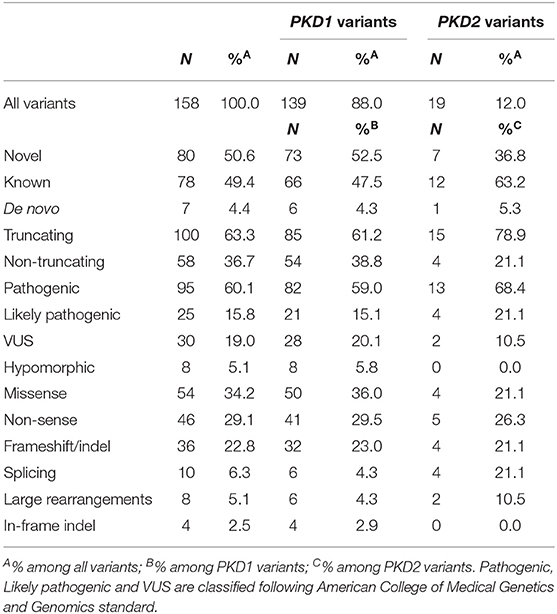
Table 3. Types of 158 distinct possible pathogenic variants detected in PKD1 and PKD2 genes.
All the 158 variants detected in PKD1 and PKD2 genes that we classified as P, LP, VUS, and H are listed in Supplementary Table 3, whereas the variants classified as LB and B are not reported. The mutations were spread over almost all exons, without hot spot regions. Seventeen mutations were detected in two or more families not known to be related.
Ten index patients showed large rearrangements (6 in PKD1 and 4 in PKD2), four of which were not previously described. These rearrangements could be detected by MLPA only, whereas our NGS analysis was unable to reveal them with enough accuracy. Among PKD1 deletions, we detected one single whole gene deletion, a deletion of exon 40, and four intragenic multiple exon deletions. In PKD2, a deletion of the whole gene was detected in two patients, whereas the deletion of exons 8 and 9 was shown in other two cases.
The molecular visualization of uncertain and hypomorphic variants performed by PyMol revealed alteration in only two of them. After searching for hydrogen bonds within 5 residues around the variants, we detected a bond loss for aminoacid changes p.(Ala3958Pro) (previously undescribed and classified as LB according to ACMG) and p.(Arg3277Cys) (previously reported as H by Rossetti et al., 2009, but classified as LB by ACMG) in Polycystin-1, as shown in Supplementary Figure 1. These results suggest a possible pathogenic role for these variants. Based on this prediction and thanks to segregation analysis we re-classified these two variants as VUS and H, respectively.
Genotype-Phenotype Analyses
The relationship between affected genes and type of mutation with main clinical features is presented in Supplementary Table 4. A significant association was detected with PKD1 truncating, PKD1 non-truncating and PKD2 mutations vs. kidney transplantation (p = 0.0006), antihypertensive treatment before 35 years (p = 0.0008), urological events before 35 years (p = 0.02).
A significantly different mean age distribution of disease onset (p = 0.0004), diagnosis (p = 0.0008) and ESRD (p = 0.04) was found among the cases with mutations in PKD1 and PKD2. Similar results were obtained by comparing the patients with PKD1 truncated mutations, PKD1 non-truncated and PKD2 mutations. ADPKD-PKD1T patients had an onset of the disease, respectively >10 years and >19 years earlier than ADPKD-PKD1NT and ADPKD-PKD2, as well as a 7 and 16 years earlier diagnosis, respectively. Moreover, ADPKD-PKD1T patients experienced ESRD 9 and >13 years earlier than ADPKD-PKD1NT and ADPKD-PKD2, respectively. These genotype-phenotype correlations are in line with previously published cohorts (Cornec-Le Gall et al., 2013; Hwang et al., 2016).
We found that 23.3% of our patients were without an apparent family history of polycystic kidney disease. Mutations in PKD1 or PKD2 were detected in 31 (68.9%) and five (16.1%) of these cases, respectively (Table 4). We demonstrated that the mutation arose de-novo in three patients, one case showed a somatic mosaicism, and five patients were carriers of two variants, partially explaining the absence of family history for the disease. The remaining 14 patients without family history, resulted negative for PKD1 or PKD2 mutations. Among them, after clinical revision, 11 were submitted to additional cystogenes re-sequencing and one of them resulted mutated in both allele of PKHD1, whereas three were PKHD1 heterozygous carriers. We did not observe significant clinical differences between patients with positive and negative family history.
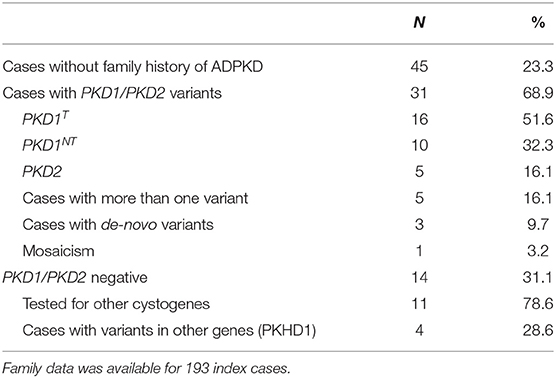
Table 4. Patients without family history of ADPKD.
Probands With More Than One Variant
Although ADPKD is typically a late-onset disease, some patients show an early and severe phenotype: ADPKDVEO (diagnosed in utero or before 18 months) and ADPKDEO (diagnosed before 15 years). Mutations in multiple PKD genes or biallelic PKD mutations can often explain these phenotypes (Bergmann et al., 2011; Audrézet et al., 2016), proving that additional PKD alleles in trans and/or de novo exert an aggravating effect and contribute to early and severe disease expression in polycystic kidney disease. In our series, 30 index patients (18.4%) had either a prenatal identification of renal cysts or clinical symptoms that allowed a diagnosis earlier than 15 years of age (Table 1).
Segregation analysis, when performed in patients who were found to carry more than one possible pathogenic variant, demonstrated the presence of compound heterozigosity in 6 and of a de novo variant in two (Table 5). Among patients with two variants, there were 4 ADPKDVEO and 3 ADPKDEO. Patients who carried two variants in cis are not reported.
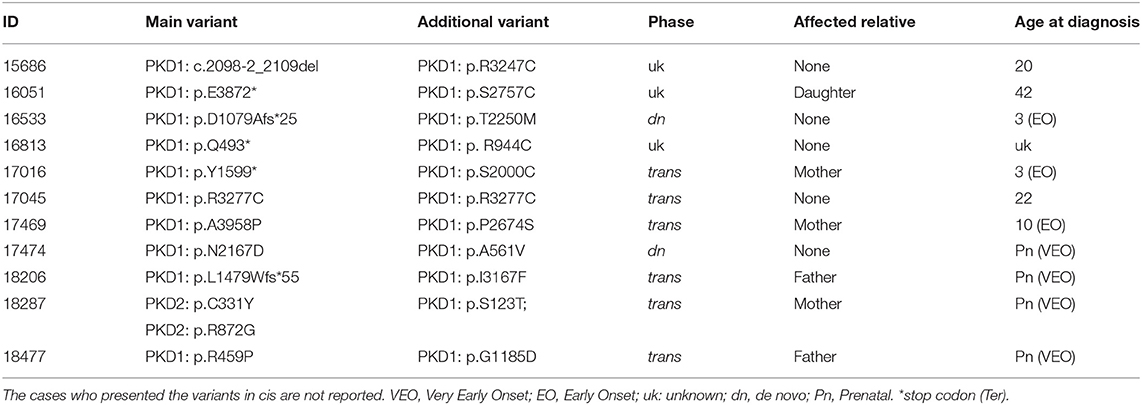
Table 5. Probands with more than one variant.
Based on familial segregation or pathogenicity information, these variants were classified as main and additional. In patients with an early onset of disease, whenever the additional variant was detected in trans, we re-classified it as hypomorphic, as it likely contributed to worsening the phenotype. In six families, we were able to diagnose a possible biallelic disease, resulting from the presence of two variants in trans (either in the same or different genes).
In four families, one variant was transmitted from a typically affected parent to a child with early onset disease or to an affected pregnancy, and a second hypomorphic variant was transmitted by an unaffected parent. (1) The p.(Ser2000Cys) (absent in GnomAD but with benign computational predictions) was identified in a child with disease onset at 3 years, in trans with a truncating mutation. (2) The p.(Pro2674Ser) variant is frequent, but it was identified in a child with early onset disease, in trans with a main variant, p.(Ala3958Pro), which was present in her typically affected mother. (3) In family 18206, two consecutive pregnancies were interrupted after sonographic detection of renal cysts. The father had a diagnosis of ADPKD and carried a frameshift mutation; molecular analysis of fetal DNA identified a second variant transmitted by a healthy mother, p.(Ile3167Phe), which was initially defined as possibly pathogenic (Rossetti et al., 2002), but is relatively frequent in the general population; we propose that it may represent a hypomorphic allele. (4) In family 18477, a pregnancy was interrupted after sonographic detection of renal cysts and PKD1 analysis revealed two variants: p.(Arg459Pro), transmitted by the affected father, and p.(Gly1185Asp), present in the mother, who had no renal cysts; a healthy child was born from a second pregnancy and he was found to carry the p.(Gly1185Asp) only. Although the p.(Arg459Pro) is extremely rare (never reported before, absent in GnomAD), it is likely benign according to ACMG guidelines; we cannot exclude that it might be in cis with an unidentified pathogenic variant.
In family 17045, a homozygous missense variant in PKD1 was detected. The proband showed an equivocal disease presentation and negative family history, but the first clinical hypothesis, at 22 years of age, was ARPKD. As PKHD1 screening resulted negative, ADPKD genes were studied and homozygosity for the known hypomorphic variant p.(Arg3277Cys) was identified (Rossetti et al., 2009). Interestingly, PyMol analysis, as well additional bioinformatics tools, predicted a possible pathogenicity for this variant. MLPA did not identify any deletion and no consanguinity was recorded.
Two patients showed ADPKDEO and ADPKDVEO, respectively, and both were carriers of a de novo P/LP PKD1 variant and a second heterozygous variant. Patient 16533 was a 15 years old boy who showed an episode of macroematuria at the age of three; abdominal ultrasound showed multiple cysts in both kidneys and the number of cysts and total kidney volume progressed over time. Family history was negative and he was found to carry a de novo pathogenic (frameshift) mutation and the p.(Thr2250Met) variant, inherited from his healthy father. The latter variant is relatively frequent in the general population and was already reported as a possible hypomorphic allele in previous studies (Irazabal et al., 2011). In family 17474, renal cysts were detected prenatally and confirmed after birth, parents are healthy (no cysts detected at renal ultrasound) and non-consanguineous. PKD1 sequencing identified a likely pathogenic variant, p.(Asn2167Asp), absent in parents, and a second maternally inherited variant, p.(Ala561Val). This is extremely rare (never reported before, absent in GnomAD), but with benign computational predictions, and it was classified as hypomorphic. We cannot formally test if variants in these two patients are in trans, but we presume they contributed to the severe clinical expression.
In family 18287 we detected a possible bilineal inheritance, with variants in both PKD1 and PKD2 (Figure 1). Two pregnancies were interrupted due to a prenatal finding of polycystic kidney disease at ultrasound examination at 20 and 13 gestational weeks, respectively. The mother was 33 year old; she had multicystic bilateral disease without affected family members, and showed a de novo missense variant p.(Cys331Thr) in PKD2. The father was a healthy 44 years old man with no signs of kidney cystic disease at ultrasound, and showed a variant in PKD1, p.(Ser123Thr), and a second variant in PKD2, p.(Arg872Gly). Both fetuses inherited the maternal PKD2 missense variant, in addition to the paternal p.(Ser872Gly) variant in PKD1, while only one fetus inherited the p.(Arg872Gly) PKD2 variant. The analysis of PKHD1 performed on the first fetus showed no mutations. We suggest that: 1) the PKD2 p.(Cys331Thr) variant is pathogenic, since it is de-novo in a patient with a renal cystic disease and is transmitted to both fetuses; 2) the p.(Ser123Thr) variant in PKD1 is hypomorphic, since it does not cause renal disease in the father (age 44 years), but worsens the renal phenotype when co-inherited with a PKD2 mutation; 3) the missense variant p.(Arg872Gly) in PKD2, already described as disease-causing in HGMD database (Neumann et al., 2013), is likely benign, since it is present in a healthy man and does not segregate with disease in the fetuses.
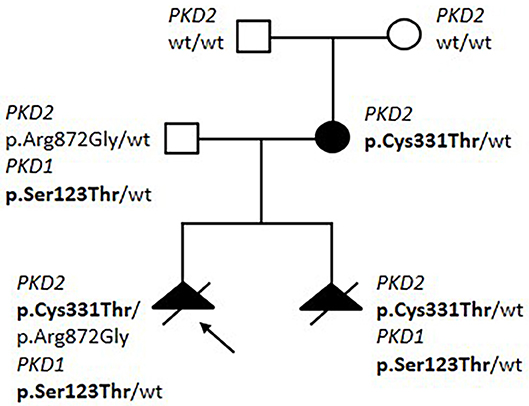
Figure 1. Pedigree of Family 18287 with bilineal inheritance of PKD1/PKD2 variants. Variants segregating with disease are shown in bolt.
Bilineal inheritance of PKD1 and PKD2 variants is an extremely rare finding and, as expected, disease associated with the presence of two distinct mutations appeared to be more severe than the disease associated with either mutation alone (Pei et al., 2001; Elisakova et al., 2018).
Detection of Mosaicism
The high sensitivity of NGS technology allowed us to detect two cases of mosaicism.
Case 1. A 53-year-old male proband was first evaluated at the age of 34, when he had an episode of hematuria. Abdominal US documented bilateral multiple renal cysts without hepatic involvement. He started treatment for arterial hypertension at the age of 40 years and he underwent to hemodialysis at 48 years old. He received a kidney transplant 2 years later. The molecular analysis showed the heterozygosis for c.2986-2_2987del splicing mutation in PKD1: this four-nucleotide deletion between intron12 and exon13 was predicted to alter an acceptor site, affecting splicing. The proband's mother was also diagnosed with ADPKD at the age of 67 years, when she performed an ultrasound for abdominal pain, although she showed a milder phenotype (renal and hepatic cysts but normal renal function at 69 years) and no other family members were reported to be affected. The c.2986-2_2987del mutation was not detected by targeted Sanger sequencing in the mother. NGS of whole PKD1 was later performed on maternal DNA and the mutation c.2986-2_2987del was detected in 156 among 2,193 reads, revealing a 6.9% of mosaicism in peripheral blood. Of note, the mutation was not sufficient for the germinal variant calling (threshold defined by our bioinformatics pipeline was 10%), but it was readable by IGV software (Figure 2A).
Case 2. A 63-year-old female patient, diagnosed at 46 years and without familiarity for the disease, showed no heterozygous mutations or rearrangements in PKD1 and PKD2 genes. However, a 2bp deletion (c.9965_9966del) in exon 30 of PKD1 gene was detected in 40 among 252 (15.9%) reads. Sanger sequencing by using two different gene-specific LR-PCR primers pair and two different sequencing primers pair confirmed the presence of the mosaicism (Figure 2B).
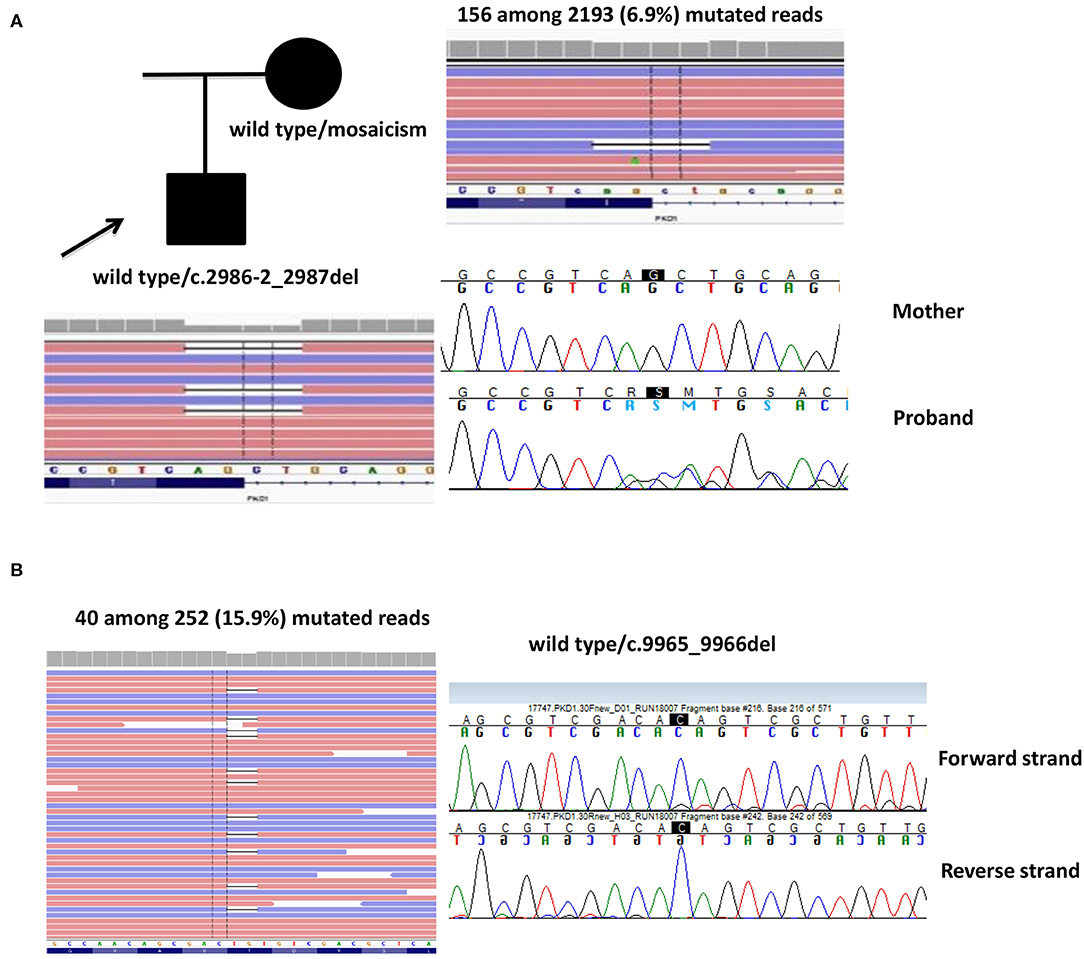
Figure 2. Two cases of mosaicism. (A) Family 16321, where the affected mother showed 6.9% of mutated PKD1 in peripheral blood. (B) Family 17747, where the proband showed 15.9% of mutated PKD1. The reads are visualized by Integrative Genome Viewer (IGV) software.
Patients Without PKD1 and/or PKD2 Mutations
In 74 index patients, neither pathogenic/likely pathogenic variants nor rearrangements were identified in both PKD1 and PKD2, although 24 of them showed a VUS. Family data were recorded for 42 cases, and 28 had a family history of polycystic disease. Among families, disease progression was typical in at least eight, while it was very mild and/or atypical (based on imaging studies) in the remaining 14 families. The median age of disease onset was 30 years; six patients had reached ESRD and three underwent renal transplantation. After re-evaluation of the clinical phenotype, 28 of these patients were tested for an additional NGS-panel that included the recently described genes associated with ADPKD and ADPLD, PKHD1 and some additional cystogenes. Six patients showed variants in one or more of these genes, as reported in Table 6.

Table 6. Molecular defects in additional cystogenes detected in 25 PKD1/PKD2-negative cases.
Patient 17316 carried compound heterozygous mutations in PKHD1, hence a diagnosis of ARPKD could be posed. She was a 31 years old woman who received a diagnosis of polycystic kidneys at 17. At 30, after her first pregnancy, she had proteinuria, multiple cysts in both kidneys and a mild reduction of GFR. No family members were reported to be affected by polycystic kidney and ADPKD gene testing resulted negative. An abdominal computed tomography scan showed, in addition to the known renal cysts, segmental dilatation of the intrahepatic bile ducts compatible with Caroli disease. PKHD1 analysis identified two mutations in compound heterozygosity, p.(Thr36Met) and p.(Cys2422Gly). MRI is shown in Figure 3A.
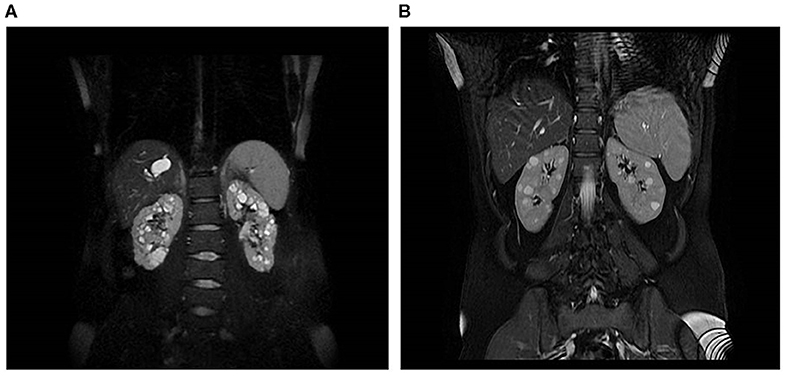
Figure 3. Kidney and liver images of two patients. (A) MRI of a 31-year-old woman with compound heterozygous mutations in PKHD1, who received a diagnosis of polycystic kidneys at 17. (B) MRI of a 45-year-old woman, who carried two heterozygous variants in PKHD1, p.(His3124Thr), and PMM2, p.(Gly42Arg).
Three additional patients showed heterozygous variants in PKHD1 (MLPA excluded large intragenic deletions/duplications). Monoallelic mutations in PKHD1 have been described to cause multiple liver cysts and/or increased kidney echogenicity in up to 10% of carriers, but were not associated with an increased risk of renal cysts (Gunay-Aygun et al., 2011). Patient 17254 carried a definitely pathogenic variant, p.(Arg124*): she was a 50 year old woman, renal cysts were identified when she was 15, but the disease was non progressive, she had no hepatic cysts and no family history. The PKHD1 mutation likely represents a spurious finding. Also patient 16672, a 42 year old man, found out to have renal cysts at the age of 40, together with multiple hepatic cysts; he carried a heterozygous variant of unknown significance, p.(Arg3913Cys).
Finally, patient 16542 was diagnosed with PKD at the age of 30 years, after the onset of arterial hypertension (MRI is shown in Figure 3B); she had normal renal function at last evaluation (45 years). Her father died at the age of 79, he had type 2 diabetes requiring insulin treatment, renal and hepatic cysts detected at ultrasonography and normal renal function. The patient carried a heterozygous variant of unknown significance in PKHD1, p.(His3124Tyr), defined as likely pathogenic in ClinVar, and a missense variant p.(Gly42Arg) in PMM2, classified as likely pathogenic. Recessive mutations in PMM2 were reported as associated to hyperinsulinemic hypoglycemia (HI) and PKD (Cabezas et al., 2017). The study of larger cohorts of patients will be able to define if the combination of variants in distinct recessive genes can be relevant for cysts progression.
Patient 17654 was diagnosed with polycystic kidney disease at the age of 61 on routine abdominal US examination. He has renal and hepatic cysts but normal renal function. His 89-year-old mother has renal and liver cysts, and a nephrocalcinosis. An uncle, who died at the age of 85, had kidney cysts and mild renal insufficiency. Molecular analysis showed a variant of uncertain significance in ALG8, p.(Leu149Arg), which is extremely rare in the general population and has a pathogenic computational verdict with no benign predictions. ALG8 was recently described as a very rare cause of ADPLD, but at least one patient had renal cysts at a young age (Besse et al., 2017). In our family, the p.Leu149Arg variant was present in the proband's mildly affected mother. ALG8 may be the cause of polycystic disease in this family, although a functional assay would be needed to prove the involvement of this variant beyond doubt.
Patient 18607 was diagnosed with polycystic disease at the age of 49, with cysts mainly affecting the liver. Family history was not known, but she carried a non-sense mutation p.(Tyr423*) in PRKCSH, a gene associated to polycystic liver disease-1 (PCLD1) with or without kidney cysts (Fedeles et al., 2011). The same mutation, p.(Tyr423*), was already reported in a family with autosomal dominant polycystic liver disease (Li et al., 2003).
Discussion
Genetic testing in the clinical management of ADPKD is not widespread for several reasons. First, analysis of PKD1 is challenging due to its segmentally duplicated region. Second, the genetic heterogeneity and the extreme allelic heterogeneity (>1.500 different pathogenic variants described for PKD1) require analysis of the coding regions of all candidate genes, and Sanger-based single gene approaches are costly and cumbersome. Third, the presence of many non-truncating alleles and potentially hypomorphic low-penetrance variants pose a further challenge for clinical interpretation. However, genetic testing is increasingly required for diagnosis, prognosis and treatment decision. Furthermore, molecular testing can provide genetic stratification for clinical trials and may guide clinical management.
In the present study, we validated a targeted-NGS method, which was adjusted in a diagnostic setting, and we applied it to analyze a cohort of 212 ADPKD patients. Our protocol is based on high-throughput simultaneous sequencing of PKD1 and PKD2 after LR-PCR of coding regions, followed by a masked reference genome alignment. Samples without PKD1/PKD2 mutations were submitted to MLPA and, when negative, tested by targeted-NGS for mutations in newly described ADPKD and ADPLD genes, ARPKD and additional cystogenes.
Our protocol overcomes the technical issues of PKD1 analysis, provides a reliable and less expensive molecular test for both genes, and resulted highly accurate. The use of previously validated primers for gene-specific enrichment, the introduction of an additional LR-PCR in order to minimize the risk of allele-drop out, and a specific alignment pipeline for avoiding miss-alignment with pseudogenes, make our genetic test specific and robust. Nevertheless, we cannot exclude that few variants were missed due to non-amplification of one allele.
Overall, we detected a pathogenic/likely pathogenic variant in 63.9% (122 among 191) of our index patients, 100 (82.0%) of which showed a genetic defect in PKD1, 17 (14%) in PKD2 and 5 (4%) in other cystogenes. In addition, 26 probands (13.6%) presented variants in these genes classified as uncertain significance.
Several studies have demonstrated that patients with PKD1 mutations predicted to truncate the protein have a more severe phenotype than patients with non-truncating mutations and patients with PKD2 mutations. In addition, hypomorphic alleles are reported in patients with milder kidney disease and in normal individuals, but can cause a severe early onset disease if they are present in trans with a pathogenic variant. Hence, we also included gene and type of detected variant in the nosology of our patient cohort (Cornec-Le Gall et al., 2013; Heyer et al., 2016; Hwang et al., 2016). In our study, we detected a higher prevalence of ADPKD-PKD1T cases in the confirmation cohort (89.3%) in comparison to that in the diagnostic cohort (76.4%), according to the different clinical phenotype of patients in the two groups. Moreover, our overall results confirm that patients with PKD1 mutations predicted to truncate the protein experienced ESRD 9 years earlier than patients with PKD1 non-truncating mutations and >13 years earlier than patients with PKD2 mutations. In addition, our ADPKD-PKD1T cases showed an onset of the disease significantly earlier than ADPKD-PKD1NT and ADPK-PKD2 (>10 and >19 years, respectively), as well as a significant earlier diagnosis (7 and 15 years, respectively). These data emphasize the need to combine clinical information with gene and allele data to achieve useful prognostic predictions. The incomplete penetrance of some PKD alleles and a dosage effect must be taken into account.
Among 158 distinct variants here detected, 80 (50.6%) were previously unreported, confirming the high allelic heterogeneity of these genes, which harbor many clinically significant private variants. Differentiating pathogenic from neutral changes remains a big challenge, since 36.7% of the variants here detected were non-truncating, 19% were classified as variants of uncertain significant and 5.1% were likely hypomorphic alleles. Our additional bioinformatics analysis using PyMOL helped us to better define the potential pathogenicity of some VUS and H variants. However, variant interpretation remains the major bottleneck in NGS analysis and data sharing is essential, in order to collect more families where segregation analysis can be performed. It is notable that, when strictly following ACMG guidelines, the only non-truncating PKD1/PKD2 variants that reach the status of “likely pathogenic” are either described in previous studies or segregate in affected family members. We did our best to organize and perform segregation analysis in as many families as possible. Functional studies to evaluate PKD1 and PKD2 variants would be extremely useful but, unfortunately, are currently unavailable.
A further key point is the distinction between neutral and hypomorphic alleles and the degree of their penetrance. This is relevant in order to select potential kidney donors and to assess the risk of biallelic disease when a hypomorphic variant is present in trans with a fully inactivating PKD1 allele. Indeed, some ADPKD and ARPKD patients can clinically overlap with early manifestation in ADPKD and late onset in ARPKD. Mutations in PKD1 and PKD2 can also be inherited in a recessive way with at least one PKD1 or PKD2 hypomorphic allele (Bergmann, 2017). In our study, eleven patients (9.4% of mutated) showed more than one variant. Segregation analysis indicated biallelic disease in five patients, digenic in one, de-novo variant with unknown phase in two, and segregation analysis was not possible in three patients. Seven patients with very early onset disease and 23 patients with early onset were present in our series, and 23.3% (7 out 30) of them exhibited two variants, typically compound heterozygosis of a pathogenic and a hypomorphic allele.
We found a further clinical phenocopy of ARPKD, due to homozygosity of the PKD1 hypomorphic allele p.(Arg3277Cys), in a patient with negative family history (ID 17045). This variant is reported as an incompletely penetrant mutant allele that resulted in occasional cyst development in heterozygotes and more severe PKD in homozygosity (Rossetti et al., 2009). Our data confirmed that the dosage/threshold of functional PKD1 protein might be critical for cyst initiation. Of note, the in silico analysis by PyMol indicated that this variant results in a bond loss in Polycystin-1.
A case of ADPKDVEO caused by digenic disease with PKD1 and PKD2 genetic defects was also detected. Digenic disease involving PKD1 and PKD2 genes are rarely described, and it usually results in more severe clinical phenotype than in cases with mono-allelic variants, but not in VEO (Pei et al., 2001; Gainullin et al., 2015). The co-inheritance of a pathogenic allele in PKD2 (from the affected mother) and a hypomorphic variant in PKD1 from the unaffected father, likely caused a severe phenotype in this family. To our knowledge, this is the first report of bilineal inheritance causing prenatal onset of polycystic kidneys.
We found one patient with biallelic mutations in PKHD1 among probands negative for PKD1/PKD2 mutations, confirming the significant overlap between ARPKD and ADPKD. ARPKD is usually a severe early onset disease, but there is wide variability in severity even among patients carrying the same PKHD1 mutations, emphasizing the importance of modifying genes and potential environmental factors (Gunay-Aygun et al., 2010). In a sporadic patient with adult-onset polycystic kidneys, the diagnosis of ARPKD should be considered. Such cases highlight the importance of the molecular diagnosis: knowing the genetic cause of the disease allows an adequate clinical management and a proper genetic counseling.
NGS has the advantage of deep sequencing and allows the detection of mosaicism, undetectable by Sanger sequencing. Somatic mutations acquired in the early stage of embryonic postzygotic development might cause mosaicism in the parent who later transmits the mutation to the offspring through germ cell. One case of mosaicism arising from this mechanism was detected in our study: the heterozygous PKD1 mutation detected in the affected proband was inherited from the less severely affected mother, who showed only 6.8% of mutated reads in her peripheral blood. An additional case of somatic mosaicism has been detected, with 15.9% of mutated reads in PKD1 gene. No offspring was available to test the heritability of this genetic defect. The incidence of mosaicism events in the human body is underestimated, but a mosaicism should be considered in patients with mild disease and healthy parents or with unclear pattern of transmission. In addition, apparently de-novo mutations might indeed be the consequence of somatic/germline mosaicism in an unaffected parent. In current diagnostic protocols, targeted deep sequencing of the mutation detected in the index case should be performed in the unaffected parents. Our NGS protocol represented a useful diagnostic tool for the detection of somatic mosaicism.
In conclusion, accurate and cost-effective diagnostic genetic tests are needed in order to drive clinical management of polycystic kidney disease. NGS technology represents a powerful approach for a better understanding of this disorder, overcoming at least part of the technical difficulties of PKD2/PKD1 molecular analysis. However, interpretation of results remains challenging and requires interdisciplinary efforts as well as broad genetic and phenotypic data sharing.
Data Availability Statement
The datasets generated for this study can be found in the https://databases.lovd.nl/shared/genes/PKD1; https://databases.lovd.nl/shared/genes/PKD2 (Fokkema et al., 2011).
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
VM: study design, analysis and interpretation of experimental data, statistical analyses, manuscript preparation, and critical revision of the manuscript. SB: study design, technical analysis and interpretation of experimental data, and manuscript preparation. CG: patients' recruitment, clinical data collection and interpretation, genetic counseling, and manuscript preparation. IC: patients' recruitment and their clinical management, clinical data collection and interpretation, and manuscript preparation. RM, CC, MP, and SD: technical analysis and interpretation of experimental data. VA and RC: clinical data collection. EA: clinical data collection, genetic counseling. AM: technical analysis and interpretation of experimental data, and bioinformatics analyses. FF and EG: patients' recruitment, clinical data collection and interpretation, and manuscript preparation. EM, FM, and AP: patients' recruitment, clinical data collection and interpretation. NS: performed MRI of patients. AW and MS: patients' recruitment, clinical data collection and interpretation, and genetic counseling. GL: patients' recruitment and their clinical management, clinical data collection and interpretation, critical revision of the manuscript and corresponding author. All authors read and approved the manuscript for submission.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Pasquale Chieco for the contribution to statistical analysis. We thank Marinella Cenci and Luciana Gargano for technical contribution, and Cecilia Carletti for the help with the figures. We are grateful to the patients and their families for participation.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00464/full#supplementary-material
Footnotes
1. ^Integrative Genome Viewer (IGV). http://www.broadinstitute.org/igv
2. ^Human Genome Variation Society. http://www.hgvs.org/mutnomen
3. ^VarSomeClinical platform. https://varsome.com
4. ^Mutation database PKDB. https://pkdb.mayo.edu
5. ^Leiden Open Variations PKD database. https://databases.lovd.nl/shared/genes/PKD1; https://databases.lovd.nl/shared/genes/PKD2
6. ^ClinVar. http://www.ncbi.nlm.nih.gov/clinvar
7. ^PyMOL. www.pymol.org/2
References
1000 Genomes Project Consortium, Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. doi: 10.1038/nature15393
Audrézet, M.-P., Corbiere, C., Lebbah, S., Morinière, V., Broux, F., Louillet, F., et al. (2016). Comprehensive PKD1 and PKD2 mutation analysis in prenatal autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 27, 722–729. doi: 10.1681/asn.2014101051
Audrézet, M. P., Cornec-Le Gall, E., Chen, J. M., Redon, S., Quéré, I., Creff, J., et al. (2012). Autosomal dominant polycystic kidney disease: comprehensive mutation analysis of PKD1 and PKD2 in 700 unrelated patients. Hum. Mutat. 33, 1239–1250. doi: 10.1002/humu.22103
Barua, M., Cil, O., Paterson, A. D., Wang, K., He, N., Dicks, E., et al. (2009). Family history of renal disease severity predicts the mutated gene in ADPKD. J. Am. Soc. Nephrol. 20, 1833–1838. doi: 10.1681/asn.2009020162
Bergmann, C. (2017). Recent advances in the molecular diagnosis of polycystic kidney disease. Expert Rev. Mol. Diagn. 17, 1037–1054. doi: 10.1080/14737159.2017.1386099
Bergmann, C., von Bothmer, J., Ortiz Brüchle, N., Venghaus, A., Frank, V., Fehrenbach, H., et al. (2011). Mutations in multiple PKD genes may explain early and severe polycystic kidney disease. J. Am. Soc. Nephrol. 22, 2047–2056. doi: 10.1681/ASN.2010101080
Besse, W., Dong, K., Choi, J., Punia, S., Fedeles, S. V., Choi, et al. (2017). Isolated polycystic liver disease genes define effectors of polycystin-1 function. J. Clin. Invest. 127, 1772–1785. doi: 10.1172/JCI90129
Borràs, D. M., Vossen, R. H. A. M., Liem, M., Buermans, H. P. J., Dauwerse, H., van Heusden, D., et al. (2017). Detecting PKD1 variants in polycystic kidney disease patients by single-molecule long-read sequencing. Hum. Mutat. 38, 870–879. doi: 10.1002/humu.23223
Bullich, G., Domingo-Gallego, A., Vargas, I., Ruiz, P., Lorente-Grandoso, L., Furlano, M., et al. (2018). A kidney-disease gene panel allows a comprehensive genetic diagnosis of cystic and glomerular inherited kidney diseases. Kidney Int. 94, 363–371. doi: 10.1016/j.kint.2018.02.027
Cabezas, O. R., Flanagan, S. E., Stanescu, H., García-Martínez, E., Caswell, R., Lango-Allen, H., et al. (2017). Polycystic kidney disease with hyperinsulinemic hypoglycemia caused by a promoter mutation in phosphomannomutase 2. J. Am. Soc. Nephrol. 28, 2529–2539. doi: 10.1681/ASN.2016121312
Carrera, P., Calzavara, S., Magistroni, R., den Dunnen, J. T., Rigo, F., Stenirri, S., et al. (2016). Deciphering variability of PKD1 and PKD2 in an Italian cohort of 643 patients with Autosomal Dominant Polycystic Kidney Disease (ADPKD). Sci. Rep. 6:30850. doi: 10.1038/srep30850
Connor, A., Lunt, P. W., Dolling, C., Patel, Y., Meredith, A. L., Gardner, A., et al. (2008). Mosaicism in autosomal dominant polycystic kidney disease revealed by genetic testing to enable living related renal transplantation. Am. J. Transplant. 8, 232–237. doi: 10.1111/j.1600-6143.2007.02030.x
Cornec-Le Gall, E., Alam, A., and Perrone, R. D. (2019). Autosomal dominant polycystic kidney disease. Lancet 393, 919–935. doi: 10.1016/S0140-6736(18)32782-X
Cornec-Le Gall, E., Audrézet, M.-P., Chen, J.-M., Hourmant, M., Morin, M.-P., Perrichot, R., et al. (2013). Type of PKD1 mutation influences renal outcome in ADPKD. J. Am. Soc. Nephrol. 24, 1006–1013. doi: 10.1681/asn.2012070650
Cornec-Le Gall, E., Audrézet, M.-P., Rousseau, A., Hourmant, M., Renaudineau, E., Charasse, C., et al. (2016). The PROPKD score: a new algorithm to predict renal survival in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 27, 942–951. doi: 10.1681/asn.2015010016
Cornec-Le Gall, E., Olson, R. J., Besse, W., Heyer, C. M., Gainullin, V. G., Smith, et al. (2018). Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am. J. Hum. Genet. 102, 832–844. doi: 10.1016/j.ajhg.2018.03.013
Cornec-Le Gall, E., Torres, V. E., and Harris, P. C. (2017). Genetic complexity of autosomal dominant polycystic kidney and liver diseases. J. Am. Soc. Nephrol. 29, 13–23. doi: 10.1681/asn.2017050483
Den Dunnen, J. T., and Antonarakis, S. E. (2000). Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum. Mutat. 15, 7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N
Eckardt, K. U., Alper, S. L., Antignac, C., Bleyer, A. J., Chauveau, D., Dahan, K., et al. (2015). Autosomal dominant tubulointerstitial kidney disease: diagnosis, classification, and management - a KDIGO consensus report. Kidney Int. 88, 676–683. doi: 10.1038/ki.2015.28
Eisenberger, T., Decker, C., Hiersche, M., Hamann, R. C., Decker, E., Neuber, S., et al. (2015). An efficient and comprehensive strategy for genetic diagnostics of polycystic kidney disease. PLoS ONE 10:e116680. doi: 10.1371/journal.pone.0116680
Elisakova, V., Merta, M., Reiterova, J., Baxova, A., Kotlas, J., Hirschfeldova, K., et al. (2018). Bilineal inheritance of pathogenic PKD1 and PKD2 variants in a Czech family with autosomal dominant polycystic kidney disease - a case report. BMC Nephrol. 19:163. doi: 10.1186/s12882-018-0978-2
Fedeles, S. V., Tian, X., Gallagher, A. R., Mitobe, M., Nishio, S., Lee, S. H., et al. (2011). A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation. Nat. Genet. 43, 639–647. doi: 10.1038/ng.860
Fokkema, I. F. A. C., Taschner, P. E. M., Schaafsma, G. C. P., Celli, J., Laros, J. F. J., and den Dunnen, J. T. (2011). LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 32, 557–563. doi: 10.1002/humu.21438
Gabow, P. A. (1993). Autosomal dominant polycystic Kidney Disease. N. Engl. J. Med. 329, 332–342. doi: 10.1056/NEJM199307293290508
Gainullin, V. G., Hopp, K., Ward, C. J., Hommerding, C. J., and Harris, P. C. (2015). Polycystin-1 maturation requires polycystin-2 in a dose-dependent manner. J. Clin. Invest. 125, 607–620. doi: 10.1172/JCI76972
Grantham, J. J., Mulamalla, S., and Swenson-Fields, K. I. (2011). Why kidneys fail in autosomal dominant polycystic kidney disease. Nat. Rev. Nephrol. 7, 556–566. doi: 10.1038/nrneph.2011.109
Gunay-Aygun, M., Font-Montgomery, E., Lukose, L., Tuchman, M., Graf, J., Bryant, J. C., et al. (2010). Correlation of kidney function, volume and imaging findings, and PKHD1 mutations in 73 patients with autosomal recessive polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 5, 972–984. doi: 10.2215/CJN.07141009
Gunay-Aygun, M., Turkbey, B. I., Bryant, J., Daryanani, K. T., Gerstein, M. T., Piwnica-Worms, K., et al. (2011). Hepatorenal findings in obligate heterozygotes for autosomal recessive polycystic kidney disease. Mol. Genet. Metab. 104, 677–681. doi: 10.1016/j.ymgme.2011.09.001
Hateboer, N., Dijk, M. A. V., Bogdanova, N., Coto, E., Saggar-Malik, A. K., San Millan, J. L., et al. (1999). Comparison of phonotypes of polycystic kidney disease types 1 and 2. Lancet 353, 103–107. doi: 10.1016/S0140-6736(98)03495-3
Heyer, C. M., Sundsbak, J. L., Abebe, K. Z., Chapman, A. B., Torres, V. E., Grantham, J. J., et al. (2016). Predicted mutation strength of nontruncating PKD1 mutations aids genotype-phenotype correlations in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 27, 2872–2884. doi: 10.1681/ASN.2015050583
Hoefele, J., Mayer, K., Scholz, M., and Klein, H. G. (2011). Novel PKD1 and PKD2 mutations in autosomal dominant polycystic kidney disease (ADPKD). Nephrol. Dial. Transplant. 26, 2181–2188. doi: 10.1093/ndt/gfq720
Hwang, Y.-H., Conklin, J., Chan, W., Roslin, N. M., Liu, J., He, N., et al. (2016). Refining genotype-phenotype correlation in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 27, 1861–1868. doi: 10.1681/asn.2015060648
Irazabal, M. V., Huston, J. III, Kubly, V., Rossetti, S., Sundsbak, J. L., Hogan, M. C., et al. (2011). Extended follow-up of unruptured intracranial aneurysms detected by presymptomatic screening in patients with autosomal dominant polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 6, 1274–1285. doi: 10.2215/CJN.09731110
Kinoshita, M., Higashihara, E., Kawano, H., Higashiyama, R., Koga, D., Fukui, T., et al. (2016). Technical evaluation: Identification of pathogenic mutations in pkd1 and pkd2 in patients with autosomal dominant polycystic kidney disease by next-generation sequencing and use of a comprehensive new classification system. PLoS ONE 11:e0166288. doi: 10.1371/journal.pone.0166288
Kurashige, M., Hanaoka, K., Imamura, M., Udagawa, T., Kawaguchi, Y., Hasegawa, T., et al. (2015). A comprehensive search for mutations in the PKD1 and PKD2 in Japanese subjects with autosomal dominant polycystic kidney disease. Clin. Genet. 87, 266–272. doi: 10.1111/cge.12372
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. doi: 10.1038/nature19057
Li, A., Davila, S., Furu, L., Qian, Q., Tian, X., Kamath, P. S., et al. (2003). Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. Am. J. Hum. Genet. 72, 691–703. doi: 10.1086/368295
Neumann, H. P., Jilg, C., Bacher, J., Nabulsi, Z., Malinoc, A., et al. (2013). Epidemiology of autosomal-dominant polycystic kidney disease: an in-depth clinical study for south-western Germany. Nephrol. Dial. Transplant. 28, 1472–1487. doi: 10.1093/ndt/gfs551
Obeidova, L., Elisakova, V., Stekrova, J., Reiterova, J., Merta, M., Tesar, V., et al. (2014). Novel mutations of PKD genes in the Czech population with autosomal dominant polycystic kidney disease. BMC Med. Genet. 15, 41–52. doi: 10.1186/1471-2350-15-41
Pei, Y., Obaji, J., Dupuis, A., Paterson, A. D., Magistroni, R., Dicks, E., et al. (2009). Unified criteria for ultrasonographic diagnosis of ADPKD. J. Am. Soc. Nephrol. 20, 205–212. doi: 10.1681/ASN.2008050507
Pei, Y., Paterson, A. D., Wang, K. R., He, N., Hefferton, D., Watnick, T., et al. (2001). Bilineal disease and trans-heterozygotes in autosomal dominant polycystic kidney disease. Am. J. Hum. Genet. 68, 355–363. doi: 10.1086/318188
Porath, B., Gainullin, V. G., Cornec-Le Gall, E., Dillinger, E. K., Heyer, C. M., Hopp, K., et al. (2016). Mutations in GANAB, encoding the glucosidase IIα subunit, cause autosomal-dominant polycystic kidney and liver disease. Am. J. Hum. Genet. 98, 1193–1207. doi: 10.1016/j.ajhg.2016.05.004
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Rossetti, S., Chauveau, D., Walker, D., Saggar-Malik, A., Winearls, C. G., Torres, V. E., et al. (2002). A complete mutation screen of the ADPKD genes by DHPLC. Kidney Int. 61, 1588–1599. doi: 10.1046/j.1523-1755.2002.00326.x
Rossetti, S., Consugar, M. B., Chapman, A. B., Torres, V. E., Guay-Woodford, L. M., Grantham, J. J., et al. (2007). Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 18, 2143–2160. doi: 10.1681/asn.2006121387
Rossetti, S., Hopp, K., Sikkink, R. A., Sundsbak, J. L., Lee, Y. K., Kubly, V., et al. (2012). Identification of gene mutations in autosomal dominant polycystic kidney disease through targeted resequencing. J. Am. Soc. Nephrol. 23, 915–933. doi: 10.1681/asn.2011101032
Rossetti, S., Kubly, V. J., Consugar, M. B., Hopp, K., Roy, S., Horsley, S. W., et al. (2009). Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int. 75, 848–855. doi: 10.1038/ki.2008.686
Stenson, P. D., Mort, M., Ball, E. V., Evans, K., Hayden, M., Heywood, S., et al. (2017). The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 136, 665–677. doi: 10.1007/s00439-017-1779-6
Tan, A. Y., Michaeel, A., Liu, G., Elemento, O., Blumenfeld, J., Donahue, S., et al. (2014). Molecular diagnosis of autosomal dominant polycystic kidney disease using next-generation sequencing. J. Mol. Diagn. 16, 216–228. doi: 10.1016/j.jmoldx.2013.10.005
Torres, V. E., Harris, P. C., and Pirson, Y. (2007). Autosomal dominant polycystic kidney disease. Lancet 369, 1287–1301. doi: 10.1016/S0140-6736(07)60601-1
Trujillano, D., Bullich, G., Ossowski, S., Ballarín, J., Torra, R., Estivill, X., et al. (2014). Diagnosis of autosomal dominant polycystic kidney disease using efficient PKD1 and PKD2 targeted next-generation sequencing. Mol. Genet. Genomic Med. 2, 412–421. doi: 10.1002/mgg3.82
Keywords: polycystic kidney disease, ADPKD, PKD1, PKD2, NGS, cystogenes
Citation: Mantovani V, Bin S, Graziano C, Capelli I, Minardi R, Aiello V, Ambrosini E, Cristalli CP, Mattiaccio A, Pariali M, De Fanti S, Faletra F, Grosso E, Cantone R, Mancini E, Mencarelli F, Pasini A, Wischmeijer A, Sciascia N, Seri M and La Manna G (2020) Gene Panel Analysis in a Large Cohort of Patients With Autosomal Dominant Polycystic Kidney Disease Allows the Identification of 80 Potentially Causative Novel Variants and the Characterization of a Complex Genetic Architecture in a Subset of Families. Front. Genet. 11:464. doi: 10.3389/fgene.2020.00464
Received: 04 December 2019; Accepted: 15 April 2020;
Published: 07 May 2020.
Edited by:
Jordi Pérez-Tur, Superior Council of Scientific Investigations (CSIC), SpainReviewed by:
Miranda Durkie, Sheffield Children's NHS Foundation Trust, United KingdomChiara Di Resta, Vita-Salute San Raffaele University, Italy
Copyright © 2020 Mantovani, Bin, Graziano, Capelli, Minardi, Aiello, Ambrosini, Cristalli, Mattiaccio, Pariali, De Fanti, Faletra, Grosso, Cantone, Mancini, Mencarelli, Pasini, Wischmeijer, Sciascia, Seri and La Manna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gaetano La Manna, gaetano.lamanna@unibo.it
†These authors have contributed equally to this work