Abstract
There is mounting evidence of an associative link between inflammatory bowel disease (IBD) and clinical depression. In the first major treatise on the eponymous disease, Burrill Crohn himself noted that: “The number of cases of ileitis that have been rescued from institutions for the treatment of mental diseases emphasises not the personality but the end results of the drain of the disease upon the psychic constitution of the sufferer.” In the 70 years since that prescient statement, a high incidence of neuropsychiatric symptoms (depression, anxiety, cognitive fatigue, and sleep disorders) in patients with IBD has been frequently observed. Since patients with depression have significantly increased rates of relapse, surgery, hospitalisation, and suicide, recognising and treating depression is of paramount importance. In this narrative review, the authors will trace some of the biochemical connections between intestinal inflammation and neuropsychiatric symptoms and focus on strategies to manage both. Additionally, the authors offer a cautionary reflection on the extant need for widespread screening for depression among patients with IBD.
INTRODUCTION
In the first major treatise on the eponymous disease, Burrill Crohn himself noted that: “The number of cases of ileitis that have been rescued from institutions for the treatment of mental diseases emphasises not the personality but the end results of the drain of the disease upon the psychic constitution of the sufferer” (emphasis added).1 In the 70 years since that prescient statement, a high incidence of neuropsychiatric symptoms (depression, anxiety, cognitive fatigue, and sleep disorders) in patients with inflammatory bowel disease (IBD) has been consistently observed.2-4 As with IBD, depression lacks clear biomarkers for definitive diagnosis, and frequently involves complex treatment regimens. Unfortunately, there is no parallel imaging technique to colonoscopy for the brain that is comparably simple and widely available. As a result, diagnosis and treatment of depression presents unique challenges alone, and those challenges are compounded both by systemic inflammation and by various medications. Since patients with depression have significantly increased rates of relapse, surgery, hospitalisation,5 and suicide,6 recognising and treating depression is of paramount importance. In this narrative review, the authors will trace some of the biochemical connections between intestinal inflammation and neuropsychiatric symptoms and focus on strategies to manage both.
COMMON THREADS: CYTOKINES
To illuminate the connections between the immune system, peripheral inflammation, and neuropsychiatric effects, it is instructive to observe the psychological effects when systemic inflammation is induced by an immediate immune challenge. One common method for inflammation induction is injection of Salmonella abortus equi endotoxin.7 In healthy volunteers, endotoxin rapidly activates the host-defence system, inducing both TNFα and IL-6 (see below), resulting in a constellation of disrupted neurological functions referred to as ‘sickness behaviour’. Among the disruptions are extraintestinal symptoms well known to legions of those with IBD: anxiety, depressed mood, fatigue, and memory impairment. In the context of treating IBD, it is important to note that both the anxiety and depressed mood normalised in parallel with decreases in the cytokine concentrations. Similar mood effects are seen with Salmonella Typhii vaccine8 and Bacillus Calmette–Guérin vaccine,9 with concomitant elevation of the same cytokines. It is also worth remembering that these were healthy volunteers, subjected to a short-duration, low-dose immune challenge in which both the cytokine levels and neuropsychiatric symptoms typically resolve in a timescale of hours. By contrast, patients with IBD have chronic immune activation, and frequently higher circulating amounts of those same cytokines, even in remission.10 Patients with IBD are, therefore, subject to prolonged alterations in brain function leading to anhedonia, anxiety, and cognitive fatigue.2-4
Perhaps the most insightful example for this discussion is exogenous IFNα, used for both chemotherapy and hepatitis C. There are two advantages to using IFNα therapy as a comparative study. First, given the widespread use of IFNα, there is a large dataset with robust documentation of systemic effects and mechanisms in both animals and humans. Second, owing to repeated and higher dosings over weeks and months, IFNα treatment more closely aligns with the chronic inflammatory state typically presented in IBD. As above, IFNα induces a rapid and sustained increase in other inflammatory cytokines, particularly TNFα and IL-6.11 Significantly, up to 50% of patients being treated with IFNα experience major depressive symptoms, with 80% reporting sickness behaviour.12,13 While those percentages are initially alarming, numerous large studies of patients with IBD have estimated between 20 and 45% of patients with IBD experienced at least some depressive symptoms,14 and elevated rates of depressive disorder of 15% or higher.2
TNFα
TNFα has been ascribed a central role in chronic inflammation not only in IBD, but also rheumatoid arthritis (RA), plaque psoriasis, psoriatic arthritis, ankylosing spondylitis, and systemic lupus erythematosus. TNFα interacts with the Type 2 receptor, leading to an inflammatory cascade via NF-κB activation. NF-κB is a transcription factor for an enormous number of inflammatory genes,15,16 including virtually every molecule discussed in this review, including itself.
Along with, and because of, the dysregulation of so many critical factors for neurotransmission, it is little surprise that circulating TNFα is strongly associated with a variety of psychiatric effects, including depression, anxiety, cognitive fatigue, and sleep disturbances.7,8,11,17-21
Serum TNFα levels have been found to be significantly elevated in patients in the acute phase of major depressive disorder (MDD).17 While levels decreased during antidepressant treatment, they remained elevated in the chronic phase of MDD. Similarly, post-mortem examination of individuals with depression has found elevated levels of TNFα in a variety of areas of the brain.18 Perhaps unsurprisingly, both TNFα protein and mRNA were also elevated in the prefrontal cortex of teenage suicide victims,19 as well as in plasma of some adults attempting suicide.20 In clinical trials, systemic TNFα infusion was tested for proapoptotic activity with certain cancers.22 Many patients developed dose-dependent, reversible, attentional deficits, memory disorders, fatigue, confusion, and in some cases neurotoxicity.
In 1998 the U.S. Food and Drug Administration (FDA) approved infliximab for Crohn’s disease, the first biologic inhibitor for TNFα, heralding a new age in the treatment of IBD. Almost overnight, anecdotal reports of rapid antidepressant effects surfaced, with later imaging studies demonstrating observable changes in brain function in 48 hours,21 and biochemical changes within 24 hours.23 As a result, in 2013, a clinical trial of infliximab for treatment-resistant depression was undertaken.24 While inhibition of TNFα was not shown to be uniformly effective, it was effective in a subgroup of patients with higher levels of C-reactive protein (CRP). This trial suggested that depression is not purely an inflammatory process, but is certainly exacerbated by existing inflammation, and treatable by suppressing inflammatory cytokines, adding to the results observed in IBD, RA,
and psoriasis.25
Notably, antidepressant effects of TNF blockade are not exclusive to infliximab, nor even to antibodies. Thalidomide, which acts as a small-molecule inhibitor of TNFα, also shows antidepressant effects.26
IL-6
Along with TNFα, IL-6 is elevated in virtually all autoimmune inflammatory disorders, and has a strong association with depression.27 Among its pleiotropic roles are induction of CRP,28 weakening of tight junctions at the intestinal epithelial barrier and the blood–brain barrier (BBB),28 and differentiation of T helper cells into Th17 (IL-17 producing) cells. IL-6 expression is controlled, at least partially, by NF-κB. In Crohn’s patients, IL-6 is highly upregulated in active disease, and is strongly downregulated in infliximab responders.29 In animal studies, IL-6 has been shown to directly pass through the BBB and into the brain.30As with TNFα, average IL-6 levels have been found to be elevated in the brains of successful suicides and in the cerebrospinal fluid of attempters. There is a rich literature demonstrating the strong relationship between IL-6 and dopamine in the symptoms of anhedonia,27,28,31,32 a core symptom of MDD. Even a comparably low level of IL-6 is strongly tied to motivational effort,32 and as such is a likely contributor to the high degree of treatment nonadherence observed in IBD patients.33
Unlike TNFα, where exogenous cytokine was tested only on ill patients, the effects of IL-6 have been directly tested by subcutaneous injection of the recombinant protein into healthy volunteers.34 Within 4 hours, self-reported measures of concentration, self-reliance, and ‘high spirits’ had all dropped significantly, while fatigue, sadness, and anxiety had all increased. Sleep analysis of this same cohort showed a significant decrease in time spent in the rapid eye movement phase of sleep. As emphasised above, these were healthy volunteers with a single low-dose acute stimulus over a short time period. As with TNFα, it appears that a significant portion of patients with IBD do not have sufficient biochemical adjustment to increased IL-6 levels, and thereby experience more chronic versions of the short-term effects listed above, manifested in sleep and mood disorders.
Given early success in blocking TNFα and subsequent focus on more gut-specific inflammation (vedolizumab), IL-6 inhibitors have not yet been used extensively in IBD despite successful clinical trials. However, studies in RA have shown that blockage of the IL-6 signalling (either antibodies to IL-635 or to the IL-6 receptor36) has positive effects on depression and anxiety symptoms, as well as improvements to sleep.
IL-17A
Of the cytokines commonly associated with both IBD and depression, IL-17A is particularly enigmatic. Th17 cells are induced either by IL-6 or by IL-23, leading to the production of IL-17A. Similar to IL-6, IL-17A is directly involved in weakening both the intestinal epithelial barrier and the BBB. In the brain, IL-17A is associated with a host of neurological diseases, including multiple sclerosis, ischaemic brain injury, and Alzheimer’s disease.37 IL-17A also appears to directly induce microglia into the reactive state, leading to significant monoamine neurotransmitter issues (see below).38 One IL-17A inhibitor in particular, Ixekizumab, demonstrated parallel results to those mentioned above in improving depressive symptoms in patients with plaque psoriasis.39 Despite initial failure as a therapeutic target in Crohn’s disease, IL-17A is still a potentially useful biomarker because IL-17A levels are associated with antidepressant effectiveness40 and even with infliximab response.41
Other Targets
While not as frequently implicated in depression, both IL-12 and IL-23 have been associated with depressive symptoms. Inhibition of both IL-12/IL-23 (ustekinumab)42 or IL-23 alone (guselkumab)43 improve depression and anxiety scores. Somewhat more surprising is the finding that vedolizumab, an α4β7-integrin inhibitor acting virtually exclusively in the gut, also ameliorates depression and anxiety, and improves sleep quality.44In fact, a 2016 meta-analysis showed that inhibition of virtually any single cytokine involved in inflammation improved symptoms of depression.25
TRANSMISSION TO THE BRAIN
There are three distinct pathways for transmission of inflammation from the intestines to the brain: the neural, the humoral, and the cellular. It is very likely that all three are in play with IBD.
In the neural pathway, cytokines bind directly to afferent fibres of the vagus nerve, transmitting the signal directly to the brain. Since this signalling does not require any molecular transport across the BBB, the transmission is fastest. There is a wealth of data on direct vagus stimulation (albeit not from the intestines) for treatment-resistant depression.45
In the humoral pathway, cytokines and other molecular miscreants directly cross the BBB into the brain, either passively or by active transport. This is perhaps the best studied of the three pathways, because BBB leakage is implicated in any number of neurological disorders, and it becomes more pronounced with age;46while active transport of cytokines and other immunomodulators is partly circadian, and is associated with sleep disruptions.47 It is worth noting that the epithelial barriers of the intestine, the retina, and the brain are similarly constituted and disrupted by many of the same factors.48 For example, Calarge et al.49 were recently able to show that increased intestinal permeability is directly associated with depressive symptoms, specifically in unmedicated adolescents. This aligns well with other data demonstrating that both IL-6 and IL-17 alter the integrity of the BBB from the periphery, while activated microglia also act to disrupt the BBB and express cytokines and chemokines from the inside.50
At least partially, the humoral pathway opens the door to the cellular pathway, both by loosening the tight-junction regulation and by increased expression of monocyte chemotactic protein (MCP-1/CCL20) in brain endothelial cells, thereby permitting cell trafficking of monocytes and macrophages into the brain.51
EFFECTS ON MONOAMINE NEUROTRANSMITTERS
Mechanistically speaking, the effects of inflammatory cytokines represent a form of ‘perfect storm’ for effects on the brain, and specifically on the synthesis and availability of monoamine neurotransmitters. Figure 1 and 2 illustrate a number of the pathways leading to neurological effects of inflammation.
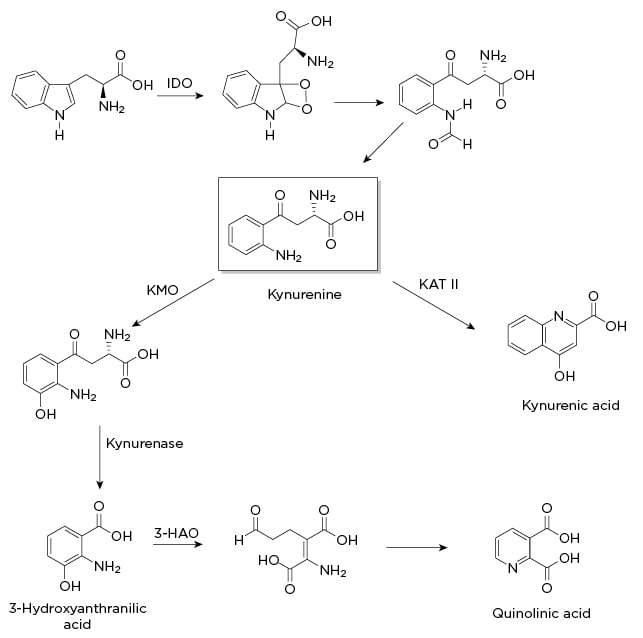
Figure 1: Indole-2,3-dioxygenase pathway.
3-HAO: 3-hydroxyanthranilic acid oxidase; IDO: indole-2,3-dioxygenase; KAT II: kynurenine aminotransferase II; KMO: kynurenine 3-monooxygenase.
Figure 2: Inflammation-altered pathways in neurotransmission.
Synthesis of 5HT, MT, DA, and NE are slowed by lowered availability of BH4 and B6. BH4 is a cofactor in the hydroxylation of TRP, PHE, and TYR. B6 is a cofactor in the decarboxylation of DOPA to form DA, and the decarboxylation of 5HTP to form 5HT. Reuptake of DA by the DAT and of 5HT by the SERT are increased.
Activated microglia express three enzymes responsible for increased glutamate and glutamatergic signalling: 1) IDO1 catabolises TRP to QA. QA is an agonist at the NMDAR, leading to GLU release. 2) GCPII cleaves NAAG to N-acetylaspartate (not shown) and GLU. As a result, NAAG signalling at both the mGlu3 and the NMDAR are decreased. 3) GLS hydrolyses GLN to GLU. Removal of excess GLU by the GLT1/EAAT2 is also downregulated (not shown).
Impaired or downregulated pathways are shown with dotted arrows.
5HT: serotonin; 5HTP: 5-hydroxytryptophan; BH4: tetrahydrobiopterin; B6: pyridoxyl-5’-phosphate; DA: dopamine; DAT: dopamine transporter; DOPA: L-3,4-dioxyphenylalanine; EAAT2: excitatory amino acid transporter 2; GCPII: glutamate carboxypepsidase-II; GLN: glutamine; GLS: glutaminase; GLT1: glutamate transporter-I/; GLU: glutamate; IDO1: Indole-2,3-dioxygenase; mGlu3: metaboglutamate-3 receptor; MT: melatonin; NAAG: N-acetylaspartylglutamate; NE: norepinephrine; NMDAR: N-methyl-D-aspartate receptor; PHE: phenylalanine; QA: quinolinic acid; SERT: serotonin transporter; TRP: tryptophan; TYR: tyrosine.
The Indole-2,3-dioxygenase pathway: The Monoamine Crossroads
One of the most well documented alterations in neurotransmitter levels is brought about by indole-2,3-dioxygenase (IDO1), a uniquely destructive nexus in the synthesis, signalling, and release of monoamines. As shown in Figure 1, this enzyme catabolises tryptophan, thereby inhibiting serotonin synthesis, increases excitotoxic glutamatergic signalling and glutamate release, and in turn hinders dopamine release. When it was discovered, IDO1 was thought to be an intestinal variation of the tryptophan-2,3-dioxygenase (expressed primarily in the liver). Later studies demonstrated that IDO1 is induced by cytokines and transcribed by NF-κB. One proposed rationale for this particular enzyme is an evolutionary defence against pathogens by lowering the amount of available tryptophan, with depressive symptoms restricting social behaviour and thereby transmission of infectious agents.52
Overexpressing this defence mechanism has the unfortunate consequence of also depleting not only available serotonin, but by extension melatonin, the monoamines most commonly associated with depressive symptoms and sleep disorders. This diversion may help explain the blunted response to selective serotonin reuptake inhibitors (SSRI) found in many patients with IBD, since SSRI drugs act downstream of neurotransmitter synthesis. While most studies of IDO1 are focussed on depletion of serotonin, it is important to note that melatonin is an important inhibitor of MMP-9,53 an upregulated enzyme that causes epithelial barrier damage. As such, depletion of melatonin likely plays a role in both intestinal and BBB permeability. Infliximab treatment strongly downregulates the expression of IDO1, and this may be a significant explanation for the antidepressant effects of anti-TNF treatment, as well as its effects on sleep quality.29
Additionally, in animal models, inhibition of IDO1 by 1-methyltryptophan had comparable effects on depressive behaviour to treatment with infliximab, suggesting that IDO1 may be a viable drug target for inflammation-mediated depression.54 There are multiple clinical trials investigating IDO1 inhibitors as add-on therapy for cancer treatments, so there may well be an approved drug in this class in the next several years.
Glutamate–N-methyl-D-aspartate receptor
As a continuation of the effects of IDO1, a primary catabolite of tryptophan is quinolinic acid (QA; Figure 1). QA is an N-methyl-D-aspartate receptor (NMDAR) agonist, leading to increased glutamate signalling, release of additional glutamate, and inhibition of the release of dopamine. QA binds preferentially to NMDAR in the forebrain, in areas critical to mood, memory, and sleep regulation.55 Multiple studies have shown that the cognitive fatigue in IBD is postsynaptic in origin, and it therefore seems likely that NMDAR signalling plays a critical role.21 One of the purported sites for the rapid antidepressant action of ketamine in treatment-resistant depression is the NMDAR. In humans with treatment-resistant depression, low-dose ketamine as an add-on therapy dropped levels of both TNFα and IL-6,56 while one animal study showed it also lowered the levels of QA and microglia.57 It remains to be seen how effective ketamine will be in patients with IBD, but the studies above suggest a more complex and useful anti-inflammatory role in addition to its use as an antidepressant.
Excitotoxicity in the immediate term is most likely manifested by anhedonia and ‘brain fog’. However, in the long term, excitotoxicity destroys neurons, specifically dopaminergic neurons. This destruction is exactly the type of process thought to lead to Parkinson’s disease. Notably, IBD has long had a particularly strong association with development of Parkinson’s disease,58 and NMDAR glutamate signalling could be a significant contributor. In support of this hypothesis, consider that IDO1 catabolites are increased in the cerebrospinal fluid of Parkinson’s patients,59 and animal models of Parkinsonism are partially induced with QA.60
Not only is glutamate release and signalling increased thanks to QA, but also by upregulations of glutamate carboxypeptidase II61 and glutaminase.62 By hydrolysing either N-acetylaspartylglutamate or glutamine, additional glutamate is released, augmenting the glutamatergic (excitotoxic) responses. Increased glutamate is indeed visible in the brain by proton magnetic resonance spectroscopy.63 Along with increased release, signalling, and synthesis of glutamate, the primary reuptake transporter (EAAT2/GLT1) is downregulated.64Added to that, decarboxylation of glutamate to GABA by GAD1 is likely regulated by NF-κB, and requires vitamin B6 as a cofactor (see below). With these enzymes combined, glutamate excitotoxic signalling almost certainly plays a central role in the neuropsychiatric comorbidities of IBD.
Tetrahydrobiopterin and Neopterin
Along with upregulation of IDO1, cytokines also are associated with diversion of the catabolic synthesis of tetrahydrobiopterin (BH4), a critical cofactor for hydroxylation of phenylalanine, tyrosine, and tryptophan, the rate-limiting step en route to the catecholamines and serotonin/melatonin. Depletion of BH4 actually occurs in two ways. First, large upregulation of the first enzyme in the BH4 synthesis (GTP cyclohydrolase) creates a synthesis bottleneck, overwhelming the second enzyme in the pathway.65 As a consequence, the critical BH4 precursor is unhelpfully diverted to neopterin. Second, cytokines strongly induce nitric oxide synthase and reactive oxygen species, both of which require BH4 as a cofactor,66 depleting the available supply. Both processes decrease the availability of monoamine neurotransmitters, and are associated with anxiety, fatigue, and anhedonia. In one particularly elegant study by Felger et al.,67 reverse microdialysis of L-DOPA into the brains of rhesus monkeys reversed the symptoms of IFNα-induced depression, suggesting that hydroxylation (controlled by BH4 levels) was the dominant issue, and subsequent decarboxylation and vesicular transport (via VMAT2) might be functioning normally.
Pyridoxyl-5’-Phosphate: Vitamin B6
The final step in the synthesis of serotonin from 5-hydroxytryptophan, dopamine from L-DOPA, and GABA from glutamate is decarboxylation by the amino acid decarboxylases. As with the hydroxylases discussed above, these enzymes are dependent on the availability of a cofactor: in this case, pyridoxal-5’-phosphate (the active form of Vitamin B6), of which patients with IBD are frequently deficient.68 Studies on RA suggest that deficits in active B6 are not an absorption issue, but rather alterations in processing and metabolism.69While there are considerably less data available, it has been reported that tryptophan metabolites inhibit the phosphorylation of pyridoxal.70 A recent animal study showed that NF-κB knockout mice had a downregulation of AOX1, the principal metabolic enzyme responsible for converting pyridoxal-5’-phosphate to pyridoxic acid.71 While this finding does not prove AOX1 is upregulated by NF-κB, it certainly fits the pattern of NF-κB playing a central controlling role in inflammatory responses in the brain.
CONCLUSIONS AND FINAL THOUGHTS
Reflecting the mounting evidence, the American College of Gastroenterology (ACG), the
British Society for Gastroenterology (BSG), the American Gastroenterological Association (AGA), the World Gastroenterology Organisation (WGO), and the Crohn’s & Colitis Foundation have all added some level of psychiatric assessment and management to their respective clinical guidelines. This is an important step toward successfully treating patients with IBD. However, as an example, the ACG recommendation (strong recommendation, very-low level of evidence)72,73 points out a larger problem in the need for broad screening. As an illustration, in a 2017 report, >4,000 patients were asked to self-evaluate, with only 20% of patients with Crohn’s disease and 14% of patients with ulcerative colitis claiming to be depressed. However, by using a short questionnaire (Patient Health Questionnaire depression scale [PHQ-8]), an alarming 38% of patients with Crohn’s disease and 32% of patients with ulcerative colitis in the same cohort met the criteria for depression.5 Given the antidepressant effects of some treatments (see above), the actual percentages in treatment-naïve patients could be considerably higher. Since depression has such strong effects on motivation (including adherence with diagnostic and treatment protocols),74 devastating effects on disease progression, and significant increases to mortality, the need for psychiatric screening as part of disease management cannot be overstated.
There are two additional problems to be addressed in screening and reporting. First, the phrase “antidepressant usage” found in many studies is wholly insufficient. This phrase is comparable to using “IBD medication” in gastroenterology. Different antidepressant classes are as mechanistically dissimilar from each other as aminosalicylate and infliximab, possibly even more so. Given that some specific SSRI drugs (mirtazapine) are reported to worsen systemic inflammation,75 while at least one norepinephrine–dopamine reuptake inhibitor (bupropion) is reported to lessen it,76 such granular detail is critical.
Second, as mentioned in the introduction, there is a dearth of reliable biomarkers for depression and anxiety. As a result, diagnosis often relies on various questionnaires that are not entirely comparable. It would be wise of the advisory bodies of IBD treatment to recommend a diagnostic tool for depression alongside the aforementioned need for its diagnosis.
Using the statistics quoted above, roughly one in three patients with IBD is potentially depressed, and possibly as high as one in six has considered, or is considering, suicide as a result of that depression within the past year.77 For a typical gastroenterologist, that translates to seeing multiple patients per week that could be actively suicidal. Would their approach change if aware that the next patient was slowly (or rapidly) losing the battle against depression exacerbated by IBD? One very small scale report from the Jackson County, Missouri, USA, Medical Examiner’s Office might provide some insight.78 Of 12 Crohn’s patients that died from 2008 to 2010 (average age: 45 years), there were five suicides, three accidental drug overdoses, and one case of liver failure from alcohol abuse. Only two of 12 died of IBD complications. As the authors rightly point out, these numbers certainly will not scale to the whole population, and treatments have certainly improved in the past decade. However, this paper serves as a sobering reminder that the intestines are not the only battlefront for IBD patients, nor their caregivers.






