


OBJECTIVE—Cardiac fibrosis is an important component of diabetic cardiomyopathy. Peroxisome proliferator–activated receptor γ (PPARγ) ligands repress proinflammatory gene expression, including that of osteopontin, a known contributor to the development of myocardial fibrosis. We thus investigated the hypothesis that PPARγ ligands could attenuate cardiac fibrosis.
RESEARCH DESIGN AND METHODS—Wild-type cardiomyocyte- and macrophage-specific PPARγ−/− mice were infused with angiotensin II (AngII) to promote cardiac fibrosis and treated with the PPARγ ligand pioglitazone to determine the roles of cardiomyocyte and macrophage PPARγ in cardiac fibrosis.
RESULTS—Cardiomyocyte-specific PPARγ−/− mice (cPPARγ−/−) developed spontaneous cardiac hypertrophy with increased ventricular osteopontin expression and macrophage content, which were exacerbated by AngII infusion. Pioglitazone attenuated AngII-induced fibrosis, macrophage accumulation, and osteopontin expression in both wild-type and cPPARγ−/− mice but induced hypertrophy in a PPARγ-dependent manner. We pursued two mechanisms to explain the antifibrotic cardiomyocyte-PPARγ–independent effects of pioglitazone: increased adiponectin expression and attenuation of proinflammatory macrophage activity. Adenovirus-expressed adiponectin had no effect on cardiac fibrosis and the PPARγ ligand pioglitazone did not attenuate AngII-induced cardiac fibrosis, osteopontin expression, or macrophage accumulation in monocyte-specific PPARγ−/− mice.
CONCLUSIONS—We arrived at the following conclusions: 1) both cardiomyocyte-specific PPARγ deficiency and activation promote cardiac hypertrophy, 2) both cardiomyocyte and monocyte PPARγ regulate cardiac macrophage infiltration, 3) inflammation is a key mediator of AngII-induced cardiac fibrosis, 4) macrophage PPARγ activation prevents myocardial macrophage accumulation, and 5) PPARγ ligands attenuate AngII-induced cardiac fibrosis by inhibiting myocardial macrophage infiltration. These observations have important implications for potential interventions to prevent cardiac fibrosis.
Progressive cardiac fibrosis found in diabetic cardiomyopathy and postmyocardial infarction remodeling and during pressure overload can result in diastolic dysfunction leading to reduced myocardial contractility and, ultimately, heart failure (1–4). Both angiotensin II (AngII) and aldosterone promote cardiac hypertrophy, inflammation, and fibrosis, whereas their antagonism improves cardiac function and decreases mortality in heart failure (5–9). However, little is known about the cellular mechanism of these effects, and the prevalence of and mortality from heart failure continues to increase and is particularly common in subjects with diabetes (10). We have shown that osteopontin (OPN), a secreted inflammatory glycophosphoprotein, plays a pivotal role in cardiac fibrosis (11) and is often increased in the tissues of diabetic mouse models (11–13). Ventricular OPN expression is increased during heart failure, paralleling the increase in atrial natriuretic peptide (ANP) expression, and AngII prominently upregulates OPN in cardiac cells (12,13). Moreover, OPN knockout (KO) mice develop less cardiac fibrosis than control mice (11) and are partially protected against streptozotocin-induced diabetic cardiomyopathy (14).
Peroxisome proliferator–activated receptors (PPARs) are ligand-activated transcription factors that control expression of key genes that modulate pathways involved in fat, lipid, and glucose metabolism; inflammation; cell cycle; and immune responses (15). Synthetic PPARγ ligands are insulin sensitizers and have profound anti-inflammatory effects, one of which is to decrease OPN expression in vascular and renal cells (16). Therefore, we hypothesized that PPARγ agonists can attenuate myocardial fibrosis by reducing OPN expression in cardiomyocytes and/or macrophages, thereby decreasing cardiac inflammatory responses that can result in fibrosis. To test this hypothesis, we analyzed the effect of a PPARγ agonist on cardiac hypertrophy and fibrosis in an AngII-stimulated model using both cardiomyocyte- and macrophage-specific PPARγ KO mice and their wild-type (WT) littermate controls.
RESEARCH DESIGN AND METHODS
Generation of PPARγ-deficient mice.
Cardiomyocyte-specific PPARγ KO (cPPARγ−/−) mice were bred by mating homozygous PPARγflox/flox DBA/2J mice (gift from Dr. F. Gonzalez) (17) with C3H transgenic mice expressing Cre recombinase under the control of the myosin light chain-2v (MLC2v-Cre) promoter (18). The F1 generation was backcrossed with homozygous PPARγflox/flox DBA mice to fix the PPARγflox/flox genotype, and successive generations were mated with their littermates. Macrophage-specific PPARγ KO (mPPARγ−/−) mice (kindly provided by Dr. P. Tontonoz, University of California Los Angeles) were generated by crossing C57Bl/6 transgenic mice expressing Cre recombinase under the control of the lysozyme promoter (LysM-Cre) into homozygous PPARγflox/flox C57Bl/6 mice (19,20). Mouse genotypes were determined by PCR (see the online appendix, available at http://dx.doi.org/10.2337/db07-0924).
Animal procedures.
Male mice (3 months of age) were fed chow diet (Harlan 8604) or chow diet supplemented with 2 mg pioglitazone/1g diet and, 2 days after starting diet, were infused with PBS or a pressor dose of AngII/PBS (2.5 μg · kg−1 · min−1; Calbiochem) for the indicated treatment intervals by subcutaneous implantation of osmotic minipumps (DURECT) (21–23). Systolic blood pressure (SBP) was measured weekly with a tail-cuff system (11). WT and cPPARγ−/− mice were assessed by echocardiography before minipump implantation and after a 6-week AngII infusion, as previously described (24). Heart rates were determined during echocardiography (under anesthesia) and during SBP measurements. No differences were detected between any of the groups. All animal procedures used in this study were approved by the University of California Los Angeles Animal Research Committee.
Morphometric analysis, cardiac fibrosis area, and macrophage content.
Coronal heart sections were cut at the ventricle equator and fixed in 10% paraformaldehyde/PBS, paraffin embedded, and stained with Massons’ trichrome to detect collagen expression. Microscope images were displayed on a high-resolution monitor, digitized by a video frame grabber (PCVISION Plus; Imaging Technology, St. Laurent, Canada) running on an IBM-compatible computer, and areas positively staining for collagen were quantified with a morphometric analysis program (Image Pro; Media Cybernetics, Silver Spring, MD). Contiguous high-power fields comprising an entire left ventricular section were analyzed for each sample. Cardiac fibrosis area (blue area) was expressed as a percentage relative to the entire cardiac cross-sectional area. Cardiomyocyte diameters were determined from 100 random fibers by a single operator blinded to the study protocol, essentially as previously described (25).
Adiponectin overexpression.
Adenovirus expressing mouse adiponectin (Ad-APN) was generated from the full-length cDNA (26), subcloned with an Adenovirus Expression Vector Kit (Takara Biomedical), propagated in HEK293 cells, and purified and quantified with BD Adeno-X Virus Purification and Rapid Titer kits (BD Biosciences). Adenovirus expressing green fluorescent protein (Ad-GFP) was similarly constructed as a control. Male WT and cPPARγ−/− mice (3 months of age) were injected via tail vein with 1 × 108 plaque-forming units of virus 5 days before the start of a 6-week AngII infusion on chow diet. Plasma adiponectin levels were measured by ELISA (K1002–1; B-Bridge).
Cardiac gene expression.
RNA was isolated from ventricular heart sections with Trizol reagent (Gibco-BRL, Rockville, MD) and subsequently processed with RNeasy Mini Kits and RNase-Free DNase (QIAGEN) to remove genomic DNA from the resulting mRNA samples. Cardiac mRNA samples were reverse transcribed with random hexamers and MultiScribe RT polymerase (Applied Biosystems). Real-time quantitative RT-PCR was performed using an ABI PRISM 7700 to measure gene expression (see online appendix for specific primer/probe sets). Gene expression values were normalized against glyceraldehyde-3-phosphate dehydrogenase.
Adult mouse cardiomyocyte analyses.
Adult mouse cardiomyocyte (AMCM) cultures were prepared according to Alliance for Cellular Signalling protocols (27). Immunohistochemistry was performed by standard procedures with specific antibodies for PPARγ (SA-206; Biomol) and myosin (MF 20; Developmental Studies Hybridoma Bank). Macrophage migration assays were performed in 96-well chambers (CytoSelect CBA-105; Cell Biolabs) according to the manufacturer's instructions. Briefly, clarified conditioned media from a 48-h AMCM culture was placed in the lower compartment of the chamber, and a 100-μl aliquot of a murine J774A.1 macrophage suspension (5 × 105 cells/ml) in myocyte culture medium was added to the upper compartment. After 4.5 h, migratory cells were detected with CyQuant GR Dye (Molecular Probes). Western blot analyses of AMCM protein (20 μg) were performed with a mouse-OPN–specific antibody (MPIIIB; Developmental Studies Hybridoma Bank). Stimulation experiments were performed with WT and cPPARγ−/− AMCMs, which were serum-deprived for 1 h, and then stimulated with AngII and aldosterone (1 μmol/l each) as indicated.
Statistical analysis.
Data are presented as means ± SEM. Data presented in Figs. 2, 4, and 5 were analyzed by one-way ANOVA due to their three-treatment design, using Tukey-Kramer multiple comparisons tests to determine treatment differences between individual means. Genotype differences between matched treatment groups were analyzed using two-tailed Student's t tests. In cases of unequal variance or non-normal distribution, nonparametric Mann-Whitney two-sample tests or Kruskal-Wallis one-way ANOVAs with Dunn post hoc analyses were performed to determine significant differences between individual means. A P value ≤0.05 was considered statistically significant for all tests. Data presented in Tables 1 and 2 and Fig. 3 were analyzed by two-way ANOVAs due to their 2 × 2 factor design, and Bonferroni post hoc analyses were used to identify differences between specific groups. In cases of unequal variance or non-normal distribution, data were analyzed using a linear regression approach in which the continuous data variables were sorted in ascending order and then assigned rank values (k = 1, 2 …, n), with tied values averaged from the start to the end of the run. Ranked values were transformed into percentiles using the relationship k/(n + 1) and then transformed into quantiles of the standard normal distribution using the inverse cumulative normal function, resulting in a symmetric standard normal distribution with mean zero and variance one. Normal scores were regressed on indicator variables for treatment, genotype, and genotype-treament interaction. The genotype-treatment interaction variable was based on the product of genotype times treatment. For each outcome variable, the Bonferroni multiple test procedure was performed to provide t tests for all possible combinations of treatment and genotype. Bonferroni P values ≤0.05 per number of tests were considered statistically significant for all analyses.
RESULTS
Cardiomyocyte-specific PPARγ KO (cPPARγ−/−) mice have cardiac hypertrophy.
cPPARγ−/− mice were created by crossing MLC2v Cre mice, which selectively delete floxed target genes in ventricular cardiomyocytes starting at the early stages of cardiac development (28), into a homozygous PPARγflox/flox mouse background. Consistent with the fact that cardiac tissue contains several cell types in addition to including cardiomyocytes, cPPARγ−/− mice ventricles expressed only 37% less PPARγ mRNA than those of WT littermates (Fig. 1A). However, PPARγ mRNA was not detectable in AMCMs isolated from cPPARγ−/−, and confocal microscopy demonstrated complete ablation of cardiomyocyte PPARγ protein expression (Fig. 1B). These data suggest that there is substantial PPARγ expression by other myocardial cell types. Cardiac PPARα and PPARδ mRNA expression were not different in cPPARγ−/− and WT mice (data not shown), suggesting that there were no substantial changes in cardiac PPARα and PPARδ mRNA expression to compensate for the loss of cardiomyocyte PPARγ expression.
cPPARγ−/− mice demonstrated normal reproduction and life expectancy with no spontaneous pathological phenotype, despite mild cardiac hypertrophy associated with increased ventricular atrial natriuretic peptide (ANP) expression (Table 1). Conversely, the PPARγ ligand pioglitazone increased ventricular heart–to–body weight ratios and ventricular ANP expression in WT, but not in cPPARγ−/−, mice (Table 1). Thus, both cardiomyocyte PPARγ deficiency and activation resulted in cardiac hypertrophy. Cardiomyocyte PPAR deficiency was also associated with differences in cardiomyocyte diameter and ventricular brain natriuretic peptide (BNP) expression, while pioglitazone treatment altered cardiomyocyte diameter and ventricular BNP mRNA expression and produced genotype-specific effects on heart–to–body weight ratios and ventricular ANP mRNA expression (Table 1, two-way ANOVA).
Neither cardiac fibrosis nor transforming growth factor (TGF)-β1 or fibronectin gene expression, assayed as surrogate markers of cardiac fibrosis, was significantly different in cPPARγ−/− versus WT mice (Fig. 2A–D). However, cPPARγ−/− mice demonstrated a 2.9-fold increase in cardiac expression of OPN, a proinflammatory monocyte chemoattractant, and a 1.4-fold increase in cardiac expression of CD68, a marker of macrophage accumulation (Fig. 2E and F), suggesting that cPPARγ−/− mice may have significantly increased basal myocardial inflammation, which could promote enhanced myocardial injury during exposure to cardiac stress.
AngII augments cPPARγ−/− versus WT mice cardiac hypertrophy and fibrosis.
To evaluate the role of cPPARγ in cardiac stress responses, mice were infused with AngII for 6 weeks at a dose known to cause hypertension, cardiac hypertrophy, and fibrosis without significant changes in plasma glucose or lipid levels (11,29). AngII infusion increased ventricular heart–to–body weight ratio and cardiomyocyte diameter in both genotypes, although cPPARγ−/− mice demonstrated significantly greater increases than their WT littermates, despite similar SBP increases in both groups (Table 1). Ventricular ANP and BNP mRNA expression was also significantly increased by AngII in both genotypes but did not significantly differ between genotypes (Table 1). Cardiac fibrosis and myocardial expression of TGF-β1 and fibronectin, two fibrosis-related genes, also significantly increased with AngII infusion in both genotypes. However, whereas AngII induced nearly twofold more cardiac fibrosis in cPPARγ−/− than in WT mice (Fig. 2A and B), myocardial TGF-β1 and fibronectin mRNA expression were similarly induced in both genotypes (Fig. 2C and D), suggesting that increased TGF-β1 and fibronectin expression was not responsible for the increased cardiac fibrosis found in AngII-infused cPPARγ−/− mice. AngII also increased myocardial OPN and CD68 expression in both genotypes but more so in cPPARγ−/−mice, which had increased basal expression of these genes (PBS infusion, Fig. 2E and F). However, no changes in heart structure or function were detected by echocardiography in either genotype after AngII administration (data not shown).
Pioglitazone attenuates AngII-induced cardiac fibrosis.
To determine the ability of cPPARγ activation to attenuate AngII-induced cardiac hypertrophy and fibrosis, mice were AngII-infused with or without simultaneous treatment with the PPARγ ligand pioglitazone. Cardiomyocyte PPARγ deficiency was associated with increases in heart–to–body weight ratios and cardiomyocyte diameters in AngII-infused mice. Pioglitazone treatment increased heart–to–body weight ratios and cardiac BNP mRNA expression in AngII-infused mice but did not alter SBP, cardiomyocyte diameters, or ventricular ANP mRNA expression (Table 1, two-way ANOVA). No genotype-specific effects of pioglitazone were detected in AngII-infused mice. Pioglitazone treatment also similarly attenuated AngII-induced cardiac fibrosis (Fig. 2A and B) and myocardial expression of fibrosis-related (Fig. 2C and D) and proinflammatory (Fig. 2E and F) genes in both cPPARγ−/− mice and their WT littermate controls, suggesting that these antifibrotic and anti-inflammatory effects of pioglitazone are cardiomyocyte PPARγ–independent.
Adiponectin does not attenuate AngII-accelerated cardiac fibrosis.
We hypothesized that the antifibrotic effect of pioglitazone on AngII-induced cardiac fibrosis could be mediated through increases in plasma adiponectin because adiponectin overexpression has been reported to attenuate cardiac hypertrophy in mice (30) and because pioglitazone treatment increased plasma adiponectin threefold in AngII-infused cPPARγ−/− and WT mice (AngII 10.2 ± 2.6 μg/ml vs. AngII+Pio 33.9 ± 17.0 μg/ml; P = 0.0003). Mice were thus injected with Ad-APN or Ad-GFP 5 days before the start of a 6-week AngII infusion. Ad-APN markedly increased plasma adiponectin levels, reaching a peak of 50 times baseline (10-fold pioglitazone induction) at 2 weeks postinjection and declining to 10-fold baseline by study end. Ad-APN attenuated AngII-induced heart–to–body weight ratio increases in WT mice (5.85 ± 0.18 Ad-GFP vs. 5.21 ± 0.15 Ad-APN; P < 0.05) but not in cPPARγ−/− mice (6.48 ± 0.35 Ad-GFP vs. 6.35 ± 0.18 Ad-APN; P = 0.79). However, Ad-APN did not attenuate AngII-induced cardiac fibrosis (Fig. 3A and B) or reduce myocardial OPN (Fig. 3C) or CD68 (Fig. 3D) mRNA expression in cPPARγ−/− or WT mice. Thus, the increased systemic adiponectin levels after pioglitazone treatment were not responsible for attenuation of AngII-induced cardiac fibrosis and OPN and CD68 expression previously observed in cPPARγ−/− mice with pioglitazone treatment.
PPARγ−/− cardiomyocytes support increased macrophage chemotaxis.
Because cPPARγ−/− mice demonstrated significantly more myocardial expression of OPN and CD68 than WT mice (Fig. 2E and F) and because macrophages have been implicated in cardiac fibrosis (31), we hypothesized that cPPARγ−/− OPN expression could promote monocyte accumulation within the myocardium that could in turn promote cardiac injury in response to proinflammatory stimuli. We found that AMCMs isolated from cPPARγ−/− mice expressed significantly more OPN protein than WT AMCMs (Fig. 4A). AMCM cultures were stimulated with AngII and aldosterone to simulate the cardiomyocyte environment during in vivo AngII infusion because AngII markedly induces adrenal aldosterone secretion. Both cPPARγ−/− and WT AMCMs demonstrated increased OPN mRNA expression when cultured with AngII and aldosterone, but the addition of pioglitazone prevented OPN induction only in WT AMCMs (Fig. 4B). Moreover, cPPARγ−/− AMCMs expressed over fivefold more OPN than WT AMCMs under all culture conditions, and conditioned media from cPPARγ−/− AMCMs stimulated with AngII and aldosterone induced 1.5-fold greater macrophage migration than WT AMCM supernatant (Fig. 4C). These in vitro data support in vivo observations that increased OPN expression in cPPAR−/− cardiomyocytes contributes to increased cardiac macrophage accumulation.
Attenuation of fibrosis by pioglitazone requires macrophage PPARγ.
Because AngII markedly enhanced cardiomyocyte OPN expression, cardiomyocyte-mediated macrophage chemotaxis (Fig. 4B and C), and in vivo macrophage recruitment (32), we hypothesized that pioglitazone could attenuate fibrosis via anti-inflammatory actions on macrophages. Macrophage-specific PPARγ KO (mPPARγ−/−) mice, previously described by Babaev et al. (33), were thus analyzed using the same treatment protocol to address the role of macrophage PPARγ in cardiac fibrosis. After 6 weeks, PBS-infused mPPARγ−/− and WT mice demonstrated similar cardiac fibrosis, and AngII-infused mPPARγ−/− and WT mice demonstrated similar cardiac fibrosis increases, which were substantially greater than in cPPARγ−/− and cPPARγ+/+ mice (data not shown), likely due to the different genetic backgrounds of these mice. To study the effect of pioglitazone on similar levels of cardiac fibrosis in these two mouse models, mPPARγ mice were studied after 2 weeks of AngII infusion (Table 2 and Fig. 5A), at which time they developed cardiac hypertrophy and fibrosis levels comparable with those of cPPARγ mice that received 6 weeks of AngII infusion (Table 1 and Fig. 3A). After 2 weeks, PBS-infused mPPARγ−/− and mPPARγ+/+ mice had similar heart–to–body weight ratios and cardiomyocyte diameters (Table 2), cardiac fibrosis and myocardial OPN and CD68 mRNA expression (Fig. 5A–D), and circulating monocyte levels (data not shown). Pioglitazone increased AngII-mediated cardiac hypertrophy independent of genotype in mPPARγ mice (Table 2, two-way ANOVA), but reduced AngII-mediated cardiac fibrosis and OPN and CD68 gene expression in mPPARγ+/+ but not in mPPARγ−/− mice (Fig. 5A–D). This data suggests that macrophage mPPARγ was required for the antifibrotic actions of pioglitazone observed in AngII-infused mPPARγ+/+ (Fig. 5A) as well as cPPARγ−/− and cPPARγ+/+ mice (Fig. 3A). Thus, the macrophage appears to be a key mediator of fibrosis in the heart.
DISCUSSION
Although PPARγ ligands can promote edema by increasing renal sodium reabsorption to exacerbate heart failure (34), the metabolic and anti-inflammatory effects of these agents are implicated in cardiovascular protection (35). Indeed, a recent analysis of patients developing congestive heart failure with thiazolidinediones suggests that correction of heart failure is not associated with worsening of ejection fraction, which usually occurs with repeated bouts of congestive heart failure (36). We used an AngII-dependent cardiac fibrosis model in this study 1) because AngII is a potent profibrotic factor (37) and patients with type 2 diabetes may have increased renin-angiotensin-aldosterone system (RAAS) activity due to increases in adipose-derived angiotensinogen (38) and 2) because, importantly, PPARγ ligand effects on fibrosis could be studied in the absence of plasma glucose and lipid changes (11,29). We report herein that PPARγ ligands attenuate cardiac fibrosis via a mechanism that requires monocyte, but not cardiomyocyte, PPARγ. cPPARγ−/− mice had no apparent pathology, except for a mild cardiac hypertrophy, in the absence of an external stress. However, AngII infusion nearly doubled cardiac fibrosis and OPN expression in cPPARγ−/− mice relative to WT littermates. Isolated cPPARγ−/− AMCMs had more than a fivefold increase in OPN mRNA expression versus cPPARγ+/+ AMCMs, and conditioned media of cPPARγ−/− AMCMs demonstrated 1.5-fold more monocyte chemoattractant activity than cPPARγ+/+ AMCMs. Increased cPPARγ−/− cardiomyocyte OPN expression may contribute to increased monocyte accumulation both in the basal state and following AngII infusion, when inflammation plays a prominent role in cardiac fibrosis. However, noncardiomyocyte-driven inflammation appears critical to the cardiac fibrosis process in this model because pioglitazone attenuated cardiac fibrosis and ventricular OPN, TGF-β1, fibronectin, and CD68 mRNA expression in cPPARγ−/− and cPPARγ+/+ mice, but not in mPPARγ−/− and mPPARγ+/+ mice. Taken together, our data suggest that 1) mechanisms contributing to initiation of cardiac hypertrophy are different from those contributing to initiation of cardiac fibrosis, 2) cardiomyocyte PPARγ activation or deficiency contributes to cardiac hypertrophy, 3) inflammation is a significant driver of AngII-induced cardiac fibrosis, 4) both cardiomyocyte and macrophage PPARγ regulate cardiac macrophage infiltration, and 5) PPARγ ligands attenuate AngII-induced cardiac fibrosis by inhibiting macrophage infiltration into the myocardium.
Since pioglitazone treatment prominently increased plasma adiponectin, we also examined whether increased plasma adiponectin might explain the antifibrotic effect of pioglitazone, independent of cardiomyocyte PPARγ activity. Cardiac hypertrophy and mortality, induced by aortic constriction or AngII infusion, is rescued by adenoviral adiponectin in adiponectin-deficient mice (30). However, whereas pioglitazone improved AngII-induced cardiac fibrosis and increased plasma adiponectin in both WT and cPPARγ−/− mice, adiponectin overexpression had no effect on cardiac fibrosis in either genotype and attenuated cardiac hypertrophy only in WT mice. Thus, adiponectin does not appear to impact fibrosis. The question of why cardiomyocyte PPARγ was required for attenuation of cardiac hypertrophy by adiponectin requires further investigation.
Cardiac PPARγ deficiency, in this study and previous reports, is associated with cardiac hypertrophy and greater hypertrophic responses to stress (39,40). Similar to Duan et al. (40), we found that PPARγ activation induced cardiac hypertrophy, which was not detectable by echocardiography and which occurred without spontaneous systolic or diastolic dysfunction. Cardiac hypertrophy in their model was associated with activation of the nuclear factor κB pathway (40). In their study, however, the PPARγ ligand rosiglitazone induced cardiac hypertrophy in both the WT and KO animals, leading the authors to conclude that cardiomyocyte PPARγ only partially mediated the effect. They proposed that rosiglitazone stimulated the p38 mitogen-activated protein kinase pathway to activate cardiac hypertrophy through a mechanism distinct from that mediated by cardiomyocyte PPARγ (40). Cardiac phenotype differences between the present study and that of Duan et al. may result from genetic differences in the two models (MLC2v Cre vs. αMHC Cre) or differences in pioglitazone versus rosiglitazone effects. Neither thiazolidinedione has been shown to cause cardiac hypertrophy in humans (41,42).
RAAS-induced inflammation contributes to the pathogenesis of cardiac fibrosis. Cardiac OPN expression is increased by activation of both cardiac AngII type 1 receptors and mineralocorticoid receptors (37). We previously reported that cardiac OPN expression is elevated in rodent models of cardiac hypertrophy and in ventricles of explanted hearts from humans receiving cardiac transplants (13). OPN promotes cardiac fibroblast attachment to the extracellular matrix (ECM), and cardiac fibroblast growth and ECM production (12,43), whereas OPN-deficient mice have attenuated cardiac fibrosis, suggesting that OPN is a key profibrotic factor in the heart (11,44,45). None of these studies differentiated between the contributions of cardiomyocyte versus macrophage OPN deficiency in attenuating cardiac fibrosis, although we reported that macrophage OPN deficiency attenuated AngII-accelerated atherosclerosis by decreasing macrophage attachment and chemotaxis and by promoting macrophage apoptosis (46).
In this study, we found that cardiac OPN expression and macrophage accumulation were markedly increased in nonstressed cPPARγ−/− mice. Although increased cardiac OPN expression could result from increased macrophage accumulation, isolated cPPARγ−/− AMCMs also expressed more OPN than WT AMCMs, suggesting that both macrophages and cardiomyocytes contribute to elevated cardiac OPN expression in cPPARγ−/− mice. We did not, however, observe increased cardiac fibrosis in nonstressed cPPARγ−/− mice, even in 1-year-old mice, indicating that the increased cardiac OPN expression was not associated with spontaneous cardiac pathology during this time frame. Because OPN is a macrophage chemoattractant, increased cardiac OPN secretion could lead to cardiac macrophage accumulation, as observed in AngII-infused mice, further increasing myocardial OPN expression and creating a vicious cycle that could escalate the cardiac fibrotic process.
This study underscores a role for macrophages in cardiac fibrosis and a possible role for PPARγ ligands in the inhibition of cardiac fibrosis. Pioglitazone decreased cardiac fibrosis, macrophage accumulation, and OPN expression in cPPARγ−/−, cPPARγ+/+, and mPPARγ +/+ mice, suggesting that macrophage accumulation and OPN expression is a major mechanism of cardiac fibrosis induced by RAAS activation. PPARγ ligands suppress macrophage OPN expression (47), attenuate macrophage expression of several pro-inflammatory genes, and inhibit monocyte chemotaxis (48). Pioglitazone did not, however, attenuate cardiac fibrosis in mice with a monocyte-specific PPARγ deficiency, suggesting that PPARγ is required for the attenuation of monocyte inflammatory behavior that leads to cardiac fibrosis. Thus, the anti-inflammatory actions of PPARγ ligands may be useful for attenuating cardiac fibrosis and may be an adjunct therapy to RAAS inhibition. However, these putative anti-inflammatory cardiac actions of PPARγ need to be confirmed in humans. Further studies are also necessary to determine whether the metabolic and insulin-sensitizing effects of PPARγ ligands are also cardiomyocyte protective in diabetes and obesity and whether weaker insulin-sensitizing PPARγ ligands that cause less edema, which are currently under development, also attenuate cardiac fibrosis.
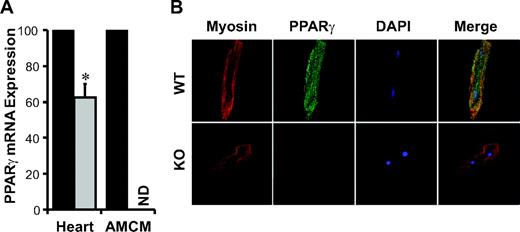
PPARγ expression in cardiac ventricles and AMCMs. A: Real-time quantitative RT-PCR of ventricular tissue of 4-month-old male WT and cPPARγ−/− mice and pooled AMCMs from male WT (black bars) and cPPARγ−/− (gray bars) hearts (n = 3/genotype; P < 0.05 by Student's t test). Values are normalized to the WT group. B: Immunohistochemistry of AMCMs of male WT and cPPARγ−/− (KO) mice. Cardiomyocytes shown are representative of at least three independent experiments. (Please see http://dx.doi.org/10.2337/db07-0924 for a high-quality digital representation of this figure.)
PPARγ expression in cardiac ventricles and AMCMs. A: Real-time quantitative RT-PCR of ventricular tissue of 4-month-old male WT and cPPARγ−/− mice and pooled AMCMs from male WT (black bars) and cPPARγ−/− (gray bars) hearts (n = 3/genotype; P < 0.05 by Student's t test). Values are normalized to the WT group. B: Immunohistochemistry of AMCMs of male WT and cPPARγ−/− (KO) mice. Cardiomyocytes shown are representative of at least three independent experiments. (Please see http://dx.doi.org/10.2337/db07-0924 for a high-quality digital representation of this figure.)

Pioglitazone treatment similarly attenuates fibrosis and pro-inflammatory gene expression in AngII-infused cPPARγ−/− and WT mice. A: Cardiac fibrosis was quantified by measuring midventricular collagen area (n = 5–13/treatment group). B: Representative images of cardiac collagen expression (blue area) in AngII- and AngII+Pio-treated mice. Ventricular TGF-β1 (n = 6–13/treatment group) (C), fibronectin (n = 5–14/treatment group) (D), OPN (n = 6–12/treatment group) (E), and CD68 (n = 5–15/treatment group) (F) mRNA expression was measured by real-time quantitative RT-PCR. Black and gray bars indicate cPPARγ WT and KO animal data, respectively. *P < 0.05 vs. WT by Student's t test; †P < 0.05 vs. PBS and ‡P < 0.05 vs. AngII by one-way ANOVA. Pio, pioglitazone. (Please see http://dx.doi.org/10.2337/db07-0924 for a high-quality digital representation of this figure.)
Pioglitazone treatment similarly attenuates fibrosis and pro-inflammatory gene expression in AngII-infused cPPARγ−/− and WT mice. A: Cardiac fibrosis was quantified by measuring midventricular collagen area (n = 5–13/treatment group). B: Representative images of cardiac collagen expression (blue area) in AngII- and AngII+Pio-treated mice. Ventricular TGF-β1 (n = 6–13/treatment group) (C), fibronectin (n = 5–14/treatment group) (D), OPN (n = 6–12/treatment group) (E), and CD68 (n = 5–15/treatment group) (F) mRNA expression was measured by real-time quantitative RT-PCR. Black and gray bars indicate cPPARγ WT and KO animal data, respectively. *P < 0.05 vs. WT by Student's t test; †P < 0.05 vs. PBS and ‡P < 0.05 vs. AngII by one-way ANOVA. Pio, pioglitazone. (Please see http://dx.doi.org/10.2337/db07-0924 for a high-quality digital representation of this figure.)

Adiponectin does not mediate the antifibrotic effect of pioglitazone in the heart. Male cPPARγ mice were infected with adenovirus overexpressing Ad-GFP or Ad-APN (n = 4–5/treatment group) 5 days before the start of a 6-week AngII infusion on chow diet. Cardiac fibrosis was quantified by measuring midventricular collagen expression area (A), and representative images of collagen expression (blue area) (B) are shown for each group. Cardiac OPN (C) and CD68 (D) mRNA expression in cPPARγ−/− and WT mice (n = 3–5/treatment group) was measured by real-time quantitative RT-PCR. AngII-mediated increases in cardiac fibrosis, OPN, and CD68 expression were not significantly attenuated in cPPARγ−/− or WT mice injected with Ad-GFP vs. Ad-APN, and adenovirus effects did not differ according to genotype (P > 0.05 by two-way ANOVA). Values for WT mice are depicted by black bars and cPPARγ−/− values by gray bars. (Please see http://dx.doi.org/10.2337/db07-0924 for a high-quality digital representation of this figure.)
Adiponectin does not mediate the antifibrotic effect of pioglitazone in the heart. Male cPPARγ mice were infected with adenovirus overexpressing Ad-GFP or Ad-APN (n = 4–5/treatment group) 5 days before the start of a 6-week AngII infusion on chow diet. Cardiac fibrosis was quantified by measuring midventricular collagen expression area (A), and representative images of collagen expression (blue area) (B) are shown for each group. Cardiac OPN (C) and CD68 (D) mRNA expression in cPPARγ−/− and WT mice (n = 3–5/treatment group) was measured by real-time quantitative RT-PCR. AngII-mediated increases in cardiac fibrosis, OPN, and CD68 expression were not significantly attenuated in cPPARγ−/− or WT mice injected with Ad-GFP vs. Ad-APN, and adenovirus effects did not differ according to genotype (P > 0.05 by two-way ANOVA). Values for WT mice are depicted by black bars and cPPARγ−/− values by gray bars. (Please see http://dx.doi.org/10.2337/db07-0924 for a high-quality digital representation of this figure.)

PPARγ−/− cardiomyocytes have increased OPN expression. A: Representative Western blot and graph of β-actin normalized OPN expression in cultured AMCMs (24 h) isolated from 10-month WT and cPPARγ−/− mice (*P < 0.05 vs. WT by Student's t test). B: OPN mRNA expression in AMCMs (n = 3/treatment group) after 24 h culture with 0.1% DMSO (control) or AngII and aldosterone (AngII+Aldo, 1 μmol/l each) with or without 1 μmol/l pioglitazone. WT values are depicted by black bars and cPPARγ−/− values by gray bars (*P < 0.05 vs. control and †P < 0.05 vs. AngII+Aldo by one-way ANOVA). C: Mouse J774A.1 macrophage migration in response to 48-h conditioned media isolated from cPPARγ−/− (KO) and WT AMCM cultures. Nonconditioned myocyte culture media supplemented with (OPN) or without (control) 20 μg/ml recombinant mouse OPN served as positive and negative controls, respectively. Data are presented as % control (*P < 0.05 vs. control; †P < 0.05 vs. WT by one-way ANOVA). Pio, pioglitazone.
PPARγ−/− cardiomyocytes have increased OPN expression. A: Representative Western blot and graph of β-actin normalized OPN expression in cultured AMCMs (24 h) isolated from 10-month WT and cPPARγ−/− mice (*P < 0.05 vs. WT by Student's t test). B: OPN mRNA expression in AMCMs (n = 3/treatment group) after 24 h culture with 0.1% DMSO (control) or AngII and aldosterone (AngII+Aldo, 1 μmol/l each) with or without 1 μmol/l pioglitazone. WT values are depicted by black bars and cPPARγ−/− values by gray bars (*P < 0.05 vs. control and †P < 0.05 vs. AngII+Aldo by one-way ANOVA). C: Mouse J774A.1 macrophage migration in response to 48-h conditioned media isolated from cPPARγ−/− (KO) and WT AMCM cultures. Nonconditioned myocyte culture media supplemented with (OPN) or without (control) 20 μg/ml recombinant mouse OPN served as positive and negative controls, respectively. Data are presented as % control (*P < 0.05 vs. control; †P < 0.05 vs. WT by one-way ANOVA). Pio, pioglitazone.

Pioglitazone treatment attenuates fibrosis and pro-inflammatory gene expression in AngII-infused mPPARγ+/+, but not mPPARγ−/−, mice. A: Cardiac fibrosis quantified by midventricular collagen area (n = 7–12/treatment group). B: Representative images of cardiac collagen expression (blue area). Ventricular OPN (C) and CD68 (D) mRNA expression were measured by real-time quantitative RT-PCR (n = 3–12/treatment group). Black and gray bars indicate WT and mPPARγ−/− mouse data, respectively. *P < 0.05 vs. WT by Student's t test; †P < 0.05 vs. PBS; and ‡P < 0.05 vs. AngII by one-way ANOVA. Pio, pioglitazone. (Please see http://dx.doi.org/10.2337/db07-0924 for a high-quality digital representation of this figure.)
Pioglitazone treatment attenuates fibrosis and pro-inflammatory gene expression in AngII-infused mPPARγ+/+, but not mPPARγ−/−, mice. A: Cardiac fibrosis quantified by midventricular collagen area (n = 7–12/treatment group). B: Representative images of cardiac collagen expression (blue area). Ventricular OPN (C) and CD68 (D) mRNA expression were measured by real-time quantitative RT-PCR (n = 3–12/treatment group). Black and gray bars indicate WT and mPPARγ−/− mouse data, respectively. *P < 0.05 vs. WT by Student's t test; †P < 0.05 vs. PBS; and ‡P < 0.05 vs. AngII by one-way ANOVA. Pio, pioglitazone. (Please see http://dx.doi.org/10.2337/db07-0924 for a high-quality digital representation of this figure.)
Cardiac hypertrophy in cPPARγ mice
| Treatment | SBP (mmHg)* | Heart–to–body wt ratio (mg/g) | Cardiomyocyte diameter (μm) | ANP/GAPDH mRNA expression | BNP/GAPDH mRNA expression | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | KO | WT | KO | Two-way ANOVA | WT | KO | Two-way ANOVA | WT | KO | Two-way ANOVA | WT | KO | Two-way ANOVA | ||||||||||
| PBS+chow diet | 111 ± 3 (9) | 109 ± 4 (8) | 3.8 ± 0.1 (9) | 4.2 ± 0.1 (8)† | Interaction | 11.4 ± 0.2 (7) | 12.7 ± 0.1 (6) | Gene, drug | 0.7 ± 0.2 (6) | 5.8 ± 0.7 (9) | Gene, drug, interaction | 0.7 ± 0.1 (9) | 1.0 ± 0.1 (8) | Drug | |||||||||
| PBS+Pio | 106 ± 3 (9) | 108 ± 2 (9) | 4.2 ± 0.1 (9)‡ | 4.0 ± 0.1 (9) | 12.1 ± 0.2 (8) | 12.5 ± 0.2 (5) | 13.1 ± 4.0 (9)‡ | 8.4 ± 2.6 (8) | 1.2 ± 0.2 (10) | 1.4 ± 0.2 (6) | |||||||||||||
| AngII+chow diet | 161 ± 5 (10) | 170 ± 5 (17) | 4.6 ± 0.2 (13) | 5.3 ± 0.2 (15)† | Gene, drug | 13.5 ± 0.2 (10) | 14.4 ± 0.2 (15)† | Gene | 21.8 ± 4.9 (8) | 40.6 ± 6.4 (9) | — | 1.7 ± 0.3 (11) | 2.2 ± 0.3 (12) | Drug | |||||||||
| AngII+Pio | 153 ± 5 (6) | 163 ± 4 (14) | 5.0 ± 0.2 (9) | 5.8 ± 0.2 (15)† | 13.8 ± 0.2 (9) | 14.8 ± 0.1 (13)† | 37.6 ± 13.6 (9) | 23.5 ± 4.8 (8) | 3.2 ± 0.5 (8) | 2.6 ± 0.2 (11) | |||||||||||||
| Treatment | SBP (mmHg)* | Heart–to–body wt ratio (mg/g) | Cardiomyocyte diameter (μm) | ANP/GAPDH mRNA expression | BNP/GAPDH mRNA expression | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | KO | WT | KO | Two-way ANOVA | WT | KO | Two-way ANOVA | WT | KO | Two-way ANOVA | WT | KO | Two-way ANOVA | ||||||||||
| PBS+chow diet | 111 ± 3 (9) | 109 ± 4 (8) | 3.8 ± 0.1 (9) | 4.2 ± 0.1 (8)† | Interaction | 11.4 ± 0.2 (7) | 12.7 ± 0.1 (6) | Gene, drug | 0.7 ± 0.2 (6) | 5.8 ± 0.7 (9) | Gene, drug, interaction | 0.7 ± 0.1 (9) | 1.0 ± 0.1 (8) | Drug | |||||||||
| PBS+Pio | 106 ± 3 (9) | 108 ± 2 (9) | 4.2 ± 0.1 (9)‡ | 4.0 ± 0.1 (9) | 12.1 ± 0.2 (8) | 12.5 ± 0.2 (5) | 13.1 ± 4.0 (9)‡ | 8.4 ± 2.6 (8) | 1.2 ± 0.2 (10) | 1.4 ± 0.2 (6) | |||||||||||||
| AngII+chow diet | 161 ± 5 (10) | 170 ± 5 (17) | 4.6 ± 0.2 (13) | 5.3 ± 0.2 (15)† | Gene, drug | 13.5 ± 0.2 (10) | 14.4 ± 0.2 (15)† | Gene | 21.8 ± 4.9 (8) | 40.6 ± 6.4 (9) | — | 1.7 ± 0.3 (11) | 2.2 ± 0.3 (12) | Drug | |||||||||
| AngII+Pio | 153 ± 5 (6) | 163 ± 4 (14) | 5.0 ± 0.2 (9) | 5.8 ± 0.2 (15)† | 13.8 ± 0.2 (9) | 14.8 ± 0.1 (13)† | 37.6 ± 13.6 (9) | 23.5 ± 4.8 (8) | 3.2 ± 0.5 (8) | 2.6 ± 0.2 (11) | |||||||||||||
Data are means ± SD (n). Because one-way ANOVAs revealed significant differences between PBS- and AngII-infused mice for all phenotypes (P < 0.05), these two groups were analyzed using separate two-way ANOVAs to identify statistically significant differences (P < 0.05) in factor level means due to genotype (gene), treatment (drug), or the interaction between genotype and treatment (interaction) for each parameter. Significant differences due to genotype
or treatment effects between specific groups were determined by Bonferroni post hoc analyses. Cardiomyocyte diameter, ANP/glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and BNP/GAPDH data were transformed prior to analysis to account for differences in normality or SDs within these groups (see research design and methods).
SBP data were analyzed by both one- and two-way ANOVA. No significant differences were detected by either method.
Cardiac hypertrophy in mPPARγ mice
| Treatment | SBP (mmHg)* | Heart–to–body wt ratio(mg/g) | Cardiomyocyte diameter (μm) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | KO | WT | KO | Two-way ANOVA | WT | KO | Two-way ANOVA | ||||||
| PBS+chow diet | 103 ± 2 (15) | 108 ± 3 (19) | 3.9 ± 0.1 (9) | 4.1 ± 0.1 (9) | 13.4 ± 0.2 (9) | 13.7 ± 0.2 (9) | |||||||
| AngII+chow diet | 137 ± 7 (7) | 148 ± 4 (12) | 5.6 ± 0.1 (9) | 5.4 ± 0.1 (12) | 14.4 ± 0.1 (9) | 14.4 ± 0.2 (12) | |||||||
| AngII+Pio | 141 ± 8 (8) | 142 ± 3 (9) | 5.9 ± 0.2 (11) | 5.9 ± 0.3 (9) | Drug† | 14.6 ± 0.1 (11) | 14.8 ± 0.2 (9) | Drug† | |||||
| Treatment | SBP (mmHg)* | Heart–to–body wt ratio(mg/g) | Cardiomyocyte diameter (μm) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | KO | WT | KO | Two-way ANOVA | WT | KO | Two-way ANOVA | ||||||
| PBS+chow diet | 103 ± 2 (15) | 108 ± 3 (19) | 3.9 ± 0.1 (9) | 4.1 ± 0.1 (9) | 13.4 ± 0.2 (9) | 13.7 ± 0.2 (9) | |||||||
| AngII+chow diet | 137 ± 7 (7) | 148 ± 4 (12) | 5.6 ± 0.1 (9) | 5.4 ± 0.1 (12) | 14.4 ± 0.1 (9) | 14.4 ± 0.2 (12) | |||||||
| AngII+Pio | 141 ± 8 (8) | 142 ± 3 (9) | 5.9 ± 0.2 (11) | 5.9 ± 0.3 (9) | Drug† | 14.6 ± 0.1 (11) | 14.8 ± 0.2 (9) | Drug† | |||||
Data are means ± SD (n). PBS- and AngII-infused groups were analyzed separately because comparision between AngII- and PBS-infused groups revealed significant increases in all phenotypes upon AngII infusion (P < 0.05; one-way ANOVA). Differences in PBS+chow diet–treated mice were analyzed with Student's t test. AngII-infused groups were analyzed with two-way ANOVAs to identify statistically significant differences (P < 0.05) in factor level means due to genotype (gene), treatment (drug), or the interaction between genotype and treatment (interaction), and Bonferroni post-test analyses were used to identify significant differences (P < 0.05) in parameter means due to genotype or treatment effects between specific groups. SBP and heart–to–body weight data from AngII-infused mice were transformed prior to analysis to account for differences in normality or SDs within these groups (see research design and methods). Both heart–to–body weight ratio and cardiomyocyte diameter values demonstrated significant differences due to treatment by two-way ANOVA, but no significant differences were detected between group means in the post hoc analyses.
SBP data were analyzed by both one- and two-way ANOVA. No significant differences were detected by either method.
Two-way ANOVA comparing AngII+chow diet and AngII+Pio data.
Published ahead of print at http://diabetes.diabetesjournals.org on 28 May 2008.
Readers may use this article as long as the work is properly cited, the use is educational and not for profit, and the work is not altered. See http://creativecommons.org/licenses/by-nc-nd/3.0/ for details.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Article Information
This work was supported by National Heart, Lung, and Blood Institute Grant HLO46916 and National Institute of Diabetes and Digestive and Kidney Diseases Grant HLO70526 to W.H. and by Takeda Pharmaceuticals North America. E.C. was supported by the German Heart Foundation. The study was partially supported by a research grant from Takeda Pharmaceuticals North America, which also provided the pioglitazone used for this study.
We thank Sarah Duong, Van Chu, Rima Boyadjian, and Longsheng Hong for excellent technical assistance.