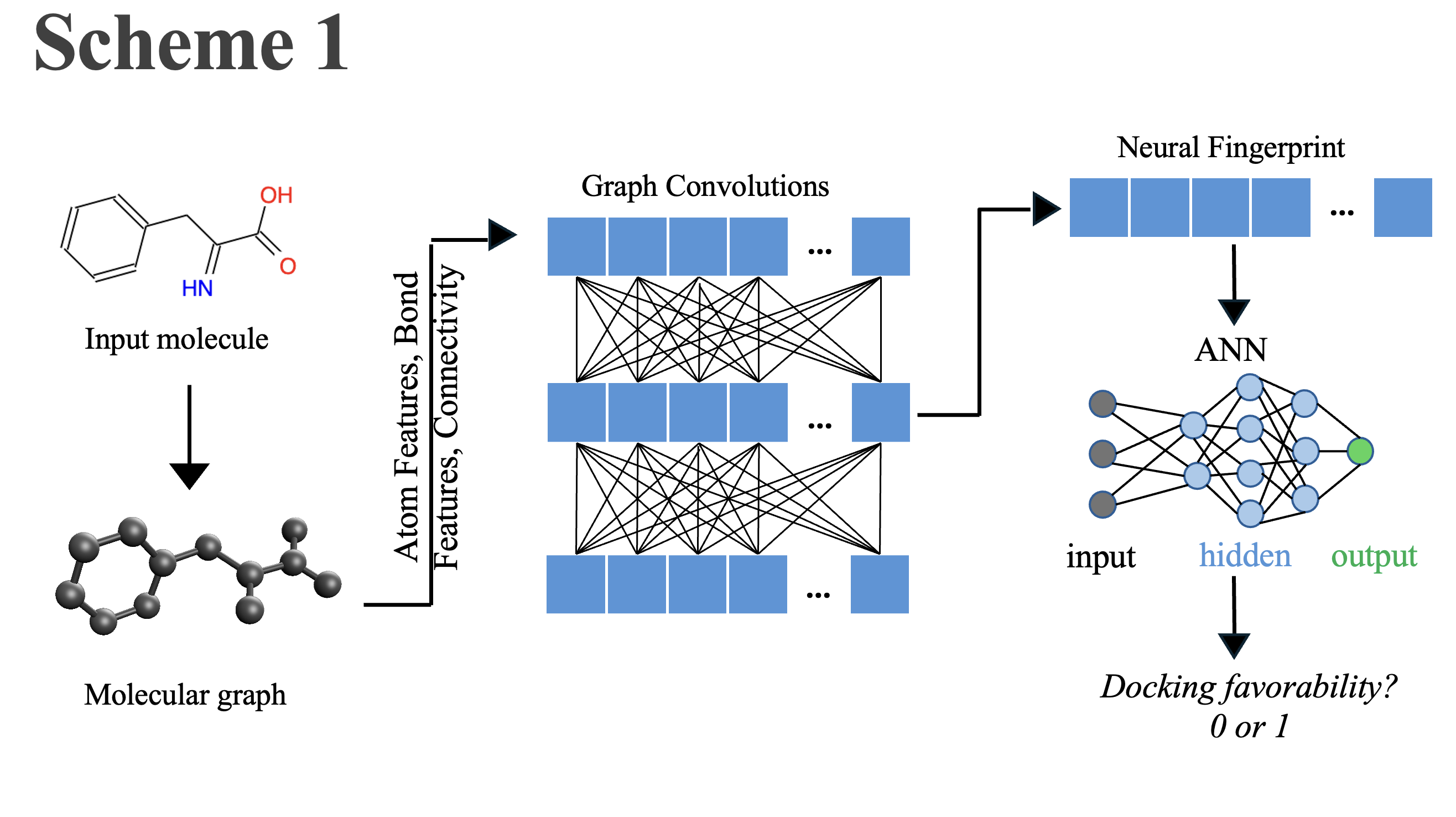
Enzyme targets
In this study, six distinct enzymes were chosen that are well regarded as potential therapeutic targets for various diseases. The following is a concise description of the structure-function of these enzymes related to their recognition as drug targets. As shown in Fig. 1, these enzymes have different folds, and the bound small molecules are diverse in terms of their structures.
Acetylcholinesterase. Abbreviated as ACEase, hereafter, is responsible for the regulation of neurotransmission through the degradation of acetylcholine (the neurotransmitter) in synapses of the nervous system [54]. Inhibitors of this enzyme are sought as they can be used as therapeutics for treatment of disease and protection against nerve agents. The X-ray crystal structure representing the inhibitor dihydrotanshinone I-bound target (PDB code 4m0e [54]) was used for this study.
Glutathione S-transferase. Abbreviated hereafter as GLTase [55], is responsible for adding electrophilic group to glutathione (the tripeptide formed with cysteine, glycine, and glutamic acid) and is responsible for detoxification. Additionally, they are involved in promoting tumor pathogenicity and chemoresistance [56]. The current study was based on the protein structure (PDB code: 1pkw [55]) in complex with glutathione (Fig. 1b).
Prostatic acid phosphatase. The prostatic acid phosphatase enzyme (abbreviated hereafter as PAPase) is responsible for the malignant growth of cells [57]. Inhibitors of this enzyme can be used as therapeutics for prostate cancer. The enzyme (PDB code: 1nd5 [57]) falls into the subclass of protein tyrosine phosphatase and is responsible for the dephosphorylation of epidermal growth factor receptor. The a-benzyl-aminobenzyl-phosphonic acid-bound structure is shown in Fig. 1c.
Protein tyrosine phosphatase 1b. Also belonging to the class of protein tyrosine phosphatase, this enzyme (abbreviated as PTPase) regulates negatively insulin [58]. This enzyme is an attractive target for type 2 diabetes and obesity and the drug (5-(3-{[1-(benzylsulfonyl) piperidin-4-yl]amino}phenyl)- 4-bromo-3-(carboxymethoxy) thiophene-2-carboxylic acid; ligand id: 527)-bound X-ray crystal structure used in the study belongs to the PDB code: 2qbp [58].
NAD(P)H:quinone oxidoreductase type 1. This oxidoreductase enzyme is responsible for protecting cells against cellular toxicity due to free radicals [59]. This enzyme (abbreviated as QR1ase) is overproduced in cancerous cells and therefore selective inhibitors of the enzyme have strong chemotherapeutic potentials. The present study is carried out on the 5-methoxy-1,2-dimethyl-3-(4-nitrophenoxymethyl)indole-4,7-dione (ligand ID: ES936)-bound X-ray crystal structure (Fig. 1e) of the enzyme bearing the PDB code: 1kbq [59].
NRH:quinone oxidoreductase. This is another oxidoreductase enzyme (abbreviated as QR2ase) involved in regulating cellular toxicity and oxidative stress [60]. The enzyme is targeted for anti-Alzheimer disease drug development. As illustrated in Fig. 1f, the X-ray crystal structure of the menadione-bound enzyme (PDB code: 2qr2) was used in the study [60].
Parameters and metrics for benchmarking
Based on the essence of compound screening, the present model was optimized to retain molecules with relatively higher binding affinities while ‘filtering’ as many molecules with relatively lower binding affinities. Therefore, the use of larger, preliminary datasets of small molecules would become feasible.
Using various metrics to benchmark, the performance of models was compared, both between the same architecture and between different architectures, e.g. convolutional fingerprint and Morgan fingerprint models. For a general metric, AUC measured the quality of the models’ predictions across different classification thresholds. To demonstrate how well these “filtration” architecture performs, increased significance was placed on the true positive rate (sensitivity). The purpose of compound screening is to retain the ligands that most favorably bind to a receptor. In turn, it is increasingly important that the number of false negatives was minimized in order to not discard any potentially promising compounds. Hence, sensitivity is maximized as much as possible without handicapping the model’s ability to be precise in its predictions.
Receiver operative characteristic and precision-recall curves. The results consisting of receiver operative characteristic (ROC) curves, precision-recall (PR) curve, are presented (Table 1). These results are based on a hyperparameter grid search undertaken only a single time to ensure a notion of randomness in the quantitative performance across all proteins.
Table 1
Relevant metrics for benchmarking obtained in the study.
| Enzyme system/ Metrics | AUC | PR-AUC |
| ACEase | 0.88 | 0.68 |
| GLTase | 0.90 | 0.68 |
| PAPase | 0.85 | 0.56 |
| PTPase | 0.94 | 0.78 |
| QR1ase | 0.91 | 0.73 |
| QR2ase | 0.86 | 0.59 |
across all proteins. Results described below are for single models chosen from the hyperparameter grid search. Parameters considered when choosing this ‘best model’ include precision, recall, and average \({\varDelta }_{\text{b}\text{i}\text{n}\text{d}}{G}^{o}\left(aq\right)\) of the predicted hits. The ROC curves of the six models (Fig. 2), with respective AUC values in Table 1.
Moreover, the precision-recall curve (Fig. 3) with corresponding AUC values have also been included in order to gauge the tradeoff between precision and recall along various thresholds. Of course, these models were constructed such that recall is maximized (Fig. 3). However, in order to
efficiently ‘filter’ out molecules with higher negative \({\varDelta }_{\text{b}\text{i}\text{n}\text{d}}{G}^{o}\left(aq\right)\)values, it is important for these models to be able to also classify the said molecules effectively.
These fundamental binary classification parameters relay two important things: the nature of the imbalanced dataset as well as the suitability of the model based on the protein being investigated. Both the ROC-AUC and PR-AUC values vary based on the protein. As a result, it is evident that the neural network architecture was able to capture the significant affinity (i.e. large negative \({\varDelta }_{\text{b}\text{i}\text{n}\text{d}}{G}^{o}\left(aq\right)\) values) for molecular substructures at a higher level for proteins such as PTPase compared to PAPase. Moreover, compared to other deep-learning-based studies,[16, 17] the ROC-AUC values seem to be more consistent across the wide array of proteins selected for this study. Although specific metrics may vary, this observation seems to substantiate the classifier’s ability to perform well at different thresholds for many different proteins.
Predictive enrichment probability. To further visualize and quantify the models’ abilities in correctly predicting those molecules that have a lower \({\varDelta }_{\text{b}\text{i}\text{n}\text{d}}{G}^{o}\left(aq\right)\)value than the predefined threshold (e.g. recall), the predictive enrichment probabilities or PEP is calculated (Eq. 4) based on the set of hits in the test dataset for each protein (Fig. 4). The PEP calculated at each Gibbs’ free
energy value lower (or equal) to the Gibbs’ free energy threshold that was used in training. The granularity of the \({\varDelta }_{\text{b}\text{i}\text{n}\text{d}}{G}^{\text{o}}\left(aq\right)\)values is found to be 0.01. Below, six graphs are shown illustrating
the PEP alongside the number of molecules in the test dataset that are hits at each Gibbs’ free energy value.
The PEPs for all proteins indicate that the neural network architecture is able to firmly grasp the molecular substructures that correlate to a lower \({\varDelta }_{\text{b}\text{i}\text{n}\text{d}}{G}^{\text{o}}\left(aq\right)\) value. Particularly, those molecules with extremely low \({\varDelta }_{\text{b}\text{i}\text{n}\text{d}}{G}^{\text{o}}\left(aq\right)\) values (Fig. 4, towards the left of the graphs) are almost always predicted correctly as hits by the neural network architecture. This is imperative in a good docking-based machine-learning model. It is necessary to filter out as many molecules while incurring the least amount of loss of tightly-bound molecules to the protein, which in this case means the loss of molecules with very low \({\varDelta }_{\text{b}\text{i}\text{n}\text{d}}{G}^{\text{o}}\left(aq\right)\)values. Without this characteristic, it would become increasingly infeasible to use such models in lieu of traditional docking techniques.
Metrics for evaluation. The pertinent parameters of all models using the neural network architecture presented in Table 2. All parameters are calculated based on final predictions using the test dataset after training and validation.
Table 2
The recall, precision, ROC-AUC, PR-AUC, and F1 score for the six protein systems studied. All values are calculated based on model predictions on the test dataset after training.
| Enzyme system/ Metrics | Recall | Precision | ROC-AUC | PR-AUC | F1 Score |
| ACEase | 0.98 | 0.30 | 0.88 | 0.68 | 0.47 |
| GLTase | 0.98 | 0.29 | 0.90 | 0.68 | 0.45 |
| PAPase | 0.94 | 0.31 | 0.85 | 0.56 | 0.47 |
| PTPase | 0.93 | 0.50 | 0.94 | 0.78 | 0.65 |
| QR1ase | 0.92 | 0.47 | 0.91 | 0.73 | 0.62 |
| QR2ase | 1.00 | 0.22 | 0.86 | 0.59 | 0.36 |
It is clear to see the effect of an imbalanced dataset on the results based on the F1 score (Eq. 5) and ROC-AUC values (Table 2). Although recall is greatly maximized, precision remains relatively low. Although that is the purpose of the architecture, there may be further optimizations that can be implemented in regard to the architecture to enable the model to more performantly discard molecules with lower \({\varDelta }_{\text{b}\text{i}\text{n}\text{d}}{G}^{\text{o}}\left(aq\right)\)values. Nevertheless, the extremely high recall attests to the architecture’s ability to retain molecules that favorably bind to the protein being investigated. Moreover, some proteins seem more favorable in investigating using this model than others (as was mentioned previously). In the case of PTPase, around 93% of molecules in the bottom 20% of \({\varDelta }_{\text{b}\text{i}\text{n}\text{d}}{G}^{\text{o}}\left(aq\right)\)values are retained whereas, based on the relatively high precision value, a good chunk of the overall dataset seems to be reduced. All in all, the architecture quantitatively performs very well at retaining molecules meaningfully.
Comparison of similarity maps. This was visually assessed by comparing the similarity maps[43] of substructures. Figure 5 illustrates the visual comparison of two molecules (top and bottom), whose similarity to the reference molecule (in the left) was assessed using two datasets: one used in the neural fingerprint-based model versus the regular docked data. As demonstrated in the figure, the neural fingerprint-based model represents a superior discrimination with the darker lines indicating greater similarities in the top molecule. In contrast, the bottom molecule represents larger dissimilarity in neural fingerprint-based training compared to the dataset obtained from the docking score alone (Fig. 5).
Comparison with Morgan fingerprints. The effectiveness of the neural fingerprint-based architecture for compound screening was compared to the commonly-used Morgan fingerprint-based[50] machine learning models. Specifically, the model and parameters used for the Morgan fingerprint-based “deep docking” study reported by Gentile et. al.[16, 17] were included in the comparative analysis. The “deep docking” algorithm used an iterative docking protocol, but the present analysis is carried out using a single iteration of this protocol. Both models calculated the threshold values to transform a continuous distribution of molecules over \({\varDelta }_{\text{b}\text{i}\text{n}\text{d}}{G}^{\text{o}}\left(aq\right)\)values into a binary classification problem. The models were compared with the same protein, ACEase, on the same overall dataset of molecules, and with the same threshold of − 10.0 kcal/mol. This value corresponded to around an 80 − 20 ratio of non-hits and hits in the dataset. Another hyperparameter grid search and training session was conducted for this comparison in regard to the neural fingerprint architecture (Table 3).
Table 3
Comparison of Precision, Recall, and Area Under the ROC Curve between both architectures. All values were calculated using the scikit-learn library and based on model predictions on randomly-sampled test datasets.
| Metrics | Neural fingerprint architecture | Morgan fingerprint architecture (Gentile et. al.[16, 17]) |
| Precision | 0.40 | 0.58 |
| Recall | 0.92 | 0.70 |
| ROC-AUC | 0.90 | 0.90 |
The precision and recall values (Table 3) seem to characterize both models appropriately. The Morgan fingerprint-based “deep docking” architecture tries to balance the two parameters as much as possible, whereas the neural fingerprint model maximizes the recall. However, combing through the results of all models trained for the “deep docking” architecture, it is apparent that the range of precision and recall seems to remain relatively constant despite changing hyperparameters (Table 3). In contrast, in the case of the neural fingerprint model, there exists significant variability in the values of these metrics. For example, while there are models that maximize recall like the one from which the results above are derived, there are also others that balance the precision and recall similarly to the Morgan fingerprint model (data not shown). One such model has a recall of 0.74 and a precision of 0.55. There are also models at the other extreme, which try to capture all the hits in the dataset without regard to a high level of precision (recall = 0.99, precision = 0.28). This represents the ability of the neural fingerprint architecture to adequately present users with the option of training for the filtering of ‘bad’ molecules, the maintenance of ‘good’ molecules, or somewhere in between.
In summary, these results demonstrated the plasticity of the neural fingerprint-based docking model and its significant ability to capture hits. Even using a smaller dataset, the algorithm was able to map chemical space with adequate efficacy.
{kind=link}