2.1 Materials
6-Hydrazinopurine, copper(II) chloride (CuCl2), and a salicylaldehyde derivative were sourced from Aladdin, Shanghai, China. MTT, JC-1, 2′,7-dichlorodihydrofluorescein diacetate (DCFH2-DA), and AO/EB were procured from Sigma-Aldrich. All biological antibodies were acquired from Abcam. Commercial sources supplied all other solvents and reagents, which were used without further purification. The 1H and 13C NMR analyses were conducted at room temperature (298 K) in DMSO-d6 using a Bruker AVANCE III HD 400 MHz spectrophotometer. X-ray crystallographic data were collected using a Rigaku HyPix diffractometer.
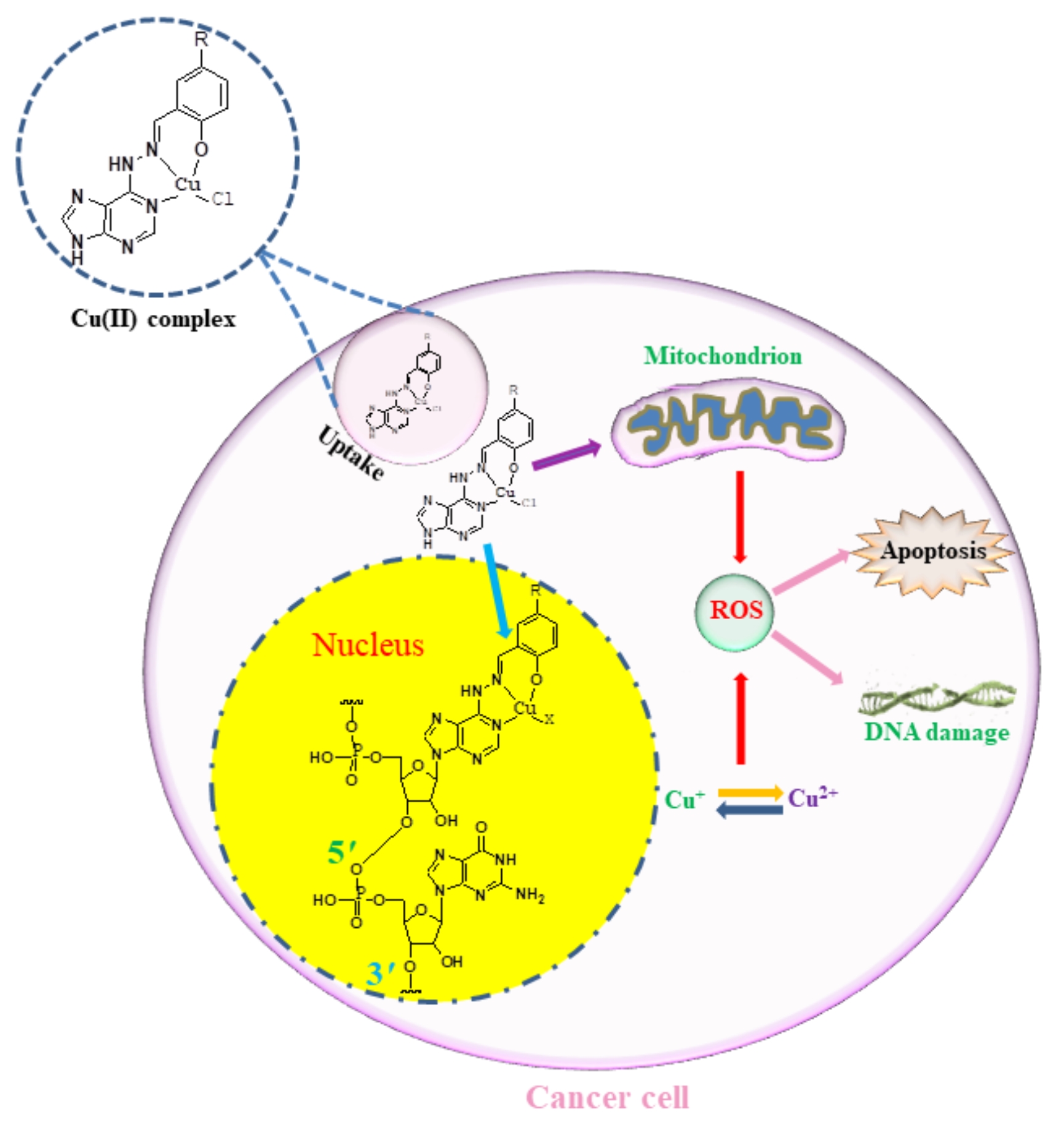
2.2 Synthesis of copper(II) complexes
The ligands based on adenine were synthesized through a reaction involving 6-Hydrazinyl-9H-purine and various aldehydes. Specifically, 6-Hydrazinyl-9H-purine (3 mmol) and the corresponding aldehyde (3 mmol) were dissolved in 10 mL of ethanol and stirred for a duration of 6 hours. The resulting product was filtered to obtain a light yellow solid. The chemical structures of these ligands were meticulously characterized using High-Resolution Mass Spectrometry (HRMS), as well as 1H and 13C Nuclear Magnetic Resonance (NMR) spectroscopy.
Subsequently, the Cu(II) complexes were synthesized by reacting the ligands (0.10 mmol) with CuCl2 (0.10 mmol) in a solvent mixture of methanol (1.5 mL) and dichloromethane (1.5 mL). The reaction was conducted under solvothermal conditions at 65°C for a period of 24 hours. Crystals suitable for X-ray diffraction analysis were carefully collected to further elucidate the molecular structure and bonding arrangements within the complexes.
L1: 1H NMR (500 MHz, DMSO-d6) δ 13.25 (s, 1H), 11.84 (s, 1H), 11.53 (s, 1H), 8.34 (d, J = 8.1 Hz, 3H), 7.34 (s, 1H), 7.25 (t, J = 7.6 Hz, 1H), 6.93–6.88 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 157.32, 152.41, 145.33, 141.38, 130.86, 119.56, 119.22, 118.77, 117.26. HRMS:m/z 255.0916 [M + H]
L2: 1H NMR (500 MHz, DMSO-d6) δ 13.20 (s, 1H), 11.82 (s, 1H), 11.32 (s, 1H), 8.31 (t, J = 19.8 Hz, 3H), 7.12 (s, 1H), 7.05 (d, J = 8.1 Hz, 1H), 6.81 (d, J = 8.2 Hz, 1H), 2.24 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 155.07, 152.37, 145.40, 141.31, 131.47, 130.75, 127.97, 118.77, 117.03, 20.31. HRMS:m/z 269.1073 [M + H]
2.3 Bioactivity
Cell cultures were maintained in Dulbecco's Modified Eagle Medium (DMEM, Gibco) supplemented with 10% (v/v) fetal bovine serum (FBS, Gibco), 100 U/mL penicillin (Sigma), 100 U/mL streptomycin (Sigma), and 2 mM glutamine (Sigma) at 37°C in a humidified atmosphere containing 95% air and 5% CO2. Cu(II) complexes were dissolved in dimethylformamide (DMF).
To assess the in vitro cytotoxicity of the Cu(II) complexes against both cancer and normal cell lines, a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT, Sigma) assay was conducted. Cells were exposed to Cu(II) complexes and free ligands at five different concentrations for 48 hours. The growth inhibitory rates were measured using an enzyme labeling instrument at a double wavelength of 570/630 nm. The growth inhibitory rates were computed using the formula: (OD control - OD test) / OD control × 100%.
2.4 Cu(II) complex C2 induced mitochondrial damage
MGC-803 cells were cultured and incubated with Cu(II) complex C1/C2 at a concentration of 4.51 µM for 6 hours. The incubation was carried out in poly-HEMA coated 6-well plates to prevent cell adhesion. Subsequently, the cells were harvested and resuspended in a fresh culture medium.
To assess mitochondrial membrane potential, 0.5 mL of JC-1 and DAPI working solution was added to the cell suspension. The cells were then incubated at 37°C for 25 minutes. Following incubation, the staining solution was carefully removed through centrifugation. The cells were rinsed three times with JC-1 staining buffer to ensure proper removal of excess staining solution.
The stained cells were promptly subjected to analysis using confocal microscopy to evaluate the changes in mitochondrial membrane potential induced by the treatment with Cu(II) complex C2.
2.5 Enhancement of Cu(II) complex C2 at intracellular ROS levels
MGC-803 cells were seeded at a density of 1 × 105 cells/well on poly-L-lysine-coated coverslips within 6-well plates using heat-deactivated complete RPMI. The cells were subsequently treated with Cu(II) complex C1/C2 at a concentration of 4.51 µM for a duration of 6 hours.
Following the treatment, the cells were washed thrice with phosphate-buffered saline (PBS) and then exposed to 25 µM H2DCFDA (Sigma, excitation/emission: 488 nm/515 nm) for 20 minutes at 37°C. After the incubation period, excess H2DCFDA was removed, and the cells were rinsed thrice with PBS.
The coverslips carrying the treated cells were then washed with ultrapure water, mounted on slides, and promptly subjected to analysis utilizing a fluorescence microscope. This methodology enabled the assessment of intracellular reactive oxygen species (ROS) levels induced by Cu(II) complex C1/C2 treatment.
2.6 Expression of γH2AX in MGC-803 cells induced by Cu(II) complex C2
MGC-803 cells were seeded on 6-well plates at a density of 1×105 cells per well. After incubation for 12 hours, the cells were treated with different concentrations of Cu(II) complex C1/C2 (4.51 µM) for 24 hours, and then the cells were incubated with rabbit γH2AX polyclonal antibody (diluted with 1% BSA) at 4°C overnight. The cells were then incubated with Alexa Fluor 488-conjugated secondary antibody for 2 hours. γH2AX expression was evaluated by confocal microscopy.
2.7 Comet assay
Following treatment with Cu(II) complex C1/C2 (4.51 µM) for 24 hours, cells were adjusted to a concentration of 106-107 cells/mL after centrifugation. A 0.5% normal melting point agarose (NMA) solution was preheated at 56℃, and 80 µL was gently applied onto a preheated glass slide. A clean cover glass was quickly placed over the agarose, and the slide was allowed to solidify at 4℃ for 10 minutes.
Subsequently, 10 µL of PBS containing 1000 cells and 75 µL of 0.5% low melting point agarose (LMA) were mixed at 37℃. The cover glass was gently removed, and the LMA-cell mixture was applied on top of the first solidified agarose layer. A clean cover glass was placed over the mixture and allowed to solidify at 4℃ for 10 minutes.
Finally, 85 µL of 0.5% LMA was preheated at 37℃ and applied to the solidified LMA layer. A cover glass was placed on top to allow solidification. The cover glass was removed, and the slide was immersed in freshly prepared cell lysate for at least 1 hour.
Post cell lysis, the slide was rinsed twice with PBS to remove excess salt. The slide was placed in a horizontal electrophoresis tank and submerged in a newly prepared alkaline electrophoresis buffer. Alkaline hydrolysis was performed for 20 minutes to allow DNA unwinding under alkaline conditions, forming single-stranded DNA and facilitating DNA breakage and migration during electrophoresis.
2.8 Cell cycle arrest by Cu(II) complex C2
MGC-803 cells were seeded at a density of 1×105 cells in 6-well plates and incubated at 37°C for 12 hours to allow cell attachment and growth. Following incubation, the cells were treated with Cu(II) complex C1/C2 at a concentration of 4.51 µM for 48 hours.
Subsequently, the cancer cells were collected in separate 1.5 mL EP tubes, and 500 µL of 70% ice-cold ethanol was added. The cells were then incubated at -20°C overnight. Following incubation, centrifugation at 1500 rpm for 5 minutes was performed to discard the ethanol. Next, the cells were treated with a staining solution of 1 mg/mL propidium iodide (PI) and 10% RnaseA and incubated in the dark for 0.5 hours.
2.9 AO/EB assay
Glass cover slides were pre-placed in a 6-well culture plate, and cell suspension was inoculated for 12 hours. Subsequently, cells were treated with Cu(II) complex C1/C2 at a concentration of 4.51 µM for an additional 24 hours.
Following the incubation, the reagent kit's instructions were followed to prepare a mixed working solution of acridine orange (AO) and ethidium bromide (EB) totaling 600 µL. This solution was carefully added to the cells in the six-well plate, and the cells were incubated in the dark for 25 minutes.
After the incubation period, the cells were washed twice with phosphate-buffered saline (PBS). The cover glass was then placed on the slide, and the samples were immediately observed under a fluorescence microscope to assess the effects of Cu(II) complex C1/C2 treatment on cellular morphology and viability.
2.10 Induced apoptosis by Cu(II) complex C2.
Flow cytometry was employed to assess the apoptosis-inducing potential of C2 in MGC-803 cells. Initially, MGC-803 cells were seeded in 6-well plates and incubated for 12 hours. Subsequently, the cells were treated with Cu(II) complex C1/C2 (4.51 µM) for 24 hours.
Following the treatment, the cells were harvested, washed three times with phosphate-buffered saline (PBS), and resuspended in 120 µL of binding buffer to achieve a final 1 × 106 cells/mL concentration. The cell suspension was treated with 5 µL of V-FITC (5 µg/mL) and incubated for 25 minutes at 37°C in a light-protected environment.
Following the incubation, 10 µL of propidium iodide was added to the cell suspension and incubated for 20 minutes at 25°C under light protection. Apoptosis was analyzed using flow cytometry, providing valuable insights into the apoptotic effects induced by Cu(II) complex C2 on MGC-803 cells.
{kind=link}