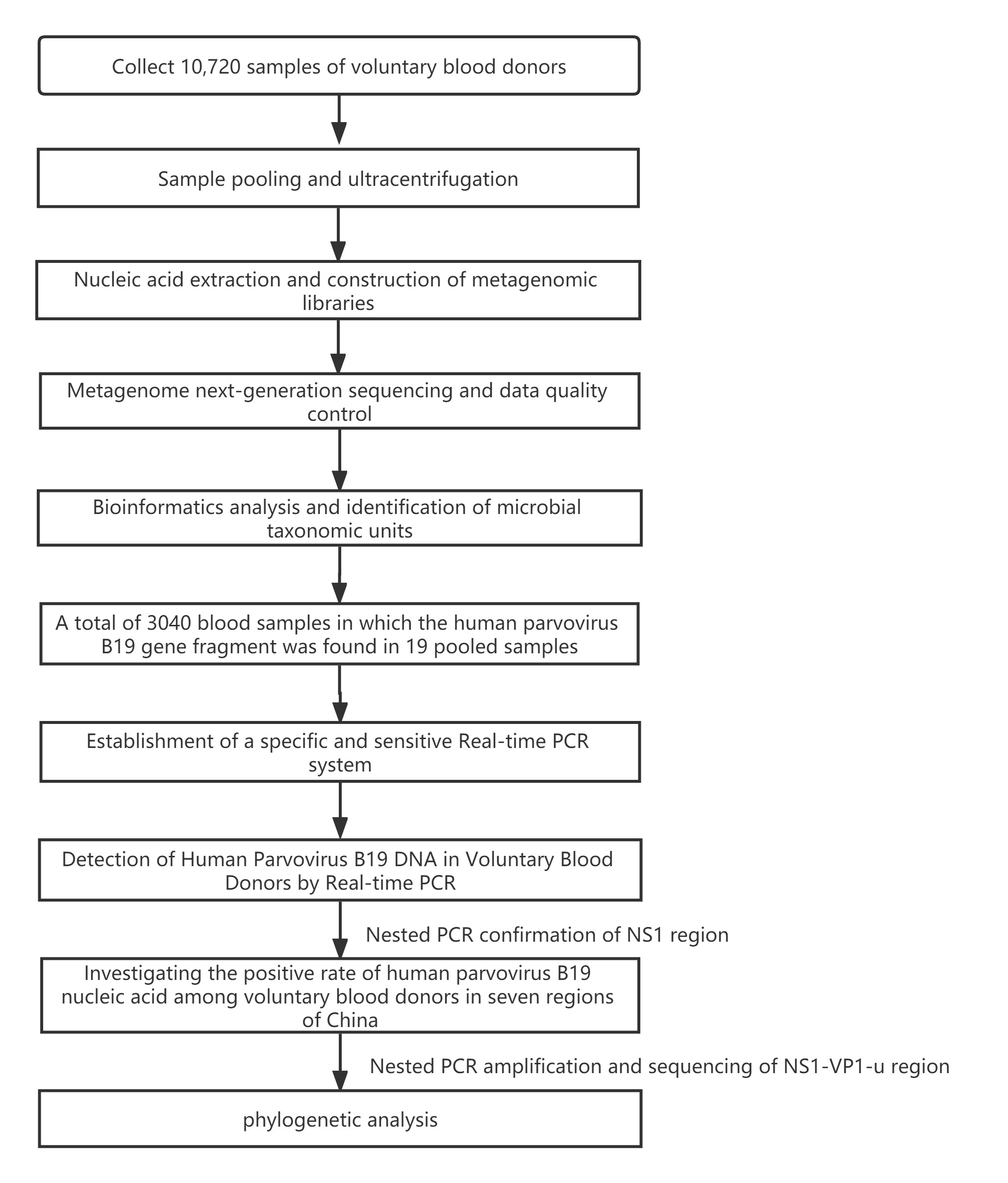
Sample collection and nucleic acid preparation
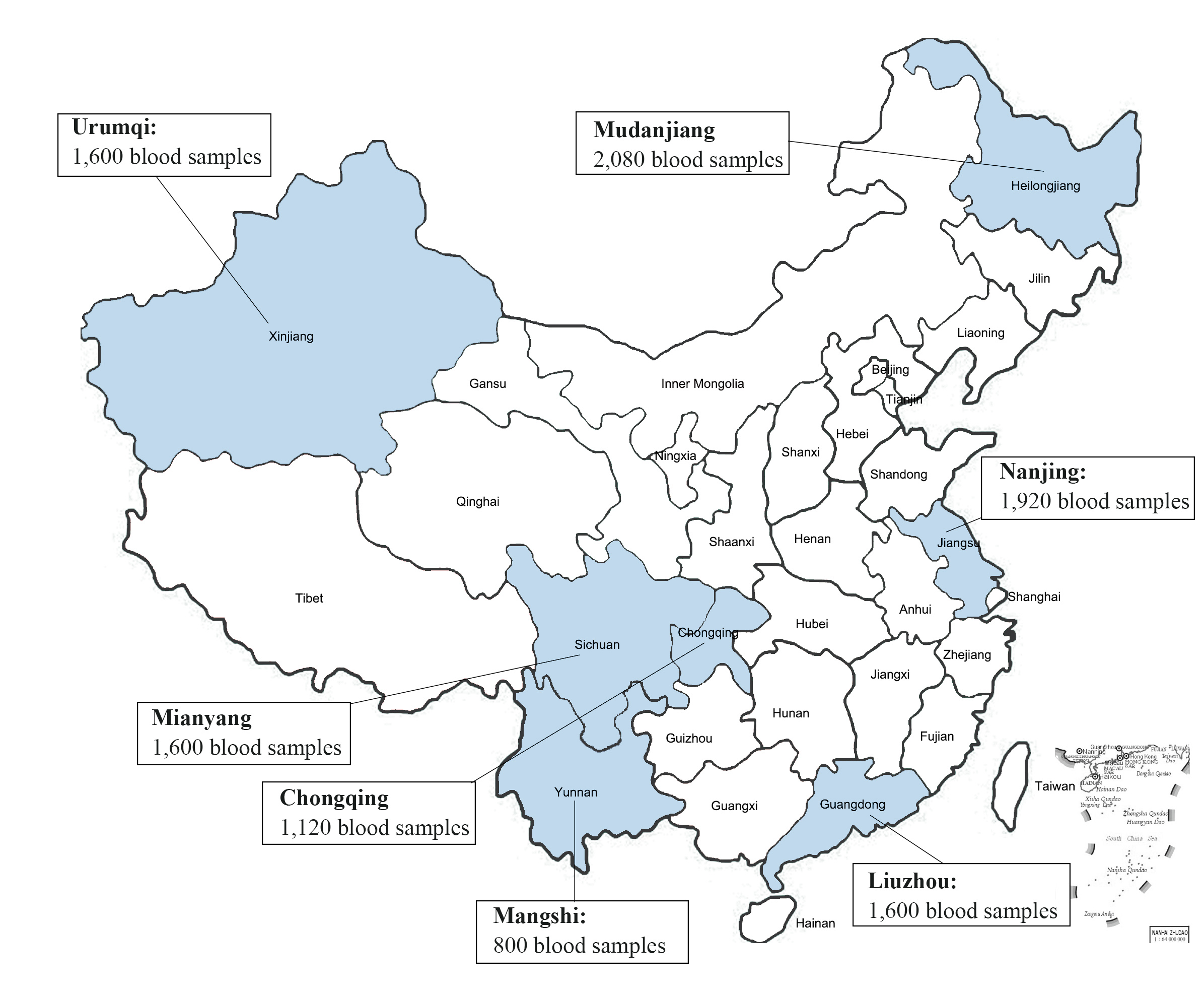
From January 1, 2012, to December 31, 2018, a total of 10,720 blood samples were randomly selectedfrom blood donors from 7 cooperative blood centers in China (The demographic information of blood donors is shown in Additional file 1). After blood samples are collected, they will be stored at -20 ℃ in blood centers, and then shipped on dry ice to the Institute of Blood Transfusion, Chinese Academy of Medical Sciences, and stored in -20 ℃ freezer, each of 10 mL (the distribution of sampling location and a corresponding number of samples are shown in theAdditional file 2). 160 (each 100 µL) blood samples werepooled for ultracentrifugation (32,000rpm, 120min, maximum centrifugal radius of 91.9mm), rinsed and resuspended with 500µl sterile PBS.
The suspensionsof the pooled samples were subjected toextraction of total DNA usingQIAamp® DNA Blood mini Kit (QIAGENCat. NO.160019269, Frankfurt, Germany), DNA concentrationwas measured by Equalbit® 1×dsDNA HS Assay Kit (VazymeCat.NO.7E302K9, Nanjing, China).The methodology employed in this study can be seen in the flow chart.(Additional file 3)
Library Construction And Metagenome Sequencing
The metagenomic library was constructed using KAPA HyperPlus Kit (KAPACat. NO.0000097583, Boston, USA) with dual-indexed Adapters (KAPACat. NO.0000093370, Boston, USA), the DNA was fragmented to 250 bp approximately by the enzyme at 37°C for 20 minutes, after end repair and A-tailing, adapter ligation, post-ligation cleanup, library amplification, and post-amplification cleanup, thelibrary was constructed.
Agilent 2100 Bioanalyzer (Agilent Technologies, Beijing, China) was used for library quality control,and qualified DNA library was sent to the Novogene company to paired-end sequence in HiSeq 4000 (Illumina, Beijing, China), and all raw data were trimmed by Trimmomatic(version 0.39)[14] to remove adapter sequence, low-quality reads, and duplicate reads.
Data analysis
After removing thehuman potential sequencesby KneadData (version0.10.0,https://github.com/biobakery/biobakery/wiki/kneaddata) using the reference genome of humans (assembly GRCh38.p13), the taxonomic labels of metagenomic sequences were assigned using kraken2(version 2.0.7)[15]and the microbereadswere surfaced. Based on the Krona algorithm, homology comparison is performed with reference sequences of bacteria, fungi, parasites, and viruses. The unclassified readswere aligned to the NCBI database using BLASTn.The best alignment hits were used to classify the reads. Subsequently, Heatmaps were generated for non-scaled, non-normalized titer data using a Pearson distance function with average linkage clustering using the program Heml (version 1.0).
B19v Dna Prevalence Inblood Donors
19 pooled plasma(from 3040 blood samples fromblood donors)containedfragments of B19V genes.These plasma samples were from 6 regions:Liuzhou(800), Mangshi (640), Mudanjiang (640), Mianyang (480), Nanjing(320) and Urumqi(160).
Dna Extraction And Nucleic Acid Amplification Technology
DNA extraction and nucleic acid amplification technology
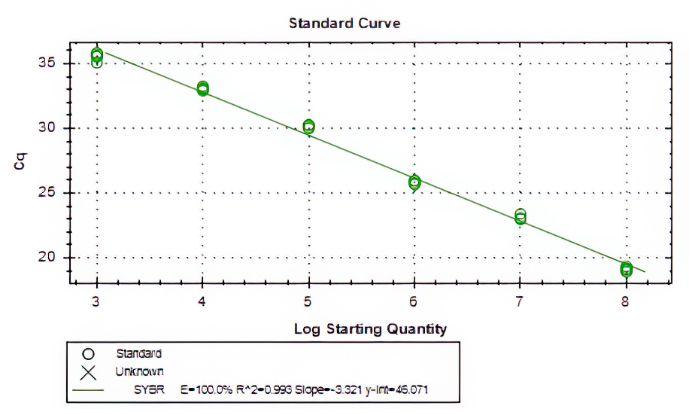
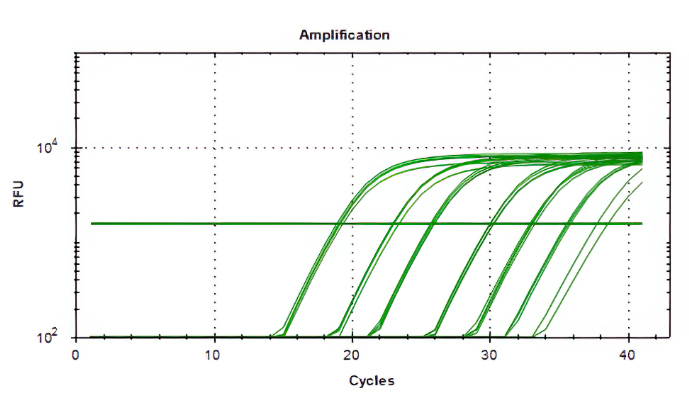
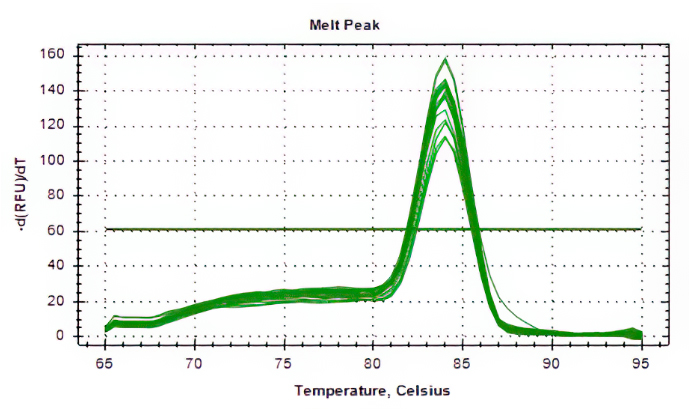
Aliquots of plasma (20ul) from 10 blood donations were pooled,then nucleic acid extraction was conducted by Magnetic Viral DNA/RNA Kit (Tiangen, Beijing, China). Screening of samples for B19V DNA was performed by 2×SG Fast qPCR Master Mix (Sangon Biotech, Shanghai, China) on fluorescence ration PCR instrument (CFX96, BIO-RAD, California, USA), and corresponding primers arelocatedin conserved regions. The sequence forward primer is 5’-TGCAGATGCCCTCCACCCA-3’ and the sequence of reverse primer is 5’-GCTGCTTTCACTGAGTTCTTC-3’. Additionally, 3µl of extracted DNA from pooled plasma was added to 17µl qPCR mixture(10µl 2×SG Fast qPCR Master Mix,0.4µl of each diluted primer, 2µl DNF Buffer) for amplification of B19 DNA. This study used the National Standard for Human Parvovirus B19 Nucleic Acids Detection Kit(National institutes for food and drug control, Cat. NO. 370023 − 201601, Beijing, China) as quantitation standards. Six quantitative standards (1×108,1×107,1×106,1×105,1×104,1×103IU/mL) were included in each PCR procedure. To avoid the fluorescence of primer dimer under high cycle number in samples with few or no target sequence, it is necessary to optimize the Tm value.
Confirmation Test And Phylogenetic Analysis
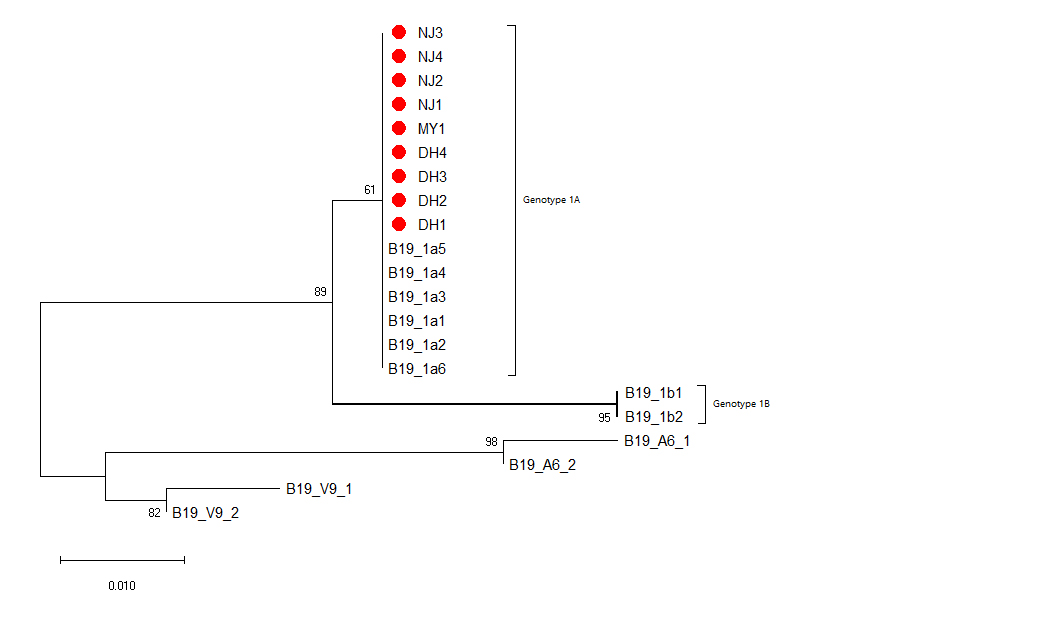
B19V DNA positive samples were confirmed by nested PCR, using primers located in the NS1 gene[16]: First round of primers (Forward:5’-CACTATGAAAACTGGGCAATAAAC-3’,Reverse:5’-AATGATTCTCCTGAACTGGTCC-3’), thesecond round of primers (Forward: 5’-ATAAACTACACTTTTGATTTCCCTG-3’, Reverse: 5’-TCTCCTGAACTGGTCCCG-3’), and then Golden Star T6 Super PCR Mix (Qsingke, Beijing, China) was used for nested PCR. Refer to the article for the reaction conditions.[17]The PCR products were sent to the SangonBiotech (Shanghai, China) for sequencing. Mega-X(version 10.0.5) was used to construct the phylogenetic evolution tree. The Genbankaccession numbers of the reference sequence are shown in the Additional file 4, and No. of bootstrap replications is set to 1000, and the model/method is set to Kimura 2-parameter.
Statistical analysis
The software IBM SPSS Statistics 23.0 was used for statistical analysis. The chi-square test was applied to calculate the correlation between the demographic characteristics, and the Pvalue less than 0.05 was used as the cutoff level for significance.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}