The interaction of acrolein with pristine and N-doped TiO2 anatase nanoparticles: A DFT study
DOI:
https://doi.org/10.17344/acsi.2016.2350Keywords:
Acrolein, TiO2, Electronic properties, Density functional theory, sensorAbstract
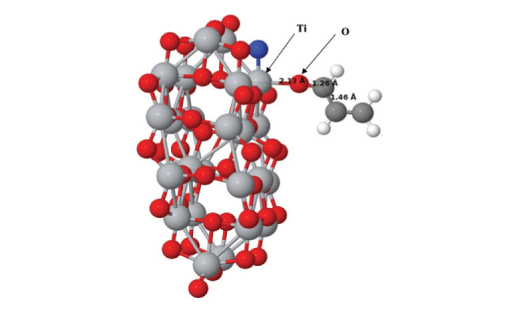
Density functional theory calculations were carried out in order to study the effects of the adsorption of acrolein molecule on the structural and electronic properties of TiO2 anatase nanoparticles. The ability of pristine and N-doped TiO2 anatase nanoparticles to recognize toxic acrolein (C3H4O) molecule was surveyed in detail. It was concluded that acrolein molecule chemisorbs on the N-doped anatase nanoparticles with large adsorption energy and small distance with respect to the nanoparticle. The results indicate that the adsorption of acrolein on the N-doped TiO2 is energetically more favorable than the adsorption on the pristine one, suggesting that the N doping can energetically facilitate the adsorption of acrolein on the N-doped nanoparticle. It means that the N-doped TiO2 nanoparticle can react with acrolein molecule more efficiently. The interaction between acrolein molecule and N-doped TiO2 can induce substantial variations in the HOMO/LUMO molecular orbitals of the nanoparticle, changing its electrical conductivity which is helpful for developing novel sensor devices for the removal of harmful acrolein molecule. The large overlaps in the projected density of states spectra reveal the formation of chemical bond between two interacting atoms. Charge analysis based on Mulliken charges indicates that charge is transferred from the acrolein molecule to the TiO2 nanoparticle.
Published
Issue
Section
License
Except where otherwise noted, articles in this journal are published under the Creative Commons Attribution 4.0 International License
