ATUALIZAÇÃO
Alterações genéticas e colesterolemia: Recentes estudos brasileiros
Neusa Forti; Luis A. Salazar; Jayme Diament; Sergio D. Giannini; Mario H. Hirata; Rosario D.C. Hirata
Faculdade de Medicina da USP, Faculdad de Medicina Universidad de La Frontera, Temuco, Chile; Faculdade de Ciências Farmacêuticas da USP; InCor do HC da FMUSP
Endereço para correspondência Endereço para correspondência Dra. Neusa Forti Instituto do Coração (InCor) Av. Dr. Enéas de Carvalho Aguiar 44 Cep 05403-000, São Paulo, Brasil E-mail: nforti@hotmail.com
A associação doença arterial coronariana (DAC) e alterações do perfil lipídico em particular a hipercolesterolemia tem sido amplamente evidenciada, seja através de estudos anatomopatológicos e experimentais, seja através de estudos epidemiológicos, clínicos e de intervenção terapêutica. Entretanto, no Brasil, as publicações referentes a aspectos clínico-epidemiológicos dessa associação são relativamente escassas e restritas a resultados obtidos em alguns grupos sociais selecionados1-5.
Por outro lado, sabe-se que a colesterolemia sofre a influência de fatores não só ligados à idade, sexo, hábitos de vida, uso de alguns medicamentos, determinadas doenças, mas também a fatores genéticos6. Dentre os últimos, destacam-se as mutações (modificações estruturais na seqüência do DNA ácido desoxirribonucléico que envolvem alterações em um único ou múltiplos nucleotídeos) e polimorfismos (mutações freqüentes nas quais o alelo mutado se apresenta com freqüência superior a 1%) que podem alterar a estrutura e função de proteínas envolvidas na síntese, homeostase e no metabolismo do colesterol, como as apolipoproteínas, receptores e enzimas. Entre as mutações e polimorfismos relacionados à elevação da colesterolemia, citam-se os que ocorrem nos genes da apolipoproteína B (APOB), da apolipoproteína E (APOE), da enzima conversora da angiotensina I (ECA) e, principalmente, dos genes do receptor de LDL (RLDL) e da HMG-CoA redutase7-12. Publicações sobre esses aspectos são ainda mais escassas no Brasil.
É finalidade deste trabalho descrever os principais resultados de investigações relacionadas a alterações genéticas e colesterolemia em indivíduos acompanhados há longo prazo em três centros de referência da cidade de São Paulo (Serviço de Prevenção Cardiológica do InCor / FMUSP, Serviço de Endocrinologia do HCFMUSP e Instituto Dante Pazzanese de Cardiologia). O estudo genético foi desenvolvido no Departamento de Análises Clínicas e Toxicológicas da Faculdade de Ciências Farmacêuticas da USP.
Alterações no gene da apolipoproteína B
O gene da apolipoproteína B (APOB) está situado no cromossomo 2. Nele já foram descritas algumas mutações e vários polimorfismos.
As principais mutações são: R3500Q (troca de arginina pela glutamina), R3500W (troca de arginina pelo triptofano), R3531C (troca de arginina por cisteína), Q3405E (troca de glutamina por glutamato) e R3480P (troca de arginina por prolina). Recentemente, foram descritas as mutações N3516K (troca de asparagina por lisina) e T3492I (troca de treonina por isoleucina). Essas alterações interferem na conformação do domínio de ligação da apo B com o receptor B/E, reduzindo a afinidade da LDL (lipoproteína de baixa densidade) por esse receptor, elevando assim os níveis circulantes de LDL. Provocam uma forma de dislipidemia conhecida como apo B-100 defeituosa familiar13-19.
Em nosso meio, Cavalli e cols20 não detectaram a mutação R3500Q ao estudarem 177 hipercolesterolêmicos (51 homens e 126 mulheres, de 58 anos em média e [colesterol ligado a LDL] > 190 mg/dL). Tanto a mutação R3500Q como a R3531C não foram detectadas em 90 indivíduos hipercolesterolêmicos familiares estudados por Salazar21.
Dentre os polimorfismos já descritos, destacam-se PvuII (exon 4 genótipos P1P1, P1P2 e P2P2), XbaI (genótipos X+X+, X+X-, X-X) e MspI (exon 26), EcoRI (exon 29 genótipos E+E+, E+E-, E-E-), Ins/Del (peptídeo sinalizador genótipos D/D, I/D, I/I), região hipervariável na extremidade 3' (3'HVR)17,22. Em relação a eles, algumas pesquisas já foram desenvolvidas em nosso meio: 1) Guzmán e col.23, para o polimorfismo XbaI (que determina os genótipos X+X+, X+X-, X-X) verificaram que os níveis plasmáticos de colesterolemia total (CT) e LDL-c se mostraram mais elevados em mulheres com o genótipo X-X-; 2) Salazar e col24,25 observaram freqüência maior do alelo Del (polimorfismo Ins/Del) em 94 mulheres sadias, de 39 a 68 anos, obesas (índice de massa corpórea > 30), acompanhando níveis mais elevados de CT e LDL-c; 3) Salazar e col26 estudaram 50 mulheres, de 48 ± 9 anos, com DAC comprovada por estudo angiográfico, em comparação a 100 mulheres, de 50 ± 6 anos, sem evidências clínicas de coronariopatia. As primeiras, além de apresentarem maiores níveis plasmáticos de CT, trigliceridemia (TG) e LDL-c, tiveram maior freqüência do genótipo X-X- (42% x 12%). Entretanto, os autores não observaram diferenças significativas em relação à freqüência dos genótipos para os polimorfismos Ins/Del e EcoRI. Esses resultados se assemelharam aos de Bydlowsky e cols27 e de Mansur e cols28, mas diferem dos achados de Scartezini e cols29. Estes últimos, em adultos jovens, encontraram associação dos genótipos X-X-/E+E+ (polimorfismos XbaI e EcoRI, respectivamente) e coronariopatia, ressaltando que os portadores desses genótipos têm 2 a 3 vezes risco maior de desenvolver coronariopatia; 4) Cavalli e col20, por sua vez, não encontraram diferenças em relação à distribuição dos genótipos e freqüência de alelos para os polimorfismos MspI, XbaI e Ins/Del ao compararem hiper e normocolesterolêmicos. Os hipercolesterolêmicos apresentaram maior freqüência de alelos menores a 43 repetições na seqüência de bases nitrogenadas para o polimorfismo 3'HVR (16,4% x 8,5%) associado a valores de colesterol mais elevados (particularmente em mulheres após a menopausa). Associação significativa entre os genótipos < 43 (3'HVR) e Del (Ins/Del) também foi verificada nos hipercolesterolêmicos; 5) Machado e cols.30, em homens com perfil lipídico considerado de alto risco para DAC, evidenciaram valores mais altos de CT, LDL-c, TG e de CT / HDL-c (colesterol ligado a HDL [lipoproteína de alta densidade]) na presença do haplótipo X+/Del construído com os genótipos dos polimorfismos XbaI e Ins/Del; 6) Machado e col31, em portadores de DAC, observaram freqüências mais elevadas dos genótipos X-X- e I/I, mas não houve diferença para os genótipos do polimorfisno EcoRI.
Alterações no gene da apolipoproteína E
O gene da apolipoproteína E (APOE) localiza-se no cromossomo 19 e codifica uma proteína de 299 aminoácidos. A apo E desempenha importante papel no catabolismo das lipoproteínas ricas em TG e no transporte reverso do colesterol, atuando principalmente como mediador do receptor de LDL. A afinidade pelo receptor depende do polimorfismo HhaI (exon 4) no gene APOE. Os 3 alelos (e2, e3 e e4) determinam a formação de 6 genótipos: E2E2, E2E3, E2E4, E3E3, E3E4 e E4E4.
Diferentes estudos evidenciaram que o alelo e4 está freqüentemente associado a maior risco de DAC, em decorrência da elevação das taxas de LDL-c e, recentemente, vem sendo associado a menor resposta terapêutica às vastatinas. Menor captação de remanescentes de Qm e VLDL, levando à disbetalipoproteinemia, ocorre na presença do genótipo E2E2. Ao genótipo E4E4, associa-se a doença de Alzheimer32-34.
O polimorfismo HhaI em mulheres nas fases pré (n=42, x=44,1 ± 3,2 anos) e pós-menopausa (n=52, x = 54 ± 7,4 anos), sem evidências clínicas de DAC também foi objeto de investigação em São Paulo, tendo-se observado: 1) maior freqüência do alelo e3, seja na fase pré ou na fase pós-menopausa (84% x 81%) mas não houve influência dos diferentes genótipos sobre a colesterolemia35,36; 2) a distribuição genotípica, foi: a) na fase pré-menopausa E2E3 10%, E3E3 69%, E3E4 21%; b) na fase pós-menopausa E2E3 10%, E3E3 63%, E3E4 25%, E4E4 2%36; 3) as mulheres obesas (fase pré e pós-menopausa), além de maiores níveis de CT e LDL-c, apresentaram maior freqüência do alelo e425.
Salazar e cols26 investigaram também o polimorfismo HhaI em 150 mulheres das quais 50 (x = 48 ± 9 anos) apresentavam coronariopatia comprovada por estudo angiográfico e as demais 100 (x = 50 ± 6 anos) eram clinicamente normais. As coronariopatas, além de taxas significativamente mais elevadas de CT, TG e LDL-c, apresentaram maior freqüência do genótipo E3E4 em relação às controles (40% x 14%). As pacientes não diferiram em relação à freqüência de E2 E3 (8% x 8%), E2 E4 (6% x 8%) e E3 E3 (46% x 70%) e, em nenhuma delas foram identificados os genótipos E2 E2 e E4 E4. Entretanto, o alelo e4 foi o mais freqüente nas coronariopatas, não havendo diferenças para e2 nem para e3, resultados muito semelhantes aos relatados em 1996 por Otta e col37 e Cavalli e col38.
Em hipertrigliceridêmicos, Guzmán e cols39 encontraram freqüência maior do genótipo E2 E4.
Alterações no gene do receptor de LDL (RLDL)
O gene do RLDL localiza-se no cromossomo 19 e codifica uma proteína de 839 aminoácidos.
a) Polimorfismos - Já foram identificados mais de 45 polimorfismos no gene do RLDL, destacando-se RsaI (extremidade 5'), StuI (exon 2), MaeIII (exon 4), TaqI (intron 4), SphI (intron 6), StuI (exon 8), HhaI (exon 11), HincII (exon 12), AvaII (exon 13), MspI (exon 15), PvuII (intron 15), MspI e NcoI (exon 18) e PstI (extremidade 3')40.
Em nosso meio, foram estudados com mais afinco os polimorfismos AvaII (genótipos A+A+, A+A-, A-A-), HincII (genótipos H+H+, H+H-, H-H-) e PvuII (genótipos P1 P1, P1 P2, P2 P2).
As investigações iniciais de Salazar e cols mostraram: 1) em mulheres na pré-menopausa (n=41, x = 44,0 ± 3,2 anos), os valores de HDL-c eram mais elevados do que na pós-menopausa (n = 53, x = 54,0 ± 7,4 anos), acompanhando os genótipos H+H+ e H-H-; 2) a frequência de A+A+ foi maior nas mulheres na pré-menopausa; 3) não houve diferenças entre os genótipos para HincII entre as mulheres na pré e na pós-menopausa; 4) no grupo de mulheres sadias, porém obesas, as freqüências dos alelos A+ e H+ foram maiores do que nas mais magras25,35,41,42.
Em prosseguimento, para análise dos polimorfismos AvaII e HincII, Salazar e cols43 selecionaram 170 indivíduos (35 homens, 115 mulheres, x = 50 anos), considerados de alto risco para DAC6, e 130 indivíduos clinicamente normais (55 homens, 75 mulheres, x = 44 anos). Os primeiros apresentaram valores médios de CT, TG e LDL-c significativamente maiores que os sadios. A freqüência dos genótipos A+A+ e H+H+ mostrou-se significativamente mais elevada nos indivíduos de alto risco (32% x 16% para A+A+ e 32% x 18% para H+H+). E, entre os indivíduos de alto risco, as taxas médias de CT e LDL-c foram maiores nos portadores de genótipo A+A+, tanto em homens como nas mulheres, estivessem elas na fase pré ou pós-menopausa. Resultados semelhantes foram observados para o genótipo H+H+. Os autores não observaram influência dos polimorfismos no perfil lipídico dos indivíduos normais.
Em 128 indivíduos considerados de alto risco para DAC6, a freqüência do genótipo P1P1 do polimorfismo PvuII foi significantemente mais elevada que a dos controles (57% x 38%), associando-se a maiores concentrações plasmáticas de CT, TG, LDL-c e VLDL-c e menores de HDL-c, tanto em homens como em mulheres (na pré e pós-menopausa). Entre os controles, na presença do genótipo P1P1 também as taxas médias de CT e LDL-c foram mais elevadas25,44.
Posteriormente, Salazar e cols26 demonstraram que mulheres, com coronariopatia comprovada, além de maiores concentrações de CT, TG e LDL-c apresentavam freqüência mais elevada dos genótipos A+A+ e P1P1 do que as clinicamente normais. Entretanto, elas não diferiram em relação ao polimorfismo HincII.
Os mesmos autores45 também demonstraram freqüência mais elevada dos genótipos A+A+, H+H+ e P1P1 em 50 indivíduos com hipercolesterolemia familiar heterozigótica (40 mulheres, 10 homens, de 50 ± 8,3 anos) em relação a 130 normolipidêmicos (85 mulheres, 45 homens, de 54,1 ± 8,8 anos): 34% x 16% para A+A+, 35% x 18% para H+H+ e 64% x 38% para P1P1.
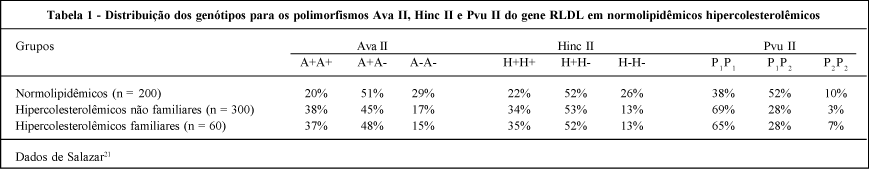
Em continuidade, Salazar21 estudou os polimorfismos AvaII, HincII e PvuII em 60 indivíduos dentre 90 hiperlipidêmicos familiares (homo e heterozigóticos, não aparentados, 25 homens, 65 mulheres) e não familiares (n = 300, 80 homens, 220 mulheres, 58 ± 12 anos) em comparação a indivíduos normolipidêmicos considerados clinicamente normais (n = 200, 95 homens, 105 mulheres, 41 ± 12 anos). A frequência dos genótipos A+A+, H+H+ e P1P1 foi significativamente mais alta tanto nos hipercolesterolêmicos não familiares como nos familiares em comparação aos controles (Fig. 1). O mesmo ocorreu em relação à freqüência relativa dos alelos A+, H+ e P1.
Comparando os valores médios das frações lipídicas nos diferentes genótipos, foram encontrados valores mais elevados de CT e LDL-c, em presença de A+A+, de H+H+, e de P1P1, tanto nos controles como nos hipercolesterolêmicos não familiares (figs. 1 e 2). O mesmo ocorreu quando associados os genótipos A+A+, H+H+ e P1P1. Essa associação foi mais freqüente nos hipercolesterolêmicos não familiares (17,3%) do que nos controles (7,5%). Os genótipos associados mais freqüentes nos dois grupos foram A+A-/H+H-/P1P1 (29%) e A+A-/H+H-/ P1P2 (17%).
Nos subgrupos de homens e mulheres em idade de risco (> 45 e > 55 anos, respectivamente) e de mulheres em fase pré e pós-menopausa, a distribuição dos diferentes genótipos e as taxas médias de CT e LDL-c exibiram comportamento semelhante ao do grupo, tanto nos hipercolesterolêmicos não familiares como nos controles. Nos hipercolesterolêmicos não familiares e nos normais, não se observou interação entre o efeito dos polimorfismos sobre os valores de CT e LDL-c e os fatores idade, sexo e fase hormonal das mulheres. Esses achados são sugestivos de que o efeito dos polimorfismos do gene RLDL sobre CT e LDL-c independe das variáveis analisadas21.
O polimorfismo Msp I (exon 15) do gene RLDL também foi estudado em nosso meio. O genótipo M-M- foi mais freqüente nos hipercolesterolêmicos familiares em relação aos controles46,47.
b) Mutações - Atualmente, mais de 900 mutações foram encontradas no gene RLDL, afetando todos os domínios da proteína e associados ao fenótipo da hipercolesterolemia familiar. Elas podem ser classificadas em 5 tipos funcionais: tipo 1 causam deficiência na síntese do receptor; tipo 2 (mais comum) aquelas que originam proteínas incapazes de serem transportadas entre o retículo endoplasmático e o Complexo de Golgi (2a) ou produzem proteínas capazes de serem transportadas mas em quantidades muito baixas (2b); tipo 3 originam proteínas que alcançam a superfície da célula, mas não têm capacidade de ligação; tipo 4 raras, produzem receptores que podem ligar LDL, mas não são interiorizadas; tipo 5 produzem receptores que têm a capacidade de ligação à LDL, são internalizados, mas não liberam as LDL nos endossomos, impedindo a reciclagem dos receptores48.
A expressão clínica das mutações no gene RLDL é variável e pode ser atribuída a: 1) diferentes efeitos das mutações sobre a afinidade de ligação do RLDL pelas partículas contendo apo B-100 ou apo E; 2) efeito funcional da mutação; 3) presença de um outro gene herdado também de forma autossômica dominante, que suprime os efeitos das mutações do gene RLDL sobre os níveis de LDL-c; 4) presença de polimorfismos genéticos49-52.
No Brasil, as primeiras caracterizações moleculares de pacientes com hipercolesterolemia familiar foram descritas em famílias de origem árabe, residentes na região de Ribeirão Preto (São Paulo). Figueiredo e cols53 detectaram a mutação Libanese (C660X) no exon 14 do gene RLDL funcionalmente 2a em 5 de 10 famílias, ao passo que Alberto e cols54 a detectaram em 9 de 31 famílias.
A identificação e caracterização do gene RLDL em pacientes com diagnóstico clínico e laboratorial de hipercolesterolemia familiar e que não apresentavam nenhum grau de parentesco nem mutação para o gene da apo B (R3500Q e R3531C) foi iniciada, em nosso meio, por Salazar e cols55. A triagem foi feita no intuito de detectar mutações nos 18 exons e região promotora do gene RLDL, pelas técnicas de PCR SSCP e sequenciamento direto do DNA56. Entretanto, a metodologia empregada não é suficiente para detectar grandes deleções ou mutações em regiões não codificantes ou regulatórias do gene RLDL21.
Os resultados obtidos em 35 pacientes já foram publicados55. Mutações do gene RLDL foram encontradas em 63% desses pacientes: 11 do tipo "missense" (substituição de um nucleotídeo, alterando um códon de modo que outro aminoácido fica presente no lugar); 2 do tipo nonsense (mutação sem sentido, resultando na expressão de uma proteína truncada) e 2 do tipo frameshift (deleção ou inserção de bases que causam um deslocamento na fase de leitura, que alteram a seqüência de aminoácidos a partir do ponto de mutação). Oito das mutações encontradas já haviam sido descritas em outras populações mas não no Brasil: E92X e C371X (nonsense no exon 4 e 8 respectivamente) e R236W, G322S, G352D, A370T, C675W e C677Y (missense, respectivamente nos exons 5, 7, 8, 8, 14 e 14). As sete outras ainda não tinham sido referidas na literatura até novembro de 2001: G(-20)R, I476P, V503G, D580H e S652R (missense, respectivamente nos exons 1, 10, 10, 12 e 14) e FsR757 e FsS828 (frameshift, nos exons 16 e 17).
Prosseguindo na investigação, Salazar21, com a mesma metodologia, incluiu em sua casuística mais 10 pacientes com hipercolesterolemia familiar, também não aparentados. Identificou mais 4 mutações: D203N (missense no exon 4) já descrita em outra população e 3 ainda não descritas na literatura. São elas a I467T, V535G e W541G (missense, respectivamente nos exons 10, 11 e 11). Paralelamente, na maioria dos 45 pacientes, fizeram-se presentes os alelos A+ (polimorfismo AvaII), H+ (polimorfismo HincII) ou P1 (polimorfismo PvuII), o que também pode explicar as taxas elevadas de CT. Esses achados contribuem para a hipótese de segregação conjunta desses polimorfismos e uma mutação funcional no gene RLDL21.
Os resultados obtidos até o momento esclarecem parcialmente as bases genéticas da hipercolesterolemia familiar no Brasil, evidenciando um padrão heterogêneo de comportamento. A investigação de mutações prossegue em mais 45 pacientes não aparentados portadores de hipercolesterolemia familiar.
Alterações em outros genes
Cavalli e cols57 analisaram o polimorfismo MboII da região 5' promotora do gene da enzima CYP3A4 (responsável pela metabolização da maioria das vastatinas) e não encontraram diferenças significativas em relação à distribuição genotípica (M+M+, M+M-, M-M-) entre grupos de hipercolesterolêmicos familiares (n = 23) e controles (n = 90).
Guzmán e cols58, por sua vez, estudaram o polimorfismo PvuII (intron 6) do gene da lipase lipoprotéica em 34 hipertrigliceridêmicos e 165 controles e não encontraram influência dos diferentes genótipos em relação às concentrações de CT e TG nos dois grupos.
Hirata e cols59 procuraram detectar diferenças em relação à freqüência dos genótipos TT, GT e GG referentes ao polimorfismo G894T do gene da eNOS (óxido nítrico sintase) em 105 indivíduos (35 com hipercolesterolemia familiar (HF) heterozigótica e 70 normocolesterolêmicos). Seus dados preliminares não mostraram associação entre HF e os diferentes genótipos do polimorfismo G894T do gene da eNOS.
O polimorfismo C677T do gene da metilenotetrahidrofolato redutase (MTHFR) foi analisado em 25 mulheres (x = 49 ± 8 anos) com clínica de DAC e em 50 mulheres sadias (x = 52 ± 7 anos): os grupos não mostraram diferenças em relação aos diferentes genótipos da MTHFR. Resultado semelhante foi obtido para o polimorfismo A1298C. Também não foram encontradas diferenças ao serem analisados 60 hipercolesterolêmicos familiares heterozigóticos (com e sem DAC) e 143 controles60-62.
Alterações genéticas e resposta terapêutica
Na prática clínica, observamos que os hipercolesterolêmicos apresentam diversidade na resposta à terapêutica com vastatinas, independentemente da dose utilizada. Esse fato pode ser atribuído a diferentes fatores (falta de adesão, interferência de fatores farmacocinéticos e farmacodinâmicos, interação de fármacos), inclusive genéticos .
Entre as alterações genéticas capazes de modificar a resposta terapêutica às vastatinas, salientam-se as mutações e/ou polimorfismos dos genes RLDL, de apoB e de apo E, de apo A I e apo A IV, da metaloproteinase stromelysin 1 (MMP-3), das enzimas CYP3A4, CETP, ECA, para oxonase, lipase hepática e de fatores envolvidos no processo inflamatório (IL-6, TNF-alfa e receptor CD14), dos genes ABC1, ABCG5 e ABCG833,34,63-65.
Entre nós, a resposta terapêutica às vastatinas foi objeto de investigação em relação ao polimorfismo Ins/Del para APOB, HhaI para APOE; AvaII, HincII e PvuII para o gene RLDL; Ins/Del para o gene da enzima conversora de angiotensina ECA; gene da HMG-CoA redutase; gene da enzima CYP3A4.
Guzmán e cols23 verificaram que hipercolesterolêmicos com genótipo I/I para o polimorfismo Ins/Del do gene APOB apresentam reduções mais acentuadas de CT quando tratados com fluvastatina. Reduções menores de CT, LDL-c e apo B em resposta à fluvastatina, foram também encontradas em investigações posteriores, nos portadores do genótipo D/D para o polimorfismo Ins/Del do gene ECA, e A+A+ (AvaII) e P1P1 (PvuII) do gene RLDL66.
Salazar e cols21,67-70 avaliaram a resposta terapêutica à atorvastatina em 25 pacientes hipercolesterolêmicos familiares e verificaram que: 1) portadores do genótipo E3E4 (n=10) apresentaram redução significantemente menor de CT, LDL-c e apo B em relação àqueles com genótipos E2E3 e E3E3; 2) a freqüência do alelo e4 foi mais elevada nos indivíduos que tiveram redução de LDL-c < 15%; 3) os portadores do genótipo D/D, em relação a I/D e I/I (ECA) também apresentaram menores reduções de CT (DD13%, ID 24%, II 30%), LDL-c (DD 12%, ID 34%, II 42%) e apo B (DD 13%, ID 23%, II 35%); 4) aqueles com genótipos A+A+ e P1P1 (polimorfismos AvaII e PvuII do gene RLDL) também apresentaram menores reduções de CT, LDL-c e apo B; 5) em relação aos genótipos determinados pelos polimorfismos HincII (exon 12) e MspI (exon 15), não ocorreram diferenças significativas.
A expressão gênica do RLDL e da HMG-CoA redutase em células mononucleares periféricas de hipercolesterolêmicos heterozigóticos tratados com atorvastatina também foi investigada por Salazar21. Naqueles que apresentaram reduções de LDL-c superiores a 15% (considerados responsivos), houve, em relação à condição basal, aumento dos valores de RNAm (ácido ribonucléico mensageiro) do RLDL (1,9x) e da HMG-CoA redutase (2,6 x). Baixos valores da expressão gênica do RLDL e menor resposta terapêutica foi verificada naqueles com genótipos A+A+ (polimorfismo AvaII) e P1P1 (polimorfismo PvuII); não ocorreram diferenças significativas entre os diferentes genótipos para HincII. Todos os seis indivíduos que não responderam à terapêutica (redução de LDL-c inferior a 15%) possuíam os genótipos A+A+ e P1P1; e na condição basal, apresentavam valores de RNAm de RLDL e HMG-CoA redutase inferiores aos do grupo de respondedores, valores esses que não se alteraram por influência da atorvastatina. Para explicar os resultados obtidos, Salazar21 aventou a hipótese da existência de alterações estruturais e/ou funcionais nas proteínas envolvidas na transcrição dos genes RLDL e HMG-CoA redutase.
Dados iniciais de Cavalli71 não mostraram influência de alterações do gene da CYP3A4 (localizado no cromossomo 7) na resposta à terapêutica com atorvastatina em 27 hipercolesterolêmicos familiares heterozigóticos e em 18 hipercolesterolêmicos não familiares.
Considerações finais
Os estudos ora descritos apontam para a influência de alterações genéticas sobre a colesterolemia e sobre a variabilidade da resposta de hipercolesterolêmicos ao tratamento com vastatinas. Deve-se ressaltar que novas mutações no gene RLDL foram detectadas em brasileiros natos portadores de hipercolesterolemia familiar. Mas, até o momento, os mecanismos pelos quais as variações genéticas influenciaram a colesterolemia e a resposta terapêutica não foram totalmente elucidados.
Considerando as dificuldades técnicas e o alto custo das determinações genéticas, a procura dessas alterações só se justifica, até o presente, em pesquisas científicas. Estas, no entanto, são necessárias não só para esclarecer o diagnóstico e permitir o desenvolvimento de novos fármacos e/ou métodos terapêuticos, mas principalmente para facilitar o aconselhamento genético das famílias afetadas. São desejáveis no futuro métodos viáveis de determinação do perfil genético que poderiam indicar a seleção de hipolipemiantes a fim de se conseguir melhor resposta terapêutica.

Recebido para publicação em 10/10/02
Aceito em 08/11/02
- 1. Manfroi WC, Marques G, Goldin JR et al. Correlação entre a extensão da aterosclerose coronária e a dislipidemia. Arq Bras Cardiol 1982; 39: 15-19.
- 2. Giannini SD, Forti N, Góis JM et al. Relações entre valores de HDL-colesterol, dos índices de risco coronário e o grau de aterosclerose avaliado por cinecoronariografia. Arq Bras Cardiol 1985; 44: 305-10.
- 3. Da Luz PL, Carvalho MEA, Cardoso RHA et al. Incidência de dislipidemia e sua relação com doença arterial coronária em populações brasileiras. Arq Bras Cardiol 1990; 54: 257-64.
- 4. Ladeia AM, Guimarães AM, Lima JC. Perfil lipídico e doença arterial coronária. Arq Bras Cardiol 1994; 63: 101-6.
- 5. Forti N, Fukushima J, Giannini SD. Perfil lipídico de indivíduos submetidos à cinecoronariografia em diferentes regiões do Brasil. Arq Bras Cardiol 1997; 68: 333-42.
-
6III Diretrizes Brasileiras sobre Dislipidemias e Diretriz de Prevenção de Aterosclerose do Departamento de Aterosclerose da Sociedade Brasileira de Cardiologia. Arq Bras Cardiol 2001; 77 (supl. III): 1-48.
- 7. Galton DJ. Genetic determinants of atherosclerosis related dyslipidemias and their clinical implications. Clin Chem Acta 1997; 257: 181-97.
- 8. Varret M, Rabes JP, Villeger L et al. Analysis of the 525 point mutation in the human LDL receptor gene database. Atherosclerosis 1999; 144: 182-3.
- 9. Davignon J, Cohn JS, Mabile L et al. Apolipoprotein E and atherosclerosis: insight from animal and human studies. Clin Chem Acta 1999; 286: 115-43.
- 10. Winkelmann BR, Hager J. Genetic variation in coronary heart disease and myocardial infarction: methodological overview and clinical evidence. Pharmacogenomics 2000; 1: 73-94.
- 11. Jeron A, Hengstenberg C, Engel S et al. The D allele of the ACE polymorphism is related to increased QT dispersion in 609 patients after myocardial infarction. Eur Heart J 2001; 22: 663-8.
- 12. Salazar LA, Cavalli SA, Hirata MH et al. Polymorphisms of the low-density lipoprotein receptor gene in Brazilian individuals with heterozygous familial hypercholesterolemia. Braz J Med Biol Res 2000; 33: 1301-4.
- 13. Myant NB. Familial defective apolipoprotein B-100: a review, including some comparisons with familial hypercholesterolemia. Atherosclerosis 1993; 104: 1-18.
- 14. Kotze MJ, Peeters AV, Langenhoven E et al. Phenotypic expression and frequency of familial defective apolipoprotein B-100 in Belgian hypercholesterolemics. Atherosclerosis 1994; 111: 217-25.
- 15. Pullinguer CR, Hennessy LK, Chaterton JE et al. Familial ligand defective apolipoprotein B: identification of a new mutation that decreases LDL receptor binding affinity. J Clin Invest 1995; 95: 1225-34.
- 16. Wenham PR, Henderson BG, Penney MD et al. Familial ligand defective apolipoprotein B-100: detection, biochemical features and haplotype analysis of the R 3531 C mutation in the UK. Atherosclerosis 1997; 129: 185-92.
- 17. Tay DY, Pan JP, Lee-Cheng GJ. Identification and haplotype analysis of apolipoprotein B-100 Arg 3500 ® Trp mutation in hyperlipidemic Chinese. Clin Chem Acta 1998; 44: 1659-65.
- 18. Bednarska-Makaruk M, Bisko M, Pulawska MF et al. Familial defective apolipoprotein B-100 in a group of hypercholesterolemic patients in Poland. Identification of a new mutation Thr 3492 Ile in the apolipoprotein B gene. Eur J Hum Genet 2001; 9: 836-42.
- 19. Gaffney D, Pullinger CR, O'Reilly DS et al. Influence of an asparagine to lysine mutation at aminoacid 3516 of apolipoprotein B on low-density lipoprotein receptor binding. Clin Chem Acta 2002; 321: 113-21.
- 20. Cavalli SA, Hirata MH, Salazar LA et al. Apolipoprotein B gene polymorphisms: prevalence and impact on serum lipid concentrations in hypercholesterolemic individuals from Brazil. Clin Chem Acta 2000; 302: 189-203.
- 21. Salazar LA. Influência de alterações genéticas do receptor do LDL no perfil lipídico e na resposta terapêutica de indivíduos brasileiros hipercolesterolêmicos. Tese de doutorado. Faculdade de Ciências Farmacêuticas da Universidade de São Paulo, 2002. 160 p.
- 22. Ludwig EH, Hopkins PN, Allen A et al. Association of genetic variations in apolipoprotein B with hypercholesterolemia, coronary artery disease and receptor binding of low-density lipoproteins. J Lipid Res 1997; 38: 1361-73.
- 23. Guzmán ECR, Hirata MH, Quintão ECR et al. Association of the apolipoprotein B gene polymorphism with cholesterol levels and response to fluvastatin in Brazilian individuals with lipid profile of high risk for coronary heart disease. Clin Chem Lab Med 2000; 38: 731-6.
- 24. Salazar LA, Diament J, Forti N et al. Polymorphisms at the apolipoprotein B-100 gene in Brazilian women in relation to body mass index. Primary prevention program. 5º Congreso Latinoamericano de Aterosclerosis, 1º Congreso de La Sociedad Ibero-latinoamericana de Aterosclerosis, Montevideo, 1999 (Abstract).
- 25. Salazar LA, Forti N, Diament J et al. Lipid levels and genetic markers for coronary artery disease in Brazilian healthy women in relation to body mass index primary prevention. 5th International Symposium Multiple Risk Factors in Cardiovascular Disease, Veneza, 1999 (Abstract).
- 26. Salazar LA, Hirata MH, Giannini SD et al. Seven DNA polymorphisms at the candidate genes of atherosclerosis in Brazilian women with angiographically documented coronary artery disease. Clin Chem Acta 2000; 300: 139-49.
- 27. Bydlowsky SP, Novak EM, Issa JS et al. DNA polymorphisms of apolipoprotein B and A I C III A IV genes in a Brazilian population: a preliminary report. Braz J Med Biol Res 1996; 29: 1269-74.
- 28. De Pádua Mansur A, Annicchino-Bizachi I, Avakian SD et al. Angiotensin converting enzyme and apolipoprotein B polymorphisms in coronary artery disease. Am J Cardiol 2000; 85: 1089-93.
- 29. Scartezini M, Zago MA, Pazin-Filho A et al. Presença do genótipo X-X- / E+E+ do gene da apo B Xba I / Eco R I é marcador de doença coronariana (DAC) em adultos jovens. Arq Bras Cardiol 1999; 73: 5.
- 30. Machado MO, Hirata MH, Bertolami MC et al. Apo B gene haplotype is associated with lipid profile of higher risk for coronary heart disease in caucasian Brazilian men. J Clin Lab Anal 2001; 15: 19-24.
- 31. Machado MO, Guzmán ECR, Hirata MH et al. Apolipoprotein B gene polymorphism in ischemic heart disease. Clin Chem 1997; 43 (supl): S 258.
- 32. Davignon J, Gregg RE, Sing CF. Apoliprotein E polymorphisms and atherosclerosis. Arteriosclerosis 1988; 8: 1-21.
- 33. Schaeffer EJ, Pedro-Botet J, Brousseau M et al. Genetic factors related to LDL lowering response to statins. XIV International Symposium on Drugs Affecting Lipid Metabolism, New York, 2001 (Abstract book p. 36).
- 34. Pedro-Botet J, Schaeffer EJ, Bakker-Arkema RG et al. Apolipoprotein E phenotypes affects plasma lipid response to atorvastatin in a gender specific manner. Atherosclerosis 2001; 158: 183-93.
- 35. Salazar LA, Ozaki AN, Hirata RDC et al. Frequency of the Hha I polymorphism of the apolipoprotein E gene in Brazilian postmenopausal women. 12th IFCC European Congress of Clinical Chemistry, Brasil, 1997 (Abstract p. 218).
- 36. Forti N, Salazar LA, Giannini SD et al. Apolipoprotein E isoforms are apparently not related to lipid modification in Brazilian postmenopausal women. Atherosclerosis 1999; 144: 106.
- 37. Otta MI, Cavalli SA, Hirata RDC et al. Apolipoprotein E genotype frequencies in hypercholesterolemic and diabetic individuals. Clin Chem 1996; 42 (supl): S 297.
- 38. Cavalli SA, Otta MI, Hirata RDC et al. Apolipoprotein E genotyping in Brazilian normocholesterolemic individuals. Clin Chem 1996; 42 (supl): S 298.
- 39. Guzmán ECR, Hirata MH, Forti N et al. Polimorfismos dos genes da lipase lipoprotéica e da apolipoproteína E estão associados com a hipertrigliceridemia primária. 9º Congresso do Departamento de Aterosclerose, Sociedade Brasileira de Cardiologia, Natal, 2001 (Abstract p. 39).
- 40. Villeger L, Abifadel M, Allard D et al. The UMD-LDR database: additions to the software and 490 new entries to the database. Hum Mut 2002; 20: 81-7.
- 41. Giannini SD, Hirata RDC, Salazar LA et al. HDL-cholesterol (HDL-c) of postmenopausal women is influenced by Hinc II polymorphism of the LDL receptor (LDL-R) gene. Atherosclerosis 1997; 134: 77-8.
- 42. Salazar LA, Giannini SD, Hirata MH et al. Influence of Ava II and Hinc II polymorphism of the LDL-receptor gene on cholesterol levels in postmenopausal women. JACC 1998; 31 (supl C): 387 C.
- 43. Salazar LA, Hirata MH, Giannini SD et al. Effects of Ava II and Hinc II polymorphisms at the LDL-receptor gene on serum lipid levels of Brazilian individuals with high risk for coronary heart disease. J Clin Lab Anal 1999; 13: 251-8.
- 44. Salazar LA, Hirata MH, Forti N et al. Pvu II intron 15 polymorphism at the LDL-receptor gene is associated with differences in serum lipid concentrations in subjects with low and high risk for coronary artery disease from Brazil. Clin Chem Acta 2000; 293: 75-88.
- 45. Salazar LA, Cavalli SA, Hirata MH et al. Polymorphisms of the low-density lipoprotein receptor gene in Brazilian individuals with heterozygous familial hypercholesterolemia. Braz J Med Biol Res 2000; 33: 1301-4.
- 46. Salazar LA, Melo CE, Cavalli SA et al. Micrométodo para extração de DNA genômico útil no diagnóstico molecular da hipercolesterolemia familiar. Rev Bras Anal Clin 2001; 33: 111-6.
- 47. Salazar LA, Hirata MH, Melo CE et al. Five polymorphic variants of the low-density lipoprotein receptor in patients with familial hypercholesterolemia from Brazil. International MEDPED Meeting, Eugendorf, 2002 (Abstract).
- 48. Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mut 1992; 1: 445-6.
- 49. Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In Scriver CR, Beaudet AL, Sly WS, Valle D (ed.). The metabolic basis of inherited disease. New York, McGraw Hill, 1995 p. 1981-2030.
- 50. Bertolini S, Canatafora A, Averna M et al. Clinical expression of familial hypercholesterolemia in clusters of mutations of the LDL receptor gene that cause a receptor-defective or receptor-negative phenotype. Arterioscler Thromb Vasc Biol 2000; 20: 1-12.
- 51. Hobbs HH, Leitersdorf E, Leffert C et al. Evidence for a dominant gene that suppresses hypercholesterolemia in a family with defective low-density lipoprotein receptors. J Clin Invest 1989; 84: 656.
- 52. Lundhal B, Leren TP, Ose L et al. A functional polymorphism of the microssomal triglyceride transfer protein (MTP-493 G/T) influences lipoprotein phenotype in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol 2000; 20: 1784-8.
- 53. Figueiredo MS, Santos JE, Alberto FL et al. High frequency of the Lebanese allele of the LDL-r gene among Brazilian patients with familial hypercholesterolemia. J Med Genet 1992; 29: 813-5.
- 54. Alberto FL, Figueiredo MS, Zago MA et al. The Lebanese mutation as an important cause of familial hypercholesterolemia in Brazil. Braz J Med Biol Res 1999; 32: 739-45.
- 55. Salazar LA, Hirata MH, Cavalli SA et al. Molecular basis of familial hypercholesterolemia in Brazil: identification of seven novel LDL-R gene mutations. Hum Mut 2002; 19: 462-3.
- 56. Salazar LA, Hirata MH, Hirata RD. Increasing the sensitivity of single-strand conformation polymorphism analysis of the LDLR gene mutations in Brazilian patients with familial hypercholesterolemia. Clin Chem Lab Med 2002; 40: 441-5.
- 57. Cavalli SA, Salazar LA, Hirata MH et al. Polimorfismo da região 5' promotora do gene da CYP 3 A 4 em indivíduos com hipercolesterolemia familiar heterozigótica. Rev Bras Cienc Farmaceut 2001; 37 (supl 1): 8.
- 58. Guzmán ECR, Hirata MH, Forti N et al. Avaliação do efeito dos polimorfismos Hha I da apolipoproteína E e Pvu II da lípase lipoprotéica em indivíduos com hipertrigliceridemia. Rev Bras Cienc Farmaceut 2001; 37 (supl 1): 7.
- 59. Hirata RD, Salazar LA, Cavalli AS et al. A method to detect the G 894 T polymorphism of the NOS 3 gene. Clinical validation in familial hypercholesterolemia. Clin Chem Lab Med 2002; 40: 436-40.
- 60. Salazar LA, Cunha AL, Forti N et al. Genetic markers in women with angiographically documented coronary artery from Brazil. Atherosclerosis 2000; 151: 228.
- 61. Salazar LA, Cunha AL, Hirata MH et al. Os polimorfismos C 677 T e A 1289 C do gene da M T H F R constituem um fator de risco para doença arterial coronariana em mulheres brasileiras? J Bras Pat Med Lab 2000; 36: 120.
- 62. Salazar LA, Santos ST, Cunha AA et al. Avaliação do efeito da mutação C 677 T do gene do M T H F R no desenvolvimento precoce de DAC em pacientes brasileiros com hipercolesterolemia familiar. Rev Soc Cardiol Est São Paulo 2001; 11: 66.
- 63. Hoffmann MM, Winkelmann BR, Wieland H. The significance of genetic polymorphisms in modulating the response to lipid lowering drugs. Pharmacogenomics 2001; 2: 107-121.
- 64. Corsini A, Bellosta S, Paoletti R. Enzymatic pathways involved in statin metabolism. XIV International Symposium on Drugs Affecting Lipid Metabolism, New York, 2001 (Abstract book p. 36).
- 65. Miettinen T, Gylling H. ABC G 5/8 genes responsible for serum plant sterol increase during statin treatment. XIV International Symposium on Drugs Affecting Lipid Metabolism, New York, 2001 (Abstract book p. 37).
- 66. Salazar LA, Hirata MH, Quintão EC et al. Lipid lowering response of the HMG-CoA reductase inhibitor fluvastatin is influenced by polymorphisms in the low-density lipoprotein receptor gene in Brazilian patients with primary hypercholesterolemia. J Clin Lab Anal 2000; 14: 125-31.
- 67. Salazar LA, Matsumoto LO, Cavalli SA et al. Polimorfismo do gene da apolipoproteína E influencia a resposta terapêutica às estatinas em indivíduos com hipercolesterolemia familiar. J Bras Patol 2001; 37: 33.
- 68. Salazar LA, Hirata MH, Forti N et al. Angiotensin I converting enzyme gene polymorphism and drug response in familial hypercholesterolemia. XIV International Symposium on Drugs Affecting Lipid Metabolism, New York, 2001 (Abstract book p. 37).
- 69. Salazar LA, Cavalli SA, Hirata MH et al. Variações genéticas no receptor de LDL, ECA e HMG-CoA redutase influenciam a resposta terapêutica na hipercolesterolemia familiar. IX Congresso do Departamento de Aterosclerose da Sociedade Brasileira de Cardiologia, Natal, 2001 (Abstract p. 38).
- 70. Salazar LA, Hirata MH, Forti N et al. Polymorphisms of low-density lipoprotein receptor gene are associated with lipid lowering response in patients with familial hipercolesterolemia. XIV International Symposium on Drugs Affecting Lipid Metabolism, New York, 2001 (Abstract book p. 119).
- 71. Cavalli SA. Estudo de alterações no gene da enzima CYP 3 A 4 que influenciam a resposta à atorvastatina em indivíduos hipercolesterolêmicos. Doutorado (qualificação), Faculdade de Ciências Farmacêuticas da Universidade de São Paulo, 2002.
Datas de Publicação
-
Publicação nesta coleção
30 Maio 2003 -
Data do Fascículo
Maio 2003
Histórico
-
Aceito
08 Nov 2002 -
Recebido
10 Out 2002