Abstract
As microRNAs (miRNAs) are important regulators of many biological processes, a series of small RNAomes from plants have been produced in the last decade. However, miRNA data from several groups of plants are still lacking, including some economically important crops. Here microRNAs from Coffea canephora leaves were profiled and 58 unique sequences belonging to 33 families were found, including two novel microRNAs that have never been described before in plants. Some of the microRNA sequences were also identified in Coffea arabica that, together with C. canephora, correspond to the two major sources of coffee production in the world. The targets of almost all miRNAs were also predicted on coffee expressed sequences. This is the first report of novel miRNAs in the genus Coffea, and also the first in the plant order Gentianales. The data obtained establishes the basis for the understanding of the complex miRNA-target network on those two important crops.
coffee; microRNA profiling; illumina sequencing
Identification of novel and conserved microRNAs in Coffea canephora and Coffea arabica
Guilherme Loss-MoraisI; Daniela C.R. FerreiraII,III; Rogério MargisIV; Márcio Alves-FerreiraII,III; Régis L. CorrêaII
ILaboratório Nacional de Computação Científica, Petrópolis, RJ, Brazil
IIDepartamento de Genética, Universidade Federal de Rio de Janeiro, Rio de Janeiro, RJ, Brazil
IIIPrograma de Biotecnologia Vegetal, Universidade Federal de Rio de Janeiro, Rio de Janeiro, RJ, Brazil
IVDepartamento de Biofísica, Universidade Federal de Rio Grande do Sul, Porto Alegre, RS, Brazil
Send correspondendce Send correspondendce to: Regis L. Corrêa Departmento de Genetica, Universidade Federal de Rio de Janeiro 21941-902 Rio de Janeiro, RJ, Brazil E-mail: regis@biologia.ufrj.br
ABSTRACT
As microRNAs (miRNAs) are important regulators of many biological processes, a series of small RNAomes from plants have been produced in the last decade. However, miRNA data from several groups of plants are still lacking, including some economically important crops. Here microRNAs from Coffea canephora leaves were profiled and 58 unique sequences belonging to 33 families were found, including two novel microRNAs that have never been described before in plants. Some of the microRNA sequences were also identified in Coffea arabica that, together with C. canephora, correspond to the two major sources of coffee production in the world. The targets of almost all miRNAs were also predicted on coffee expressed sequences. This is the first report of novel miRNAs in the genus Coffea, and also the first in the plant order Gentianales. The data obtained establishes the basis for the understanding of the complex miRNA-target network on those two important crops.
Keywords: coffee, microRNA profiling, illumina sequencing.
Introduction
There are two major classes of small regulatory noncoding RNAs (sRNAs) in plants: small interfering RNAs (siRNAs) and microRNAs (miRNAs) (Chen,2012). Both types of sRNAs are generated from double-stranded RNA (dsRNA) precursors that are processed into approximately 20-24 nucleotide (nt)-sequences by conserved proteins generically called Dicers or Dicer-like (DCL) (Hamilton and Baulcombe, 1999).
MiRNAs can control basic aspects of development, as well as the molecular responses to different types of stresses (de Lima et al., 2012). In plants, genes coding for miRNAs are generally 100-400 nt long and can be located in either the exons or introns of protein coding genes or in intergenic regions (Bartel, 2005). Mature miRNAs are initially generated from hairpin-like precursors as dsRNA duplexes. One of the strands (the guide strand) is loaded into RNA silencing complexes called RISC, while the other strand (the passenger or star strand) is usually degraded (Baumberger and Baulcombe, 2005; Qi et al., 2005). The RISC complex, containing Argonaute proteins (AGO), isthen directed to RNAs having similarity with the embedded guide sequence. Depending on the Argonaute effector protein present in the complex, targets can be repressed either by RNA degradation or by translation inhibition (Huntzinger and Izaurralde, 2011).
Although some miRNAs are known to be conserved throughout the plant kingdom, the advent of massively parallel DNA sequencing methods allowed the identification of a vast number of non-conserved genes, even in closely related plants (Rajagopalan et al., 2006; Fahlgren et al., 2007; Ma et al., 2010). To date, there are about ten thousand mature miRNA sequences of green plants (Viridiplantae) deposited in the miRBase database (miRBase) (Griffiths-Jones, 2004). However, these sequences are not evenly distributed among the taxa. For example, information from economically important plants, including the ones belonging to the genus Coffea, is almost entirely lacking.
The genus Coffea belongs to the family Rubiaceae and contains more than a hundred species. C. arabica is the only tetraploid species within the genus and probably arose through the hybridization between the diploid genomes of C. eugenioides and C. canephora (Lashermes et al., 1999). Since most of the coffee produced in the world comes from C. arabica and C. canephora, some efforts have been made to sequence and characterize transcripts from these two species (Lin et al., 2005; Mondego et al., 2011; Combes et al., 2013). However, only few conserved miRNAs have been described in C. arabica so far (Rebijith et al., 2013, Akter et al., 2014). In this work we deep-sequenced total sRNAs from C. canephora leaves and found conserved and novel miRNA genes belonging to 33 families, including two that have never been observed in other plants before.
Material and Methods
Plant material and deep sequencing
Leaves of C. canephora (conilon cultivar) were harvested at an experimental field from the Federal University of Viçosa, Minas Gerais State, Brazil. Total RNAs were extracted using the Plant RNA Reagent (Invitrogen, cat 12322-012) and were sent as ethanol precipitates to be sequenced at Fasteris Life Science Co. (Geneva, Switzerland). The small RNA library was prepared according to a modified Illumina protocol previously described (Silva et al., 2011) and sequenced using the HiSeq2000 platform. The raw data obtained from the sequenced library was deposited at the NCBIs Gene Expression Omnibus (GEO) database under the accession number GSE46617.
Data processing and filtering
Adaptor sequences were trimmed from the generated data using custom scripts. After the removal of low-quality reads and reads smaller than 16 nt and bigger than 26 nt, the high-quality raw sequences were used as queries in local BLASTN (Altschul, et al., 1997) searches against known cellular non-coding RNAs (rRNA, tRNA, snoRNA, mtRNA and cpRNA). Filtering was done using the following sequences or databases: complete chloroplast DNA from C. arabica (NC_008535); complete mitochondrial DNA from Nicotiana tabacum (NC_006581), Boea hygrometrica (NC_016741) and Mimulus guttatus (NC_018041), tRNA from A. thaliana, Populus trichocarpa, Medicago truncatula, rRNA from Asclepias syriaca, C. arabica, and snoRNA from all plant species available. The sRNAs matching with the refereed sequences without mismatches and gaps were discarded and the remaining unique sequences were used to search for conserved miRNAs.
Identification of conserved and novel miRNAs
MiRNAs were identified by three independent strategies: i) BLAST searches (Altschul et al., 1997) against the sequences deposited in the miRBase database (release 20) (Griffiths-Jones, 2004); ii) mapping sRNAs onto C. arabica and C. canephora contigs using SOAP2 software (Li et al., 2009) and iii) using the plant miRDeep tool (Yang and Li, 2011). For BLAST searches, the filtered set of unique sRNAs (all high quality reads from 16 to 26 nt, without rRNAs, tRNAs, snoRNAs, mtRNAs and cpRNAs) were used in local BLASTN searches against all plant mature sequences retrieved from the miRBase database. Only sequences that fully matched known genes from the database, without gaps and with at least 10 reads, were further processed. The remaining sequences were considered as unknown sequences. For the SOAP2 analysis, the full set of redundant reads was matched against C. arabica and C. canephora contigs/ESTs (Expressed Sequence Tags) retrieved from the Brazilian Coffee Genome Project (Mondego et al., 2011) or from a C. canephora RNA-seq database (Combes et al., 2013). The SOAP2 output was filtered with an in-house filter tool (FilterPrecursor) in order to identify candidate sequences as miRNA precursors using a mapping pattern of one or two blocks of aligned small RNAs with perfect matches (Kulcheski et al., 2011). The filtering was done with the following default parameters: minimum number of mapped reads in the candidate precursors: 10; maximum offset allowed for a single read: 5; maximum percentage of reads mapped out of columns: 25; maximum number of columns in the mapping profile: 2. Parameters used for the miRDeep analysis were: length of best perfect match: 28; type of output: 2(traditional BLAST output); Identity percentage cut-off [Real]: 0 (perfect match); maximum number of hits: 10. The selected candidate precursors were manually inspected using the Tablet software (Milne et al., 2010) to visualize the presence of the mapping pattern. The secondary structures of candidate sequences were checked with the RNA Folding/annotation tool from the UEA sRNA toolkit (Moxon et al., 2008), using default parameters. The following criteria were used to define a good miRNA candidate: no more than four unpaired nucleotides between the putative mature and star sequences, of which no more than three nucleotides were consecutive and no more than three nucleotides were without a corresponding unpaired nucleotide in the near complementary sequence within the hairpin structure (Meyers et al., 2008). Only contigs matching those rules and with at least 10 reads in the putative miRNA region were considered as miRNA precursors. Candidates were then used as queries for BLASTN searchers against plant miRBase sequences. Reads having full matches without gaps with miRBase sequences were considered as conserved miRNAs. Sequences with no matches in the database were considered as novel miRNAs and the ones having nonperfect matches were considered as variants of known miRNAs.
Prediction of miRNA targets
The prediction of the putative target genes for conserved and novel miRNAs was done with the psRNATarget software (Dai and Zhao, 2011). The search was done against the C. canephora and C. arabica contigs retrieved from the Brazilian Coffee Genome Project or against the C. canephora RNA-Seq data (Combes et al., 2013), with the following parameters: maximum expectation value: 3; multiplicity of target sites: 2; and nucleotide range of central mismatch for translational inhibition: 9-11. Candidate sequences were annotated based on BLASTN (Altschul et al., 1997) and PFam searches (Punta et al., 2012). Gene ontology terms were obtained by using the GO slimmer tool from the AmiGO toolkit (Carbon et al., 2009), using default parameters.
Digital expression analysis
The expression of target miRNAs was computed in the different libraries of the Brazilian Coffee Genome Project. The frequency of reads for each miRNA contig in each library was computed and then normalized by the number of reads in the library. The values obtained were then analyzed with the Cluster and Tree View programs (Eisen et al., 1998). Aggregation was made by hierarchical clustering, based on Spearman Rank correlation matrix. Digital blot matrix was ordered according to similarities in the patterns of gene expression and displayed as an array, where the normalized number of reads for each EST-contig in each specific library is represented in gray scale.
Results
C. canephora sRNA library
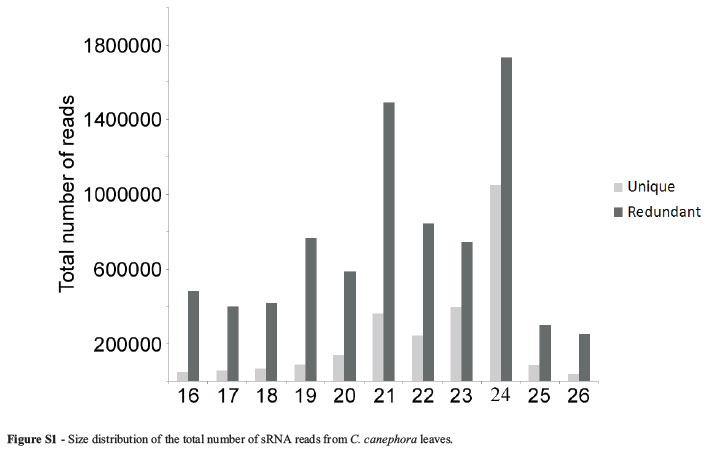
About eight million high quality reads ranging from 16 to 26 nt were obtained in the Illumina sequencing of sRNAs from C. canephora leaves (Table S1). Most of the redundant and unique reads identified were 24 nt and 21nt long, respectively (Figure S1). This pattern has already been observed in several other plant deep-sequencing libraries and is probably due to the high abundance of heterochromatic siRNAs and microRNAs, which are 24 nt and 21nt long, respectively (Nobuta et al., 2008; Lelandais-Briere et al., 2009; Wei et al., 2009; Klevebring et al., 2009; Romanel et al., 2012).
The identified sRNAs were divided into six categories: small nucleolar RNAs (snoRNAs), transfer RNAs (tRNAs), ribosomal RNAs (rRNAs), mitochondrial RNAs (mtRNAs), chloroplastidial RNAs (cpRNAs) and miRNAs (Table S1). Together, about 49.7% of all redundant sRNAs matched to snoRNAs, tRNAs, rRNAs, mtRNAs or cpRNAs, but about 46.6% of all reads could not be assigned to any of the six categories (Table S1). An interesting prominent peak of 19 nt was observed in tRNA-derived reads (Figure 1). It has recently been reported that some of tRNA-derived sequences of this size can be found in complexes with Argonaute proteins and therefore may not be merely degradation products (Loss-Morais et al., 2013). Sequences belonging to miRNAs represented about 3.6% of the total redundant reads in the library. Those sequences were found by searching against plant miRNA sequences deposited in the miRBase database release 20 (Griffiths-Jones, 2004) and by aligning all reads against expressed contigs from C. arabica and C. canephora or against RNA-Seq data from C. canephora (Combes et al., 2013) (see Methods for details).
As expected, most of the C. canephora miRNA reads were 21 nt long, but sequences of 20, 22, 23 and 24 nt were also found (Figure 1). Since there are no coffee miRNA sequences in the current release of the miRBase database, all miRNAs identified here are new for the refereed species. MiRNAs from all size classes were then separated into three categories: 1) miRNAs that are conserved in other plant species and whose sequences are identical to sequences deposited in the miRBase (47 miRNAs, in Table 1); 2) variants of known miRNAs (nine miRNAs, in Table 2 and 3) novel coffee miRNAs whose sequences are not related to any of the known families of plant miRNAs deposited in miRBase (two miRNAs, in Table 2).
Identification of conserved miRNAs in C. canephora and C. arabica
For the identification of conserved miRNAs, C. canephora high quality reads from 16 to 26 nt in size that did not match to snoRNAs, tRNAs, rRNAs, mtRNAs or cpRNAs were used as queries in local BLASTN searches against plant miRNA genes deposited in the miRBase database. Only results having full matches, without gaps and with at least 10 reads were further analyzed. A total of 250,992 reads, representing 47 unique miRNAs and belonging to 24 families, matched the plant miRBase sequences under those strict rules (Table 1). The number of miRNA mature sequences found on each family varied significantly. Most of the families (14 out of 24) were represented by only one mature sequence (Table 1). In general, families with multiple miRNA sequences had one dominant form, followed by lower expressed sequences. This is the case, for example, for the family miR159, where four mature sequences were found, with the dominant miR159a having about 44 thousand reads, while the other three members together having less than two hundred reads (Table 1). In the family miR166, however, a total of eight miRNA sequences were identified, with two of them having high read abundance (Table 1).
The precursor sequences for five of the conserved miRNAs could be found by mapping the C. canephora reads to C. canephora and C. arabica available expressed contigs (Table 1, Figure 2 and Figure 3) (Mondego et al., 2011). The precursor from miR396b, however, was the only one retrieved from C. canephora contigs (Table 1, Figure 2), while the other four (miR167b, miR172b, miR390b and miR398a) were found in C. arabica-derived sequences (Table 1, Figure 3). The star sequences for all of them, except miR167b were also found by this analysis (Figure 2 and Figure 3), increasing the confidence of our data. The identification of precursor sequences in C. arabica based on C. canephora reads also indicates that these genes are likely conserved between the two species.
Identification of non-conserved miRNAs in C. canephora and C. arabica
Since the coffee genome sequence is still publicly unavailable, the identification of non-conserved miRNAs was done by separately mapping all redundant sRNA reads from C. canephora against contigs from C. canephora and C. arabica (Mondego et al., 2011) or against C. canephora RNA-seq data (Combes et al., 2013) using the SOAP2 software (Li et al., 2009) or the miRDeep-P tool (Yang and Li, 2011). After filtering results with Perl scripts (see Methods), about 250 contigs from C. canephora and 350 contigs from C. arabica were considered as candidate precursor sequences. All contigs were then visually inspected through the Tablet software and their structures computed by a RNA hairpin folding and annotation tool (Moxon et al., 2008). Only structures following strict pairing rules were then considered as miRNA precursors. By using this strategy, eleven non-conserved miRNAs were found (Table 2). From those, nine are variants of known plant miRNA families (Table 2). Those sequences were annotated according to the miRBase best hit (Table S2). Two other genes, however, are totally unrelated to known families of miRNAs and are therefore members of two new putative families in plants (Table 2).
From the seven families of miRNAs where new members were found, six (miR159, miR393, miR479, miR5225, miR8558 and miR8697) have only one member each, while family miR482 have three sequences (Table 2). Interestingly, all members of the miR482 family identified here seem to be highly expressed in C. canephora leaves (Table 2). The precursor sequences from only four genes (miR479, miR482a, miR482c, miR5225) were found in C. canephora contigs (Table 2, Figure 2). All the others, except miR479, had their precursors found among C. arabica contigs (Table 2, Figure 3). Therefore, the miRNAs miR482a and miR482c were the only cases, in this study, where the precursor sequences were found on both species. The star sequence could also be detected for all but miR159e and miR482b, reinforcing the idea that the identified genes are processed by the canonical DCL pathway (Figure 2 and Figure 3).
The novel precursor miRNAs were found in two Coffea canephora expressed sequences (Table 2 and Figure 3). Since those genes are still not deposited in public databases, they were temporarily named as miR-Seq-1 and miR-Seq-2. Their putative miRNA star sequence could also be retrieved from the sequenced reads and their precursor sequences fit the requirements for being considered as true miRNA genes (Figure 3).
Isoforms of miRNAs found in C. canephora sRNAs
MiRNA isoforms, also known as iso-miRNAs, are a group of diverse sequences derived from a single precursor gene. They are frequently observed and are likely originated from imprecise DCL processing or post-transcriptional editing processes (Morin et al., 2008). Isoforms, however, may be loaded into Argonaute complexes and therefore exert their silencing activities (Fernandez-Valverde et al., 2010; Wang H et al., 2011). In total, 17 iso-miRNAs, derived from 11 genes, were found (Table 3). It is unlikely that the observed iso-miRNAs are sequencing artifacts, since all bases mapped to coffee transcripts have Q quality scores higher than 30 (99.9% of accuracy) (data not shown). Most precursors have only one isoform, but up to five isoforms coming from a single gene were observed for miR482c. The two miR-seq-1 isoforms showed the highest degree of sequence diversity when compared to their reference miRNA (Table 3). The expression of most iso-miRNAs, however, was significantly lower than their reference variant (Table 3). For example, miR8558 is about six hundred times more expressed than its detected isoform (Table 3). Although miRNAs miR396b, miR482a and miR482b also follow this rule, their respective isoforms have a high read abundance, indicating that they might be systematically produced in C. canephora cells (Table 3).
Targets of miRNAs
All expressed contigs from both C. canephora and C. arabica were used to search the putative targets of the identified miRNAs through the web-based psRNATarget tool (Dai and Zhao, 2011). In total, 339 and 149 probable targets were identified in C. arabica and C. canephora, respectively. From these, 442 sequences were predicted to be targeted by conserved miRNAs (Table S3) and 46 by the non-conserved ones (Table 4). All the identified targets were categorized into Gene Ontology (GO) terms to evaluate their putative functions (Figure S2). GO terms covering a broad range of biological processes were obtained, demonstrating the putative importance of coffee miRNAs in controlling several physiological aspects. GO terms related to regulation of transcriptional, development and cell differentiation, however, were by far the most enriched terms observed (Figure S2).
Several coffee sequences similar to known miRNA targets were found, including genes involved in different aspects of development (Table S3). This includes genes associated with vegetative phase change, like the Squamosa promoter binding protein (SBP/SBL)-like and Apetala-2, targets of miRNAs 156 and 172 (Wang JW et al., 2011), respectively, and cell proliferation-related genes, like NACcontaining genes, target of miR164 (Mallory et al., 2004), and WRC domain-containing Growth regulating factors, target of miR396 (Rodriguez et al., 2010). Genes involved with root and leaf formation were also identified, including Auxin Responsive Factors, targeted by miR160 (Wang et al., 2005), and Homeodomain-containing genes, targeted by the miRNA165/166 family during the establishment of leaf polarity (Rhoades et al., 2002). Biotic and abiotic stress-related genes were also observed among the putative coffee miRNA targets. For instance, the identified target of miR395, the APT sulfurylase, is known to be involved in sulfate homeostasis during sulfur starvation (Jones-Rhoades and Bartel, 2004). Another interesting example is the Plastocyanin domain-containing Copper binding protein, whose gene is regulated by miR398, which can play a role during abiotic and biotic stresses (Sunkar and Zhu, 2004).
The three members of the family miR482, together with the related sequence miR8558, accounted for almost half of the 46 predicted targets of non-conserved miRNAs (Table 4). The putative miR482-targeted genes included kinase proteins (Casein-,Calcium-and Malectin-like kinases), calcium binding proteins, ATPases, glutamine aminotransferases, disease resistance and DNA repair genes, among others. The non-conserved miRNAs miR5225 and miR-Seq-1 had only one predicted target in C. canephora and C. arabica, respectively. Curiously, both miRNAs targets are associated with the ubiquitin proteasome system (Table 4). The miRNA miR8697 was predicted to target 14 different genes that were annotated into five groups: Nucleoside diphosphate kinase Group I (NDPk_I)-like, Drought induced 19 protein (Di19), NADH dehydrogenase, NBS resistance gene and autophagy-related genes (Table 4). Four putative targets were identified for the novel miRNA miR-Seq-2. These targets, however, are all related to Pyruvate dehydrogenase E1 component subunit or secretory peroxidases (Table 4), which are involved in the glycolysis metabolic pathway and response to oxidative stress, respectively.
The digital expression of the predicted miRNA targets was also computed among C. canephora and C. arabica EST-based contigs (Mondego et al., 2011) (Figure S3). The predicted targets were in general depleted in leaf-derived libraries (LF1 for C. canephora and LV4, LV5, LV8, LV9 and RM1 for C. arabica) (Figure S3). This is the case for example for miRNAs miR165/166, miR172a and miR398a, where the high abundance of reads detected in C. canephora leaves by deep-sequencing (Table 1) is well correlated with the low accumulation of their targets in the contig-based digital analysis (Figure S3). In accordance, some of the putative miR482-targeted genes, including Calcium kinase (Contig5317), targeted by miR482a, Malectin-like receptor kinase protein (Contig3591), targeted by miR482b and alpha-crystallin-Hsps_p23-like (Contig2315), targeted by miR482c and most of the miR8697 targets are depleted in leaf-derived tissues (Figure S3).
Discussion
In this study we used a deep-sequencing approach to identify and classify miRNAs in C. canephora and C. arabica. Out of approximately 9 million reads, we have identified about 280 thousand reads corresponding to miRNAs. These sequences represented 58 unique mature miRNAs that were divided into 33 families, including two that, as far as we know, have never been described in the literature before (with provisory names miR-Seq-1 and miR-Seq-2).
Our searching pipeline involved three independent strategies: i) BLAST searches against the miRBase database, for the identification of the conserved genes, ii) alignments of the sRNA reads to contigs/ESTs from C. canephora and C. arabica through the SOAP2 software and iii) search for novel miRNAs with the plant miRDeep tool (Yang and Li, 2011). The results are robust, since very stringent rules were used in all analyses. For example, only sequences having full matches, no gaps and at least 10 reads were retrieved from the BLAST searches. Some miRNAs were probably missed by using these rules, since nucleotide polymorphisms are observed even in closely related species (Ma et al., 2010), which might explain the reduced amount of conserved miRNAs observed in Coffea compared to other plants.
The data obtained from the SOAP2 software was also filtered with stringent rules. Only contigs or ESTs having a mapping pattern similar to what would be expected for a canonical miRNA locus (i.e. having one or two similar blocks with piled up reads) and having at least 10 reads were initially selected. Then, all candidate precursor sequences were checked by RNA folding programs for identifying bona fide miRNA transcripts. Since precursor miRNAs are readily degraded by the RNA silencing machinery, such sequences are not frequently observed on EST sequencing efforts. However, some miRNAs have already been found by this strategy (Frazier and Zhang, 2011; Guzman et al., 2012). The precursor sequences from 16 of the 58 miRNAs identified could be predicted in coffee expressed sequences by this strategy, including five conserved genes, nine variants of known miRNAs and the two new putative families of miRNAs (Figures 2 and 3). Although the sRNAs were extracted and sequenced from C. canephora leaves, most of the precursors were found in C. arabica sequences. The discrepancy might be explained by the fact that the total number of available sequences from C. arabica was about two times higher than the ones from C. canephora (35,153 vs 18,007). From the 16 precursors, 10 were only found in C. arabica, four only in C. canephora and two on both species. In some cases the miRNA* sequences could not be observed, probably due to their low accumulation or imprecise DCL processing. However, the probable miRNA* sequence, with the 2-nt overhang characteristic of the DCL activity, could be found in the majority of the cases. The two novel miRNAs, miR-Seq-1 and miR-Seq-2, were found by the plant miRDeep tool in C. arabica EST-based contigs and RNA-seq data, respectively, providing extra support for the results.
The targets of almost all identified miRNAs could be predicted in coffee expressed sequences (Tables S3 and 4). Several known targets of miRNAs involved in different aspects of plant development and stress response were retrieved by this analysis. As observed for other plants (Shivaprasad et al., 2012), the new members of the family miR482 and the related gene miR8558 identified here have a wide range of presumed targets, including nucleotide binding site-leucine-rich repeat (NBS-LRR) plant innate immune receptors (Table 4). This miRNA has also been associated with the biogenesis of trans acting siRNAs (tasiRNAs) in other plants, a class of 21-nt long secondary siRNAs that are able to regulate the expression of several targets (Allen et al., 2005; Yoshikawa et al., 2005). TasiRNAs are made by the mutual action of a miRNA and the silencing amplification machinery on non-coding Trans-acting siRNA transcripts (TAS). One of the three coffee members of this family identified in this study (miR482c), and the related sequence miR8558, are 22 nt long (Table 2), the miRNA size usually associated in the biogenesis of tasiRNAs (Cuperus et al., 2010; Manavella et al., 2012). Furthermore, as also observed for other members of the family, the miR482 genes found here have several isoforms and are highly and promiscuously expressed in different types of tissues (Shivaprasad et al., 2012).
Some putative targets of non-conserved miRNAs, including the novel ones, could also be found. The miRNAs miR5225 and miR-Seq-1, for example, were predicted to target genes involved in the process of protein ubiquitination (Table 4). miR8697 has a broad-spectrum of predicted targets, including Nucleoside diphosphate kinase Group I (NDPk_I)-like, NADH dehydrogenase, Ubiquitin domain of GABA-receptor-associated protein and Drought induced 19 protein (Di19) (Table 4). NDPKs catalyze the transfer of phosphate from nucleoside triphosphates to nucleoside diphosphates. There are four isoforms of NDPKs annotated in the genome of the model plant Arabidopsis thaliana. Apart from their role in basal metabolism, some NDPK isoforms have also been associated with intracellular signaling and heat-stress responses (Hasunuma et al., 2003). The Ubiquitin domain of GABA-receptorassociated protein observed in one of the miR8697 targets belongs to a large and conserved family of proteins involved in membrane trafficking and autophagy. Autophagy has historically been attributed to the control of basal cellular functions, but can also be activated as a response against certain types of stresses (Liu and Bassham, 2012). Finally, the zinc-binding protein Di19 is known to be up-regulated in leaves and roots of A. thaliana plants under progressive drought stress (Gosti et al., 1995). The protein functions as a transcriptional factor, inducing the expression of some pathogenesis-related proteins that can buffer the drought effects (Liu et al., 2013). As most of the miRNA targets predicted, Di19 was not observed in leaf-derived libraries of the EST-based digital expression analysis (Figure S3). This correlation should be taken with caution, since the tissues used for making the EST-libraries were not taken from plants in the same conditions or developmental stages as the ones used for deep-sequencing. However, the general trend supports our target-discovery strategy. The understanding of how, where and when miRNAs interact with other genes will provide useful insights into coffee physiology, expanding both basic and applied knowledge about these economically important plants.
Acknowledgments
The authors would like to thank Dr. Marcelo Loureiro, from the Federal University of Viçosa, Brazil, for providing the C. canephora leaves used in this study. We also thank Elisson Romanel and Carlos Guerra Schrago from Federal University of Rio de Janeiro for their support in the bioinformatics analysis. This work was supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (FAPERGS) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
Internet Resources
Received: May 30, 214
Accepted: August 29, 2014.
Associate Editor: Juan Lucas Argueso Almeida
Supplementary Material
The following online material is available for this article: Table S1 -Categories of sRNA sequences, ranging from 16 to 26 nt, found in the library of C. canephora leaves.
Table S2 -BLAST results of variants of known miRNAs against the the miRBase database.
Table S3 -Targets of conserved microRNAs found in C. canephora and C. arabica.
Figure S1 -Size distribution of the total number of sRNA reads from C. canephora leaves.
Figure S2 -Gene Ontology terms of miRNA targets identified on C. canephora contig/EST libraries.
Figure S3 -Electronic northern blot of predicted miRNA targets on C. canephora and C. arabica contig/EST libraries.
This material is available as part of the online article from .
Note Added in Proof
While this manuscript was in press, a paper describing the C. canephora genome sequence came out and identified 92 conserved miRNAs by comparing with miRBase sequences (Denoeud et al., 2014). Of those, 38 miRNAs belonging to 19 families were also found in our pipeline based on expressed sequences.
Denoeud F, Carretero-Paulet L, Dereeper A, Droc G, Guyot R, Pietrella M, Zheng C, Alberti A, Anthony F, Aprea G et al. (2014) The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science 345:1181-1184.
Supplementary Data
The supplementary data is available in pdf: [Supplementary data]
- Akter A, Islam MM, Mondal SI, Mahmud Z, Jewel NA, Ferdous S, Amin MR and Rahman MM (2014) Computational identification of miRNA and targets from expressed sequence tags of coffee (Coffea arabica). Saudi J Biol Sci 21:3-12.
- Allen E, Xie Z, Gustafson AM and Carrington JC (2005) microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 121:207-221.
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W and Lipman DJ (1997) Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 25:3389-3402.
- Bartel DP (2005) MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116:281-297.
- Baumberger N and Baulcombe DC (2005) Arabidopsis ARGO-NAUTE1 is an RNA Slicer that selectively recruits micro-RNAs and short interfering RNAs. Proc Natl Acad Sci USA 102:11928-11933.
- Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B and Lewis S (2009) AmiGO: Online access to ontology and annotation data. Bioinformatics 25:288-289.
- Chen X (2012) Small RNAs in development -Insights from plants. Curr Opin Genet Dev 22:361-367.
- Combes MC, Dereeper A, Severac D, Bertrand B, Lashermes P (2013) Contribution of subgenomes to the transcriptome and their intertwined regulation in the allopolyploid Coffea arabica grown at contrasted temperatures. New Phytol 200:251-260.
- Cuperus JT, Carbonell A, Fahlgren N, Garcia-Ruiz H, Burke RT, Takeda A, Sullivan CM, Gilbert SD, Montgomery TA and Carrington JC (2010) Unique functionality of 22-nt miRNAs in triggering RDR6-dependent siRNA biogenesis from target transcripts in Arabidopsis. Nat Struct Mol Biol 17:997-1003.
- Dai X and Zhao PX (2011) psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res 39:W155-159.
- de Lima JC, Loss-Morais G and Margis R (2012) MicroRNAs play critical roles during plant development and in response to abiotic stresses. Genet Mol Biol 35:1069-1077.
- Eisen MB, Spellman PT, Brown PO and Botstein D (1998) Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA 95:14863-14868.
- Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Law TF, Grant SR, Dangl JL, et al. (2007) High-throughput sequencing of Arabidopsis microRNAs: Evidence for frequent birth and death of MIRNA genes. PLoS One 2:e219.
- Fernandez-Valverde SL, Taft RJ and Mattick JS (2010) Dynamic isomiR regulation in Drosophila development. RNA 16:1881-1888.
- Frazier TP and Zhang B (2011) Identification of plant microRNAs using expressed sequence tag analysis. Methods Mol Biol 678:13-25.
- Gosti F, Bertauche N, Vartanian N and Giraudat J (1995) Abscisic acid-dependent and -independent regulation of gene expression by progressive drought in Arabidopsis thaliana Mol Gen Genet 246:10-18.
- Griffiths-Jones S (2004) The microRNA registry. Nucleic Acids Res 32:D109-111.
- Guzman F, Almerao MP, Korbes AP, Loss-Morais G and Margis R (2012) Identification of microRNAs from Eugenia uniflora by high-throughput sequencing and bioinformatics analysis. PLoS One 7:e49811.
- Hamilton AJ and Baulcombe DC (1999) A species of small antisense RNA in post transcriptional gene silencing in plants. Science 286:950-952.
- Hasunuma K, Yabe N, Yoshida Y, Ogura Y and Hamada T (2003) Putative functions of nucleoside diphosphate kinase in plants and fungi. J Bioenerg Biomembr 35:57-65.
- Huntzinger E and Izaurralde E (2011) Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat Rev Genet 12:99-110.
- Jones-Rhoades MW and Bartel DP (2004) Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol Cell 14:787-799.
- Klevebring D, Street NR, Fahlgren N, Kasschau KD, Carrington JC, Lundeberg J and Jansson S (2009) Genome-wide profiling of populus small RNAs. BMC Genomics 10:e620.
- Kulcheski FR, de Oliveira LF, Molina LG, Almerão MP, Rodrigues FA, Marcolino J, Barbosa JF, Stolf-Moreira R, Nepomuceno AL, Marcelino-Guimaraes FC, et al. (2011) Identification of novel soybean microRNAs involved in abiotic and biotic stresses. BMC Genomics 12:e307.
- Lashermes P, Combes MC, Robert J, Trouslot P, D'Hont A, Anthony F and Charrier A (1999) Molecular characterisation and origin of the Coffea arabica L. genome. Mol Gen Genet 261:259-266.
- Lelandais-Briere C, Naya L, Sallet E, Calenge F, Frugier F, Hartmann C, Gouzy J and Crespi M (2009) Genome-wide Medicago truncatula small RNA analysis revealed novel microRNAs and isoforms differentially regulated in roots and nodules. Plant Cell 21:2780-2796.
- Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K and Wang J (2009) SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 25:1966-1967.
- Lin C, Mueller LA, Mc Carthy J, Crouzillat D, Petiard V and Tanksley SD (2005) Coffee and tomato share common gene repertoires as revealed by deep sequencing of seed and cherry transcripts. Theor Appl Genet 112:114-130.
- Liu WX, Zhang FC, Zhang WZ, Song LF, Wu WH and Chen YF (2013) Arabidopsis Di19 functions as a transcription factor and modulates PR1, PR2, and PR5 expression in response to drought stress. Mol Plant 6:1487-1502.
- Liu Y and Bassham DC (2012) Autophagy: Pathways for selfeating in plant cells. Annu Rev Plant Biol 63:215-237.
- Loss-Morais G, Waterhouse PM and Margis R (2013) Description of plant tRNA-derived RNA fragments (tRFs) associated with argonaute and identification of their putative targets. Biol Direct 8:e6.
- Ma ZR, Coruh C and Axtell MJ (2010) Arabidopsis lyrata small RNAs: Transient MIRNA and small Interfering RNA loci within the Arabidopsis genus. Plant Cell 22:1090-1103.
- Mallory AC, Dugas DV, Bartel DP and Bartel (2004), MicroRNA regulation of NAC-domain targets is required for proper formation and separation of adjacent embryonic, vegetative, and floral organs. Curr Biol 14:1035-1046.
- Manavella PA, Koenig D and Weigel D (2012) Plant secondary siRNA production determined by microRNA-duplex structure. Proc Natl Acad Sci USA 109:2461-2466.
- Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, Cao X, Carrington JC, Chen X, Green PJ, et al. (2008) Criteria for annotation of plant microRNAs. Plant Cell 20:3186-3190.
- Milne I, Bayer M, Cardle L, Shaw P, Stephen G, Wright F and Marshall D (2010) Tablet -Next generation sequence assembly visualization. Bioinformatics 26:401-402.
- Mondego JM, Vidal RO, Carazzolle MF, Tokuda EK, Parizzi LP, Costa GG and Pereira LF (2011) An EST-based analysis identifies new genes and reveals distinctive gene expression features of Coffea arabica and Coffea canephora BMC Plant Biol 11:e30.
- Morin RD, O'Connor MD, Griffith M, Kuchenbauer F, Delaney A, Prabhu AL, Zhao Y, McDonald H, Zeng T, Hirst M, et al. (2008) Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res 18:610-621.
- Moxon S, Schwach F, Dalmay T, MacLean D, Studholme DJ and Moulton V (2008) A toolkit for analysing large-scale plant small RNA datasets. Bioinformatics 24:2252-2253.
- Nobuta K, Lu C, Shrivastava R, Pillay M, De Paoli E, Accerbi M, Arteaga-Vazquez M, Sidorenko L, Jeong DH, Yen Y, et al. (2008) Distinct size distribution of endogeneous siRNAs in maize: Evidence from deep sequencing in the mop1-1 mutant. Proc Natl Acad Sci USA 105:14958-14963.
- Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, et al. (2012) The Pfam protein families database. Nucleic Acids Res 40:D290-301.
- Qi Y, Denli AM and Hannon GJ (2005) Biochemical specialization within Arabidopsis RNA silencing pathways. Mol Cell 19:421-428.
- Rajagopalan R, Vaucheret H, Trejo J and Bartel DP (2006) A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana Genes Dev 20:3407-3425.
- Rebijith KB, Asokan R, Ranjitha HH, Krishna V and Nirmalbabu K (2013) In silico mining of novel microRNAs from coffee (Coffea arabica) using expressed sequence tags. J Horticult Sci Biotechnol 88:325-337.
- Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B and Bartel DP (2002) Prediction of plant microRNA targets. Cell 110:513-520.
- Rodriguez RE, Mecchia MA, Debernardi JM, Schommer C, Weigel D and Palatnik JF (2010) Control of cell proliferation in Arabidopsis thaliana by microRNA miR396. Development 137:103-112.
- Romanel E, Silva TF, Correa RL, Farinelli L, Hawkins JS, Schrago CE and Vaslin MF (2012) Global alteration of microRNAs and transposon-derived small RNAs in cotton (Gossypium hirsutum) during Cotton leafroll dwarf polerovirus (CLRDV) infection. Plant Mol Biol 80:443-460.
- Shivaprasad PV, Chen HM, Patel K, Bond DM, Santos BA and Baulcombe DC (2012) A microRNA superfamily regulates nucleotide binding site-leucine-rich repeats and other mRNAs. Plant Cell 24:859-874.
- Silva TF, Romanel EA, Andrade RR, Farinelli L, Osteras M, Deluen C, Correa RL, Schrago CE and Vaslin MF (2011) Profile of small interfering RNAs from cotton plants infected with the polerovirus Cotton leafroll dwarf virus. BMC Mol Biol 12:e40.
- Sunkar R and Zhu JK (2004) Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 16:2001-2019.
- Wang H, Zhang X, Liu J, Kiba T, Woo J, Ojo T, Hafner M, Tuschl T, Chua NH and Wang XJ (2011) Deep sequencing of small RNAs specifically associated with Arabidopsis AGO1 and AGO4 uncovers new AGO functions. Plant J 67:292-304.
- Wang JF, Wang LJ, Mao YB, Cai WJ, Xue HW and Chen XY (2005) Control of root cap formation by microRNA-targeted auxin response factors in Arabidopsis Plant Cell 17:2204-2216.
- Wang JW, Park MY, Wang LJ, Koo Y, Chen XY, Weigel D and Poethig RS (2011) miRNA control of vegetative phase change in trees. PLoS Genet 7:e1002012.
- Wei B, Cai T, Zhang R, Li A, Huo N, Li S, Gu YQ, Vogel J, Jia J, Qi Y, et al. (2009) Novel microRNAs uncovered by deep sequencing of small RNA transcriptomes in bread wheat Triticum aestivum (L.) and Brachypodium distachyon (L.) Beauv. Funct Integr Genomics 9:499-511.
- Yang X and Li L (2011) miRDeep-P: A computational tool for analyzing the microRNA transcriptome in plants. Bioinformatics 27:2614-2615.
- Yoshikawa M, Peragine A, Park MY and Poethig RS (2005) A pathway for the biogenesis of trans-acting siRNAs in Arabidopsis Genes Dev 19:2164-2175.
- tRNA database (A. thaliana, Populus trichocarpa and Medicago truncatula), http://lowelab.ucsc.edu/GtRNAdb/#eukarya (July, 2013).
- rRNA database (Asclepias syriaca and C. arabica), http://www.arb-silva.de/browser/ (July, 2013).
- snoRNA database, http://bioinf.scri.sari.ac.uk/cgi-bin/plant_snorna/get-sequences (July, 2013).
» link - Brazilian Coffee Genome Project, http://www.lge.ibi.unicamp.br/cafe/ (July, 2013).
» link - FilterPrecursor's source code, http://code.google.com/p/filter-precursors/downloads/list
Send correspondendce to:
Publication Dates
-
Publication in this collection
03 Nov 2014 -
Date of issue
Dec 2014
History
-
Received
30 May 2014 -
Accepted
29 Aug 2014