Abstract
Intrahippocampal administration of kainic acid (KA) induces synaptic release of neurotrophins, mainly brain-derived neurotrophic factor, which contributes to the acute neuronal excitation produced by the toxin. Two protein tyrosine kinase inhibitors, herbimycin A and K252a, were administered intracerebroventricularly, in a single dose, to attenuate neurotrophin signaling during the acute effects of KA, and their role in epileptogenesis was evaluated in adult, male Wistar rats weighing 250-300 g. The latency for the first Racine stage V seizure was 90 ± 8 min in saline controls (N = 4) which increased to 369 ± 71 and 322 ± 63 min in animals receiving herbimycin A (1.74 nmol, N = 4) and K252a (10 pmol, N = 4), respectively. Behavioral alterations were accompanied by diminished duration of EEG paroxysms in herbimycin A- and K252a-treated animals. Notwithstanding the reduction in seizure severity, cell death (60-90% of cell loss in KA-treated animals) in limbic regions was unchanged by herbimycin A and K252a. However, aberrant mossy fiber sprouting was significantly reduced in the ipsilateral dorsal hippocampus of K252a-treated animals. In this model of temporal lobe epilepsy, both protein kinase inhibitors diminished the acute epileptic activity triggered by KA and the ensuing morphological alterations in the dentate gyrus without diminishing cell loss. Our current data indicating that K252a, but not herbimycin, has an influence over KA-induced mossy fiber sprouting further suggest that protein tyrosine kinase receptors are not the only factors which control this plasticity. Further experiments are necessary to elucidate the exact signaling systems associated with this K252a effect.
Herbimycin A; K252a; Cell death; EEG; Epilepsy
Braz J Med Biol Res, May 2008, Volume 41(5) 403-410
Protein tyrosine kinase inhibitors modify kainic acid-induced epileptiform activity and mossy fiber sprouting but do not protect against limbic cell death
C.M. Queiroz* and  Correspondence and Footnotes
Correspondence and Footnotes
 L.E. Mello
L.E. Mello
Departamento de Fisiologia, Escola Paulista de Medicina, Universidade Federal de São Paulo, São Paulo, SP, Brasil
 References
References
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Abstract
Intrahippocampal administration of kainic acid (KA) induces synaptic release of neurotrophins, mainly brain-derived neurotrophic factor, which contributes to the acute neuronal excitation produced by the toxin. Two protein tyrosine kinase inhibitors, herbimycin A and K252a, were administered intracerebroventricularly, in a single dose, to attenuate neurotrophin signaling during the acute effects of KA, and their role in epileptogenesis was evaluated in adult, male Wistar rats weighing 250-300 g. The latency for the first Racine stage V seizure was 90 ± 8 min in saline controls (N = 4) which increased to 369 ± 71 and 322 ± 63 min in animals receiving herbimycin A (1.74 nmol, N = 4) and K252a (10 pmol, N = 4), respectively. Behavioral alterations were accompanied by diminished duration of EEG paroxysms in herbimycin A- and K252a-treated animals. Notwithstanding the reduction in seizure severity, cell death (60-90% of cell loss in KA-treated animals) in limbic regions was unchanged by herbimycin A and K252a. However, aberrant mossy fiber sprouting was significantly reduced in the ipsilateral dorsal hippocampus of K252a-treated animals. In this model of temporal lobe epilepsy, both protein kinase inhibitors diminished the acute epileptic activity triggered by KA and the ensuing morphological alterations in the dentate gyrus without diminishing cell loss. Our current data indicating that K252a, but not herbimycin, has an influence over KA-induced mossy fiber sprouting further suggest that protein tyrosine kinase receptors are not the only factors which control this plasticity. Further experiments are necessary to elucidate the exact signaling systems associated with this K252a effect.
Key words: Herbimycin A; K252a; Cell death; EEG; Epilepsy
Introduction
Kainate binding sites have been described in the hippocampus as being restricted to the molecular layer of granule cells and to the stratum lucidum of the CA3 region, where the mossy fibers (granule cell axons) terminate (1). In vivo, intracerebral injection of kainic acid (KA) leads to selective destruction of pyramidal CA3 neurons (2) and are associated with epileptiform discharges (3). KA administration also mimics some features of human temporal lobe epilepsy (4), i.e., hippocampal neuronal loss, reactive gliosis, mossy fiber sprouting (MFS), and spontaneous recurrent seizures (5).
Intrahippocampal injection of KA increases brain-derived neurotrophic factor (BDNF) immunoreactivity in the mossy fiber terminal zone (6) and it is postulated that BDNF protein production could be related to the long-term effects of KA (7). In fact, in vitro blockade of the tyrosine kinase receptor TrkB signaling inhibits mossy fiber formation (8), which in turn is strongly induced by local application of its endogenous ligand, the BDNF molecule (9). Recently, it was demonstrated that K252a, an inhibitor of tyrosine kinase activities, delayed KA-induced murine granule cell dispersion and increased MFS without modifying perforant path evoked granule cell responses (10).
In the present study, we investigated the effects of two different protein tyrosine kinase inhibitors, K252a and herbimycin A, on the acute epileptiform activity, cell death and MFS induced by the intrahippocampal injection of KA in rats. We hypothesized that blockade of protein tyrosine phosphorylation could attenuate the epileptiform discharges in the hippocampus and, thus, reduce the associated morphological alterations in the hippocampus (as indicated by cell death and MFS).
Material and Methods
Stereotaxic surgery
Male Wistar rats weighing 250-300 g were positioned in a stereotaxic frame (David Kopf, USA) under halothane anesthesia (4% induction and 1% maintenance). After exposure of the skull, small holes were drilled above the target structure according to the bregma and a stereotaxic atlas (11). Using a silica needle (Æ = 1 mm) attached to a Hamilton syringe, KA (0.2 µL of 1 µM) or saline were slowly administered (0.2 µL/min) into the right dorsal hippocampus (intrahippocampal administration), over the hilar-CA3 border region (-3.4, -2.5, and -3.5 mm). A 5-µL Hamilton syringe, attached to another silica needle, was used for the intracerebroventricular (icv; -0.8, -1.5, -2.9 mm) administration of 5 µL (2 µL/min) herbimycin A (348 µM), K252a (2 µM) or vehicle (1% dimethylsulfoxide, DMSO). In all cases, after injection the needle was allowed to rest for 2 min to allow the adequate distribution of the solution through the structure. Thereafter, bipolar electrodes were implanted in both dorsal hippocampi (-3.6, ±3.5, -3.5 mm) and fixed with dental cement to a plastic plug (Plastics One, USA). The time between the intrahippocampal and the icv administrations was approximately 20 min and the total time of surgery was never greater than 90 min. Using this procedure, the rats were divided into 4 groups (intrahippocampal and icv administrations, respectively): saline and DMSO (SAL-DMSO; N = 5); KA and DMSO (KA-DMSO; N = 4); KA and herbimycin A (KA-HERB; N = 4); KA and K252a (KA-K252a; N = 4). The post-surgery condition of the animals was monitored carefully during the following days. Animals were manipulated in accordance with ethical guidelines and the experimental procedures were previously approved by the Ethics Research Committee of UNIFESP/EPM (CEP1078/00).
EEG recording
Immediately after the surgery, the animals were connected through a cable to a digital EEG system (Harmony, Nicolet, USA). The signal was amplified, sampled at 250 Hz and filtered under 0.1 and above 100 Hz. The recordings started 1 h after drug administration and were registered continuously for 4 h. Subsequently, the recordings were made for 2 min every 20 min during the following 72 h. The recordings were analyzed qualitatively off-line for the onset and duration of the epileptiform activity in both hippocampi. ANOVA followed by the post hoc Student-Newman-Keuls test was used for comparison of seizure-like latencies.
Histology: Nissl and neo-Timm
After 45 days (chronic phase), animals were anesthetized profoundly (thiopental, 40 mg/kg) and transcardially perfused for the neo-Timm staining as described previously (12). Brains were sectioned on a cryostat and 40-µm thick coronal slices were mounted on glass slides for processing. The slides were immersed in a 360-mL solution (21ºC) containing 240 mL gum arabic, 10.25 g citric acid, 9.45 g sodium citrate, 3.73 g hydroquinone, and 510 mg silver nitrate for approximately 45 min for neo-Timm staining. Visual inspections of the slides were used to control the reaction time. Adjacent sections were used to assess cell death using Nissl staining (0.4% cresyl-violet) and electrode placement.
Cell counting and dentate gyrus width measurements
Cell counting was performed on Nissl-stained sections as described elsewhere (13). Briefly, cell body nuclei (Æ »10 µm, for hilar cells and Æ »7 µm for cortical cells) were counted in ipsi- and contralateral hilar regions at 400X magnification over a 4 x 4 (10,000 µm2) microscope grid (Olympus BX50, USA) based on the rat stereotaxic atlas (11). The hilar region was defined by the granule cell layer borders and by two imaginary lines drawn from the apex of the dentate gyrus and the proximal end of the CA3 pyramidal cell layer. Counting was carried out in four different portions of the hilar region in three different slices of the dorsal hippocampus separated by 200 µm. Since the total area evaluated for each hilus/animal was the same, mean counting for each group was normalized based on the control group counting (SAL-DMSO = 1). Thus, cell density was reported as median and interquartile intervals. Granule cell dispersion was evaluated by the width of a line perpendicular to the cell layer in the suprapyramidal region of the dentate gyrus at two levels: the apex and the knee. Although both measurements (cell counts and dentate dispersion) do not allow comparisons with reports of other studies, they are sufficient to show differences between treatments and hemispheres, in addition to being less time-consuming. The Kruskal-Wallis test followed by the Mann-Whitney U-test was used to compare cell density and ANOVA followed by the post hoc Student-Newman-Keuls test was used for granule cell dispersion measurements.
Results
General behavior
Animals from all groups recovered from the anesthesia 4-6 min following discontinuation of 1% halothane ventilation. While animals from the SAL-DMSO group walked, groomed and slept after surgery, animals injected with KA presented tremors, clonic movements, wild-running, Racine stage V seizure (14), and status epilepticus (KA-DMSO: 3 of 4 animals; KA-HERB: 3 of 4 animals; KA-K252a: 2 of 4 animals). Notwithstanding the similar behavioral evolution between groups that received KA, animals in the KA-DMSO group presented their first Racine stage V seizure 90 ± 8 min after KA administration, while this latency was 369 ± 71 and 322 ± 63 min for KA-HERB and KA-K252a, respectively (P < 0.05; KA-DMSO vs KA-HERB or KA-K252a). Spontaneous seizures were rarely observed in the days after KA injection (only animals of the KA-DMSO group (2 of 4) presented spontaneous seizures), although it should be noted that no systematic attempt was made to address this issue.
EEG profile
Although the recordings were initiated just after electrode implantation, animals from the control group displayed a normal EEG, i.e., steady interictal spikes or epileptiform activity was never recorded in these animals. KA administration produced isolated interictal spikes, sustained synchronous activity and epileptiform discharges (Figure 1; KA-DMSO). Herbimycin or K252a co-treatment did not completely inhibit the epileptiform discharge but changed the temporal profile of this activity. Herbimycin co-treatment delayed and inhibited the spreading of the epileptic activity to the contralateral hippocampus in all animals, clearly in contrast with the pattern of seizure spread that occurred in all animals of the KA-DMSO group (Figure 1). The duration of the ipsilateral hippocampal seizure in animals co-treated with herbimycin was shorter than in the KA-DMSO group. K252a co-treatment also changed the epileptiform profile, but in a different manner. K252a was not able to inhibit the spread of seizure activity to the contralateral side but shortened the seizure discharges to less than 1 min each (Figure 1). These shorter seizure activity epochs persisted during the first 4 h of the recordings. Thus, while KA-DMSO animals showed a continuous epileptiform activity also in the hippocampus contralateral to the KA-injected site, KA-K252a animals displayed intermittent epileptiform activity in the same brain hemisphere.
Cell death and granule cell dispersion
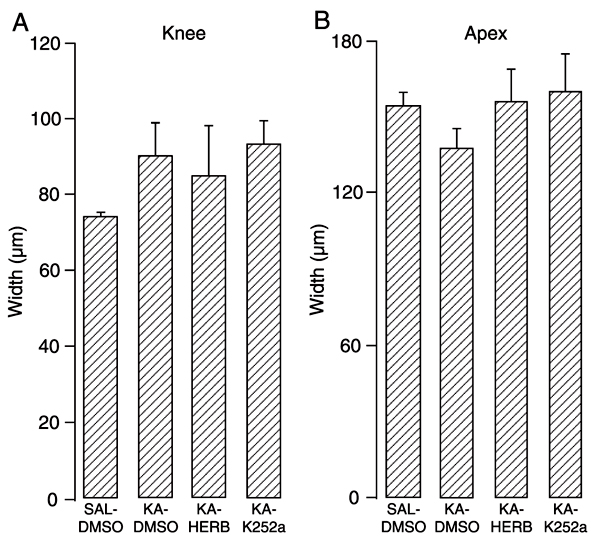
KA treatment produced massive death of hilar cells and pyramidal CA3 neurons ipsilateral to the KA injection site (Figure 2D,E). Despite an apparent preservation of CA1 pyramidal neurons, the gross architecture of this layer and of the cellular morphology was altered by KA treatment (Figure 2F). Extensive cell death was observed and quantified in the ipsilateral dorsal hilus (P < 0.01, Figure 3B). Nissl cell counting revealed no differences between the various KA-treated groups (DMSO, K252a or herbimycin), that ranged from 60-90% reduction in the cell counts. KA treatment also led to mild granule cell dispersion (Figure 2D,G,J). There was a trend in KA-treated groups to present an increased width of the suprapyramidal granule cell layer in the knee (~20%, Figure 4A) and a decreased width in the apex regions (~10%, Figure 4B) in the ipsilateral hippocampus; although, there were no distinguishable differences between granule cell layer width in either hemisphere for all groups analyzed (data not shown). There were also no alterations in cell counts in the contralateral hilar region (Figure 3A) as well as in the CA3 and CA1 regions (data not shown).
Mossy fiber sprouting
In control animals, neo-Timm staining was seen in the hilus and stratum lucidum of CA3 regions. Intragranular mossy fibers were observed in both hemispheres of the control group, mainly at the apex of the dentate gyrus (Figure 5; SAL-DMSO). These consisted of small branches emerging from granule cells towards the inner molecular layer of the dentate gyrus. KA treatment led to the appearance of small silver grains in the inner molecular layer of the dentate gyrus, mainly ipsilateral to the KA injection site (Figure 5; KA-DMSO and KA-HERB). These grains were observed in the apex and in the crests of the dentate gyrus. KA-K252a-treated animals presented less pronounced supragranular MFS in both hemispheres with a similar extent of intragranular fibers. The amount of silver grains in the inner molecular layer was more pronounced in animals from the KA-DMSO and KA-HERB groups.
Representative EEG recordings of the first epileptiform activity after intrahippocampal administration of kainic acid (KA; 0.2 µL of 1 µM) to a rat. Intracerebroventricular administration (20 min later) of herbimycin (HERB; 5 µL of 348 µM) modified KA-induced hippocampal synchronization and its spread to the contralateral hippocampus, while K252a (5 µL of 2 µM) administration reduced the duration of the epileptiform activity and modified its pattern. HPC-R = right hippocampus (site of KA injection); HPC-L = left hippocampus (contralateral side); DMSO = 1% dimethylsulfoxide. Calibration bar: 500 µV and 1 s.
Representative photomicrographs of Nissl-stained sections of hippocampal structures, ipsilateral to site of kainic acid (KA) administration in rats. KA-treated animals displayed extensive cell loss in the CA3 region (asterisks in E, H, and K). Protein tyrosine kinase inhibitors (1.74 nmol herbimycin or 10 pmol K252a) administered icv were not able to protect against cell death (compared with the SAL-DMSO group, B). Hilar cells were observed in the control group (A, arrowheads), but were absent after KA treatment (D, G, J). Mild alterations were observed in the CA1 region after KA treatment (F, I, L), compared to the SAL-DMSO group (C). mol = molecular layer of dentate gyrus; GC = granule cell layer; h = hilar region; rad = stratum radiatum; Pyr = stratum pyramidale; ori = stratum oriens. See legend of Figure 1 for experimental details.
Cell survival 45 days after intrahippocampal kainic acid (KA) administration to rats. KA treatment produced clear hilar cell death in the ipsilateral (right, B) hilus, while the contralateral (left, A) cells were preserved. Herbimycin (HERB, 1.74 nmol) and 10 pmol K252a icv treatment did not attenuate KA-induced cell death. Data are reported as median and interquartile intervals for N = 4 rats in each group except for SAL-DMSO in which N = 5. *P < 0.05 compared to SAL-DMSO group. See legend of Figure 1 for experimental details.
Granule cell dispersion 45 days after intrahippocampal kainic acid (KA) treatment of rats. The knee (A) portion of the granule cell layer was thinner than the apex (B) portion (note that the difference in scale used is almost 1.5-fold on the y-axis). Herbimycin (HERB, 1.74 nmol) and 10 pmol K252a icv administration did not modify granule cell layer width. Data are reported as means ± SEM for N = 4 rats in each group except for SAL-DMSO in which N = 5. See legend of Figure 1 for experimental details.
Representative photomicrographs of neo-Timm staining from the apex of dentate gyrus (DG). Arrowheads show the branches in the intragranular layer and arrows show silver grains in the inner molecular layer. Note that kainic acid (KA) treatment reduced intragranular staining and increased inner molecular layer staining. This morphological alteration was more intense in the ipsilateral DG. Herbimycin (HERB, 1.74 nmol, icv) attenuated staining in the inner molecular layer while 10 pmol K252a icv almost abolished the aberrant mossy fiber sprouting. See legend of Figure 1 for experimental details.
Discussion
In the present study, the tyrosine protein kinase inhibitors, herbimycin A and K252a, modified the electrographic epileptiform activity induced by intrahippocampal KA administration but did not alter the cell loss pattern. K252a, but not herbimycin, treatment reduced supragranular MFS.
Secretion of neurotrophins in the hippocampus, where nerve growth factor (NGF) and BDNF are expressed abundantly (15), is modulated significantly by neuronal activity (6,16). Accordingly, the excitotoxin kainate induces neurotrophins NGF and BDNF mRNA synthesis in the hippocampus (17) and increases their protein content, although it is still not known if this effect is related to the compound itself or to the KA-induced epileptiform activity. Intrahippocampal kainate administration leads to neuronal depolarization that, in turn, releases neurotrophins that will further depolarize neurons creating an excitatory feedback loop that may eventually lead to hyperexcitation. In fact, intrahippocampal application of BDNF elicits electrographic discharges with subsequent spontaneous seizures (18) and it can exacerbate an ongoing status epilepticus (19). Since secreted neurotrophins can powerfully stimulate protein tyrosine kinase receptors leading to phosphorylation of tyrosine residues (20), we speculate that K252a and herbimycin attenuate the electrographic seizures by decreasing tyrosine phosphorylation signaling. Our data support the role of protein tyrosine phosphorylation in the acute seizure activity triggered by KA administration.
Both protein tyrosine kinase inhibitors used in the present study were able to modify kainate-induced epileptiform activity. While herbimycin inhibited the spreading of the epileptiform activity to the contralateral hippocampus, K252a reduced intermittent burst activity. The differences between the inhibitors may be explained by their specificity to the wide variety of tyrosine substrates. In fact, herbimycin is described as having a broad spectrum of affinity for tyrosine substrates, including strong inhibitory activity on the family of the Src kinases (21). Src kinases have been described as "a hub for NMDA receptor regulation" (22) and have been involved in NMDA receptor phosphorylation during status epilepticus (23). On the other hand, bath application of K252a inhibits BDNF-induced inhibition of GABA-A receptor-mediated inhibitory postsynaptic currents in CA1 slices (24). By altering the balance between excitatory and inhibitory synaptic transmissions in kainate-induced epileptiform activity in the hippocampus, K252a could change the pattern of epileptiform burst activity (Figure 1). Independent of the mechanisms involved, the present results suggest that herbimycin is a more powerful inhibitor of kainate-induced epileptiform activity than K252a.
In contrast to previous results (25), K252a treatment did not prevent seizure-induced cell death. This difference may be explained by the administration routes used for KA injection (systemic vs intrahippocampal) and K252a treatment protocol (single icv acute injection vs 4 days of systemic injection). In the same study, the authors also did not find any EEG alterations in K252a-treated animals, probably because the last K252a injection occurred 1 week before the KA administration. In that case, long-term (4 days) pretreatment with K252a produced a neuroprotection independent of the KA-induced neuronal synchronization (25). In the present study, we observed the opposite effect, that is, attenuation of the KA-induced epileptiform activity without preventing limbic cell death. These results are consistent with the idea that K252a reduces neurotrophin-mediated neuronal excitation (20). Thus, despite reducing neuronal excitation, protein tyrosine kinase inhibitors were not able to prevent cell death and granule cell dispersion. The intrahippocampal administration of KA produced mild granular cell dispersion in the dentate gyrus (Figure 4) and the inhibitors did not attenuate this alteration. Recently, K252a was shown to retard granule cell dispersion in mice treated with KA (10), although granule cell dispersion was 300% higher than in controls (typical of mice). In our study, this dispersion was in the order of 20%, as described earlier for rats (26). These results suggest that K252a and probably herbimycin acutely affect cellular activation measured by EEG recordings but this effect is not sufficient to prevent morphological alterations.
Our results showed that K252a reduces aberrant MFS in KA-treated animals. However, herbimycin did not modify aberrant MFS in KA-treated animals, which is curious since this drug also inhibits protein tyrosine kinase and attenuated epileptiform activity (Figure 1). There are two possibilities to explain this effect. First, the inhibition of epileptiform activity by K252a was more effective than herbimycin in decreasing the levels of BDNF released during status epilepticus (6). Since BDNF is one of the major substances involved in fasciculation of mossy fibers, K252a reduced aberrant MFS by decreasing its levels in the hippocampus. A second possibility is that K252a has a specific effect over MFS per se, i.e., the binding affinity and drug kinetics make K252a a more suitable drug to inhibit aberrant MFS. In fact, it was recently shown that K252a disrupts mossy fiber pathfinding in organotypic culture of hippocampal slices, probably by inhibiting the action of endogenous neurotrophins and specifically the activation of protein tyrosine kinase receptors (9,27). Similarly, we have recently shown that herbimycin icv administration failed to attenuate aberrant MFS in pilocarpine-induced status epilepticus (13). It should be noted that the effect of neurotrophins on MFS is still controversial (28), since there are reports that show that MFS is not impaired in BDNF knock-out animals (29) as well as the fact that no "spontaneous" (i.e., aberrant) MFS is observed in animals overexpressing BDNF (30). Our current data indicate that K252a, but not herbimycin, influences KA-induced MFS and for this reason we suggest that BDNF does have an important role in the development of this plasticity.
Taken together, our results support the idea that neurotrophins can modify the epileptogenesis process (31,32) and that this effect is mediated by protein tyrosine kinase receptor signaling (33). Regarding the occurrence of spontaneous seizures as an indication of the epileptic condition, in the present experiment only 2 animals of the 4 in the KA-DMSO group presented spontaneous seizures, which suggests a possible antiepileptogenic effect of herbimycin and K252a. Herbimycin has been used unsuccessfully in the pilocarpine-induced status epilepticus model, since animals co-administered with herbimycin presented similar development of spontaneous seizure in the chronic phase of the pilocarpine model of epilepsy (13). Given that MFS was also reduced in the KA-K252a-treated animals, future experiments with K252a in animal models with more frequent seizures are encouraged.
Our current results indicate a crossroad where the use of animal models with frequent seizures might have such widespread cell damage that this cannot be counter-affected by local (intraventricular) administration of protein tyrosine kinase inhibitors. In contrast, the use of animal models with more restricted damage, such as that used here, may be more likely to be affected by local protein tyrosine kinase inhibitor administration, but might require an extensive investigation of spontaneous seizures given their scarcity in these models. The importance of reaching the basis for potential means to prevent epileptogenesis or to modify disease progression certainly warrants the investment in these laborious studies.
Acknowledgments
The authors would like to thank the valuable technical assistance carried out by Ms. Ivone de Paulo.
Address for correspondence: L.E. Mello, Departamento de Fisiologia, EPM, UNIFESP, Rua Botucatu, 862, 5º andar, E.C.B., 04023-062 São Paulo, SP, Brasil. Fax: +55-11-5579-2033. E-mail: lemello@unifesp.br
*The present address of C.M. Queiroz is Universiteit van Amsterdam, Swammerdam Institute for Life Sciences, Kruislaan 320, 1098SM, Amsterdam, The Netherlands. Research supported by FAPESP and CNPq. C.M. Queiroz was the recipient of a FAPESP fellowship (#00/10826-3). Received June 14, 2007. Accepted May 5, 2008.
- 1. Unnerstall JR, Wamsley JK. Autoradiographic localization of high-affinity [3H]kainic acid binding sites in the rat forebrain. Eur J Pharmacol 1983; 86: 361-371.
- 2. Nadler JV, Perry BW, Cotman CW. Selective reinnervation of hippocampal area CA1 and the fascia dentata after destruction of CA3-CA4 afferents with kainic acid. Brain Res 1980; 182: 1-9.
- 3. French ED, Aldinio C, Schwarcz R. Intrahippocampal kainic acid, seizures and local neuronal degeneration: relationships assessed in unanesthetized rats. Neuroscience 1982; 7: 2525-2536.
- 4. Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol 1989; 26: 321-330.
- 5. Cavalheiro EA, Riche DA, Le Gal La Salle G. Long-term effects of intrahippocampal kainic acid injection in rats: a method for inducing spontaneous recurrent seizures. Electroencephalogr Clin Neurophysiol 1982; 53: 581-589.
- 6. Wetmore C, Olson L, Bean AJ. Regulation of brain-derived neurotrophic factor (BDNF) expression and release from hippocampal neurons is mediated by non-NMDA type glutamate receptors. J Neurosci 1994; 14: 1688-1700.
- 7. Guilhem D, Dreyfus PA, Makiura Y, Suzuki F, Onteniente B. Short increase of BDNF messenger RNA triggers kainic acid-induced neuronal hypertrophy in adult mice. Neuroscience 1996; 72: 923-931.
- 8. Danzer SC, Crooks KR, Lo DC, McNamara JO. Increased expression of brain-derived neurotrophic factor induces formation of basal dendrites and axonal branching in dentate granule cells in hippocampal explant cultures. J Neurosci 2002; 22: 9754-9763.
- 9. Koyama R, Yamada MK, Fujisawa S, Katoh-Semba R, Matsuki N, Ikegaya Y. Brain-derived neurotrophic factor induces hyperexcitable reentrant circuits in the dentate gyrus. J Neurosci 2004; 24: 7215-7224.
- 10. Rougier A, Arthaud S, Zombre N, La Salle GG. Patterns of dentate granule cell responses to perforant path stimulation in epileptic mice with granule cell dispersion. Epilepsy Res 2005; 63: 119-129.
- 11. Paxinos G, Watson C. The rat brain stereotaxic coordinates 2nd edn. London: Academic Press; 1986.
- 12. Mello LE, Cavalheiro EA, Tan AM, Kupfer WR, Pretorius JK, Babb TL, et al. Circuit mechanisms of seizures in the pilocarpine model of chronic epilepsy: cell loss and mossy fiber sprouting. Epilepsia 1993; 34: 985-995.
- 13. Queiroz CM, Mello LE. Effects of herbimycin A in the pilocarpine model of temporal lobe epilepsy. Brain Res 2006; 1081: 219-227.
- 14. Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 1972; 32: 281-294.
- 15. Ernfors P, Wetmore C, Olson L, Persson H. Identification of cells in rat brain and peripheral tissues expressing mRNA for members of the nerve growth factor family. Neuron 1990; 5: 511-526.
- 16. Thoenen H. Neurotrophins and neuronal plasticity. Science 1995; 270: 593-598.
- 17. Ballarin M, Ernfors P, Lindefors N, Persson H. Hippocampal damage and kainic acid injection induce a rapid increase in mRNA for BDNF and NGF in the rat brain. Exp Neurol 1991; 114: 35-43.
- 18. Scharfman HE, Goodman JH, Sollas AL, Croll SD. Spontaneous limbic seizures after intrahippocampal infusion of brain-derived neurotrophic factor. Exp Neurol 2002; 174: 201-214.
- 19. Lahteinen S, Pitkanen A, Koponen E, Saarelainen T, Castren E. Exacerbated status epilepticus and acute cell loss, but no changes in epileptogenesis, in mice with increased brain-derived neurotrophic factor signaling. Neuroscience 2003; 122: 1081-1092.
- 20. Kafitz KW, Rose CR, Thoenen H, Konnerth A. Neurotrophin-evoked rapid excitation through TrkB receptors. Nature 1999; 401: 918-921.
- 21. Uehara Y, Hori M, Takeuchi T, Umezawa H. Screening of agents which convert `transformed morphology' of Rous sarcoma virus-infected rat kidney cells to `normal morphology': identification of an active agent as herbimycin and its inhibition of intracellular src kinase. Jpn J Cancer Res 1985; 76: 672-675.
- 22. Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci 2004; 5: 317-328.
- 23. Moussa RC, Ikeda-Douglas CJ, Thakur V, Milgram NW, Gurd JW. Seizure activity results in increased tyrosine phosphorylation of the N-methyl-D-aspartate receptor in the hippocampus. Brain Res Mol Brain Res 2001; 95: 36-47.
- 24. Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci 1997; 17: 2959-2966.
- 25. Smith-Swintosky VL, Kraemer PJ, Bruce AJ, McCants N, Maki A, Brown RW, et al. Bacterial alkaloids mitigate seizure-induced hippocampal damage and spatial memory deficits. Exp Neurol 1996; 141: 287-296.
- 26. Mello LE, Cavalheiro EA, Tan AM, Pretorius JK, Babb TL, Finch DM. Granule cell dispersion in relation to mossy fiber sprouting, hippocampal cell loss, silent period and seizure frequency in the pilocarpine model of epilepsy. Epilepsy Res Suppl 1992; 9: 51-59.
- 27. Tamura M, Koyama R, Ikegaya Y, Matsuki N, Yamada MK. K252a, an inhibitor of Trk, disturbs pathfinding of hippocampal mossy fibers. Neuroreport 2006; 17: 481-486.
- 28. Shetty AK, Zaman V, Shetty GA. Hippocampal neurotrophin levels in a kainate model of temporal lobe epilepsy: a lack of correlation between brain-derived neurotrophic factor content and progression of aberrant dentate mossy fiber sprouting. J Neurochem 2003; 87: 147-159.
- 29. Bender R, Heimrich B, Meyer M, Frotscher M. Hippocampal mossy fiber sprouting is not impaired in brain-derived neurotrophic factor-deficient mice. Exp Brain Res 1998; 120: 399-402.
- 30. Qiao X, Suri C, Knusel B, Noebels JL. Absence of hippocampal mossy fiber sprouting in transgenic mice overexpressing brain-derived neurotrophic factor. J Neurosci Res 2001; 64: 268-276.
- 31. Kokaia M, Ernfors P, Kokaia Z, Elmer E, Jaenisch R, Lindvall O. Suppressed epileptogenesis in BDNF mutant mice. Exp Neurol 1995; 133: 215-224.
- 32. Van der Zee CE, Rashid K, Le K, Moore KA, Stanisz J, Diamond J, et al. Intraventricular administration of antibodies to nerve growth factor retards kindling and blocks mossy fiber sprouting in adult rats. J Neurosci 1995; 15: 5316-5323.
- 33. He XP, Kotloski R, Nef S, Luikart BW, Parada LF, McNamara JO. Conditional deletion of TrkB but not BDNF prevents epileptogenesis in the kindling model. Neuron 2004; 43: 31-42.
Correspondence and Footnotes

Publication Dates
-
Publication in this collection
03 June 2008 -
Date of issue
May 2008
History
-
Received
14 June 2007 -
Accepted
05 May 2008