
Abstract
In more than 800 GLA gene mutations causing Fabry Disease (FD), renal involvement vary according to the α-GAL A mutation. The aim is to describe the genotype/phenotype variations of renal complications in two siblings with confirmed FD with the mutation p.Q279X in exon 6. We present a retrospective study of two venezuelan male siblings, ages 34 (patient 1) and 33 (patient 2), evaluated by general lab tests, renal ultrasound, renal scintigram , and renal biopsy. Fabry disease diagnose was made by α-galactosidase A activity determined in dried blood spot. Genomic DNA was sequenced by Sanger method. Patient 1 developed CKD grade 5 and high blood pressure, treated by hemodialysis during 8 years. Patient 2 showed GFR >60 ml/min, and proteinuria less than 600 mg/24H. Renal biopsy showed segmental sclerotic lesions and hypertrophic podocytes with vacuolated cytoplasm. Both patients received ERT every two weeks since 2003. Patient 1 died because dialysis complications (hyperparathyroidism, cardiomyopathy). The genotype/phenotype variation of the c.835C>T mutation (p.Gln279Ter. Q279X) in exon 6 of the GLA gene can express an important renal variation with a wide range of clinical manifestations that cannot be predicted, therefore, an early nephrological evaluation and periodic follow-up of these patients are necessary.
Keywords
Fabry disease; genotype/phenotype variations; exon 6
Introduction
Fabry disease (FD) (OMIM 301500) is an X-linked inherited disorder, caused by the accumulation of glycosphingolipids, predominantly globotriaosylceramide (Gb3), in organs throughout the body, as a results from deficiency in the activity of a lysosomal hydrolase called α-galactosidase A (α-GAL A.) [11. Ishii S, Taguchi A, Okino N, Ito M, Maruyama H. Determination of globotriaosylceramide analogs in the organs of a mouse model of Fabry disease. J Biol Chem. 2020; 295(17): 5577-5587. doi: 10.1074/jbc.RA120.012665
https://doi.org/10.1074/jbc.RA120.012665...
]
The increasing birth prevalence of GLA mutations, from previous estimates of 1:40,000-170,000 up to 1:1250 in newborn screening studies, reflects the existence of a majority of nonclassic mutations and variants of unknown significance. [22. Svarstad E, Marti HP. The Changing Landscape of Fabry Disease. Clin J Am Soc Nephrol. 2020;15(4):569-576. doi: 10.2215/cjn.09480819
https://doi.org/10.2215/cjn.09480819...
]
Mutations causing a virtually null enzymatic activity are associated to severe and early onset classical phenotypes, while mutations leading to a residual enzymatic activity are associated to attenuated and late onset phenotypes. In early onset variants during childhood or adolescence, classical phenotypes are characterized by early development of acroparesthesias, neuropathic pain, hypohydrosis, heat, cold and exercise intolerance, cornea verticillata, angiokeratomas, gastrointestinal symptoms and proteinuria. In adulthood, patients also suffer from sensorineural deafness and cardiac, renal and cerebrovascular manifestations. In contrast, late onset variants are characterized by the development of cardiac, renal and/or cerebrovascular manifestations in adulthood and the phenotype may be dominated for the participation of one organ, such as the heart or the kidneys. [33. Acevedo O, Gago M F, Miltenberger- Miltenyi G, Robles A, Costa M, Pereira O, et al. Natural history of the late-onset phenotype of Fabry disease due to the p.F113L mutation. Mol Genet Metab Rep. 2020;22:100565. doi: 10.1016/j.ymgmr.2020.100565
https://doi.org/10.1016/j.ymgmr.2020.100...
]
Delays in diagnosis are common in FD male patients, with a mean of 21.9 years. Cross-sectional and cohort studies have shown that renal manifestations (e.g. proteinuria or decreased glomerular filtration rate [GFR]) occur early in life in a significant proportion of children, in many females and in almost all male patients with FD. [44. Ortiz A, Oliveira J, Waldek S, Warnock D, Cianciaruso B, Wanner C. Nephropathy in males and females with Fabry disease: Cross-Sectional description of patients before treatment with enzyme replacement therapy. Nephrol Dial Transplant. 2008;23(5):1600-1607. doi: 10.1093/ndt/gfm848
https://doi.org/10.1093/ndt/gfm848...
] These manifestations progress over time and leading to an end-stage kidney disease (ESRD) in nearly all males and some female patients. Although progression to ESRD is less common in women, the median age at which patients reached ESRD (38 years) was the same in both genders. [55. Ortiz A, Cianciaruso B, Cizmarik M, Germain D, Mignani R, Oliveira P et al. End-stage renal disease in patients with Fabry disease: natural history data from the Fabry Registry. Nephrol Dial Transplant. 2010;25(3):769-775. doi: 10.1093/ndt/gfp554
https://doi.org/10.1093/ndt/gfp554...
]
To date, more than 800 GLA gene disease causing mutations have been reported in FD, being point mutations the most frequent type of disease-causing variants (70%). Small rearrangements accounts for about 28% of reported variants whereas large rearrangements accounts for about 2%. [66. Gal A, Beck M, Winchester R. Clinical utility gene card for: Fabry Disease. Eur J Human Genet. 2012;20:e1-e3. doi: 10.1038/ejhg.2011.178
https://doi.org/10.1038/ejhg.2011.178...
] There are suggestions that exons 6 and 7 are regions prone to small gene rearrangements. Renal involvement in FD may vary according to the α-GAL A mutation, for example a patient with a p.D266E mutation entered hemodialysis at 15 years old and received a kidney transplant at 16 years old; a patient with a p.S247C mutation manifested chronic renal failure stage 3 and mild proteinuria at 64 years old. One patient with the p.R301Q mutation developed renal failure and received renal transplantation at 59 years old. [77. Germain D, Shabbeer J, Cotigny S, Desnick R. Fabry Disease: Twenty novel α-Galactosidase a mutations and genotype-phenotype correlations in classical and variant phenotypes. Mol Med. 2002;8:306-312. doi: 10.1007/bf03402156
https://doi.org/10.1007/bf03402156...
]
Aim
To describe the genotype/phenotype variations of renal complications in two siblings with confirmed FD with the mutation p.Q279X in exon 6.
Methods
We present a retrospective study of two venezuelan male siblings, ages 34 (patient 1) and 33 (patient 2), blood type A+, with acroparesthesias and hypohydrosis since 9 years old. The physical evaluation showed angiokeratomas in external genitals, abdomen, buttocks and palms of hands; cornea verticillata by slit lamp, besides left ventricular hyperthrophy by electrocardiogram and echocardiogram, without arrhythmias. They were evaluated by:
-
a.- Lab Tests: hemoglobin, hematocrit, blood ureic nitrogen (BUN), serum creatinine, plasma sodium and potassium, urine exam, creatinine clearance by Jaffé method and proteinuria in 24 hours urine.
-
b.- Diagnostic method:
-
b.1.- α-galactosidase A activity determined in dried blood spot. [88. Chamoles N, Blanco M, Gaggioli D. Fabry disease: enzymatic diagnosis in dried blood spots on filter paper. Clin Chim Acta. 2001;308(1-2):195-196. doi: 10.1016/s0009-8981(01)00478-8
https://doi.org/10.1016/s0009-8981(01)00... ] Reference value > 1.86 µmol/l/h. [99. Civallero G, Michelin K, de Mari J, Viapiana M, Burin M, Coelho JC, et al. Twelve different assays on dried-blood spot paper samples for detection of patients with selected inherited lysosomal storage disease. Cli Chim Acta. 2006; 372(1-2):98-102. doi: 10.1016/j.cca.2006.03.029
https://doi.org/10.1016/j.cca.2006.03.02... ] -
b.2.- Genomic DNA was extracted from leukocytes. Each exon and flanking intron sequence of the GLA gene was amplified and PCR products were sequenced in both directions (forward and reverse) by the automated Sanger method (dideoxy terminators), using ABI 310 genetic sequencer (Applied Byosystems).
-
-
c.- Kidney biopsy by puncture
-
d.- Renal ultrasound
-
e.- Static and dynamic renal scintigram, with perfusion curve.
Compliance with Bioethics Guidelines
Informed consent was obtain from each patient for molecular study and renal biopsy, besides lab and image studies. Authors observed the Good Clinical Practice Guidelines and preserved patients identity.
Results
Table 1 summarizes lab results and molecular test.
Renal Biopsy and clinical progression
Patient 1:
Renal cortex. 18 glomeruli in the core, 8 of them globally sclerotic. 8 glomeruli were additionally evaluated and showed hypertrophic capillary tufts with isolated polymorphonuclear in capillaries. In 5/8 glomeruli segmental sclerotic lesions were observed (Figure 1) and hypertrophic podocytes exhibited clear and lacy cytoplasm. Interstitium was affected by increased fibrosis associated to moderate mononuclear inflammatory infiltrate. Proximal tubular epithelium was hypertrophic and vacuolated. Hyalinosis arteriolar was present in most arterioles.
Patient developed chronic renal failure grade 5, anuric, two years later, and began chronic hemodialysis, three times per week, 4 hours each. Kidneys were hypotrophic at the ultrasound and, due the poor diuresis renal scintigram was not possible.

Patient 1, kidney biopsy. Presence of positive PAS material, which totally or partially occludes capillary lights (arrows).
Patient 2:
Renal cortex, 16 glomeruli in the core, 3 were globally sclerotic and the rest showed moderate hypertrophic tufts with red cells and isolated mononuclears in capillaries. One glomerulus showed segmental sclerotic (Figure 2) lesion and partially collapsed capillaries. Podocytes showed hypertrophic and vacuolated cytoplasm. Mild diffuse interstitial fibrosis and focal tubular epithelium was also present. Arterial/arteriolae had focal subintimal hyalinosis.
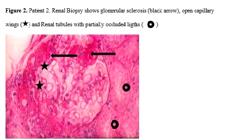
Patient 2. Renal Biopsy shows glomerular sclerosis (black arrow), open capillary wings (⋆) and Renal tubules with partially occluded ligths (🞈).
Report of static and dynamic renal scintigraphy with perfusion curve (Patient 2): Good perfusion and function in both kidneys, without elimination phase lesion. Differential function: right kidney 57%, left kidney 43% (Figure 3).

Renal ultrasound report (Patient 2): Both kidneys with normal form and size; right kidney: 94.2 mm x 41.8 mm, parenchyma 12.1 mm. Left kidney 112.5 mm x 62.4 mm, parenchyma 13.6 mm. No cyst nor pielocalicial dilatation.
The urine laboratory test showed: 1.010 density, acid pH, proteinuria 1+, granular and hyaline cylinders, crystals of amorphous urates, sparse clear cells.

Discussion
Although Gb3 accumulation is regarded as the primary pathogenic event in FD, remains unclear how the secondary pathophysiological derangements arising therefrom, at cell, tissue, and organ levels, which ultimately give rise to the clinical manifestations. However, the wide phenotypic variability observed among patients with the same GLA mutation, even when patients are male related familiarly, illustrates the complexity of the genotype-phenotype correlations in FD and the need to consider additional genetic and environmental modulating factors for their proper understanding. [1010. Oliveira J, Ferreira S. Multiple phenotypic domains of Fabry disease and their relevance for establishing genotype-phenotype correlations. Appl Clin Genet. 2019;12:35-50. doi: 10.2147/TACG.S146022
https://doi.org/10.2147/TACG.S146022...
]
Patients reported here are siblings with the p.Gln279Ter (Q279X) mutation in exon 6, showing an important renal genotype/phenotype variation demonstrated by glomerular filtration rate evolution: patient 1 required hemodialysis treatment, while patient 2 showed non nephrotic proteinuria and chronic kidney disease stage 2 (KDIGO). [1111. Gorostidi M, Santamaría R, Alcázar R, Fernández-Fresnedo G, Galcerán J, Marian Goicoechea M. Documento de la Sociedad Española de Nefrología sobre las guías KDIGO para la evaluación y el tratamiento de la enfermedad renal crónica. Nefrología. 2014;34(3):302-316. doi: 10.3265/nefrologia.pre2014.feb.12464
https://doi.org/10.3265/nefrologia.pre20...
] Nevertheless patient 1 had more glomerulosclerosis and glomerular obsolescence at biopsy than his brother. Four years later Fabry disease diagnosis, patient 1 began hemodialysis, presenting high blood pressure (HBP), requiring 3 antihypertension drugs. At theses moment it´s important to mention that all progressive chronic glomerulopathies are associated with a tubulointerstitial lesion characterized by a mononuclear inflammatory infiltrate, interstitial fibrosis and tubular atrophy, [1212. Ortiz A. Etiopatogénesis de las enfermedades glomerulares. In: Avendaño LH, Aljama García M, Arias Rodríguez M, Caramelo Díaz C, Egido de los Ríos JE, Lamas Peláez S (Editores). Nefrología Clínica. Editorial Médica Panamericana; 2003: 255-267.] that together with proteinuria greater than 1 g / day, hyperfiltration, activation of the RAAS, among others factors, would favor kidney disease progression. The magnitude of tubulointerstitial injury correlates better with the prognosis of renal function than the magnitude of glomerular damage. Factors that may contribute to interstitial damage include the passage of inflammation mediators to the postglomerular capillaries that bathe the tubules, the leakage of inflammation mediators through the glomerulus, and the toxic effect of proteinuria. Persistent proteinuria and exposure to cytokines can damage tubular epithelial cells and activate them to express cytokines and adhesion molecules and act as antigen-presenting cells to T lymphocytes.[1212. Ortiz A. Etiopatogénesis de las enfermedades glomerulares. In: Avendaño LH, Aljama García M, Arias Rodríguez M, Caramelo Díaz C, Egido de los Ríos JE, Lamas Peláez S (Editores). Nefrología Clínica. Editorial Médica Panamericana; 2003: 255-267.] The pathophysiology of Fabry nephropathy describes the accumulation of Gb3, the elevation of circulating myeloperidoxidases, the increase in oxidative stress, the action of globotriaosylphingosine metabolites (lyso Gb3), all of which would favor the secretion of bioactive mediators of the inflammatory response such as TGF-β1 and NF κβ. The result would be glomerular sclerosis on one hand, and tubulo-interstitial atrophy and fibrosis on the other, the latter favored by the "toxic" action exerted on the renal interstitium by tubular reabsorption of proteins. [1313. Herrera J, Vidau P. La Enfermedad de Fabry en pacientes en hemodiálisis: valoración del tratamiento enzimático sustitutivo. Dial Trasp. 2012;33(3):71-74. doi: 10.1016/j.dialis.2011.11.002
https://doi.org/10.1016/j.dialis.2011.11...
]
As previously mentioned, patient 1 evolved to end stage renal disease over a period of 4 years, receiving chronic hemodialysis 3 times per week, using an autologous left brachial arteriovenous fistula. Due to voluntary suspension of antihypertensive medication, he developed ischemic stroke of the left middle cerebral artery, with subsequent motor recovery, maintaining mild dysarthria of late recovery. He also developed bone mineral disease and cardiomyopathy as a complications of chronic kidney disease, and died 8 years after starting hemodialysis.
On the other hand, patient 2 was diagnosed with Fabry's disease, by renal biopsy carried out as a family investigation. At that time, he had a glomerular filtration rate of 110 ml / min, and proteinuria of 200 mg / 24H. He outcome with a slight decrease in glomerular filtration rate, maintained above 60 ml / min / 1.73m2 and proteinuria remained between 400-600 mg / 24H, during the observation period. As clinical complications, left hearing loss, secondary to traumatic brain injury, and spontaneous pneumothorax with ad integrum restoration, were reported. [1414. Villalobos J, Febres Celestina, Colina Virginia Leticia. Comportamiento de los signos vitales durante la infusión de Agalsidasa B en el tratamiento de la Enfermedad de Fabry. VITAE Academia Biomédica Digital. 2013;53. http://www.bioline.org.br/abstract?id=va13006⟨=en
http://www.bioline.org.br/abstract?id=va...
] It was no identify any other cardiovascular risk factor (heart failure, ischemic cardiomyopathy, renal stenosis, obesity, smoking, alcohol abuse) that can explain so different renal outcome in these patients. Patient 1 developed HBP as part of the glomerular disease.
Both patients received ERT since 2003 every two weeks with B agalsidase during almost 10 years and then switched to A agalsidase (no medical reasons); patient 1 developed low blood pressure, requiring suspension of antihypertensive treatment, and lost the arteriovenous fistula one year before died.
The c.835C>T mutation (p.Gln279Ter. Q279X) in exon 6 of the GLA gene was identified in four members (30 + 24, range 1-60 years) of a greek family.[1515. Koulansios K, Styhanou K, Pateinakis P, Zamanakou M, Loules G, Manou E, et al. Fabry disease due to D313Y and novel GLA mutations. BMJ Open. 2017;7:e017098. doi: 10.1136/bmj.jopen2017-017098
https://doi.org/10.1136/bmj.jopen2017-01...
] The proband´s clinical description (a 31.year-old-man) is very similar to that observed in our patient 2, but not in patient 1, because he is currently under ERT with beneficial result in regard with kidney function, proteinuria and pain. There is no description about dialysis and its complications. As we know, there is no relation between both, greek and venezuelan families.
The structure of human α-galactosidase A has been determined by X-ray crystallographic method and the amino acid substitutions responsible for the biochemical defect have been characterized. Expression and functional studies on α-galactosidase A aim to define the mutations effects on the mature protein in order to determine causative mutations and evaluate therapy options. [1616. Filoni C, CaciottiA, Carraresi L, Cavecchi C, Parini R, Antuzzi D. Functional studies of new GLA gene mutations leading to conformational Fabry disease. Biochim Biophys Acta. 2010;1802(2):247-252. doi: 10.1016/j.bbadis.2009.11.003
https://doi.org/10.1016/j.bbadis.2009.11...
] It is important to highlight that these two brothers have similar systemic characteristics, except in the results of their kidney function; patient 1 had been treated with hemodialysis for more than five years, starting at 29 years old, and developed bone mineral disease as a dialysis complication. The p.Gln279Ter (Q279X) mutation produce a stop codon located in the active site of the α-galactosidase molecule and generate a truncated protein with any residual effect, which makes these genotype/phenotype variations more interest and clinically unpredictable. The establishment of a reliable genotype-phenotype correlation provides challenges for genetic counseling, since molecular testing may allow prediction of the development of the disease. [1717. De Baere E, Beysen D, Oley C, Lorenz B, Cocquet J, De Sutter P, et al. FOXL2 and BPES: Mutational Hotspots, Phenotypic Variability, and Revision of the Genotype-Phenotype Correlation. Am J Hum Genet. 2003;72(2): 478-487. doi: 10.1086/346118
https://doi.org/10.1086/346118...
]
Although specific genotype-phenotype correlations can be established for most genotypes, but this correlation could be explained only in part of the phenotypic variability. This phenotypic variation may be determined by the influence of environmental factors and/or the influence of modifier genes. Characterizing modifier genes is important for several reasons. First, it could lead to improve diagnosis and genetic counselling by facilitating a more accurate prediction of the phenotype based on the genotype of both GLA and the modifier gene. Second, it could provide a better definition of the pathways in which the primary mutation functions and gain potentially novel insights into the molecular pathology of Fabry disease. [1818. Hilgert N, Huentelman M, Thorburn A, Fransen E, Dieltjens N, Mueller-Malesinska M. Phenotypic variability of patients homozygous for the GJB2 mutation 35delG cannot be explained by the influence of one major modifier gene. Eur J Hum Genet. 2009;17:517-524. doi: 10.1038/ejhg.2008.201
https://doi.org/10.1038/ejhg.2008.201...
]
Several strategies can be used to identify the modifier genes in human disorders. In mice, the identification can be performed by crossing parental inbred strains that carry the disease-causing mutation and show a difference in phenotype. [1616. Filoni C, CaciottiA, Carraresi L, Cavecchi C, Parini R, Antuzzi D. Functional studies of new GLA gene mutations leading to conformational Fabry disease. Biochim Biophys Acta. 2010;1802(2):247-252. doi: 10.1016/j.bbadis.2009.11.003
https://doi.org/10.1016/j.bbadis.2009.11...
] Clinical variability among patients carrying the same splicing mutation has been related to variable levels of aberrantly spliced transcripts. This phenomenon may occur especially in splicing mutations that do not affect the intronic canonical splice sites (GT/AG). This type of splicing mutation generates a variable proportion of wild-type transcript, in addition to the aberrantly spliced transcript.
The higher the proportion of the aberrantly spliced transcript, the more severe is the disease phenotype expected to be. [1919. Ars E, Torra R. Rare diseases, rare presentations: recognizing atypical inherited kidney disease phenotypes in the age of genomics. Clin Kidney J. 2017;10(5):586-593. doi: 10.1093/ckj/sfx051
https://doi.org/10.1093/ckj/sfx051...
]
In the case presented, it cannot be said that epigenetic factors do not influence, especially because there are inheritable epigenetic modifications and even if they had been raised in the same environment, factors such as diet or habits may be different. On the other hand, in patient 1 no other pathologic condition was identified, as vasculitis or rheumatology disease, that could explain the renal compromise. In family pedigree (Figure 4) we can observe a nephew 8 years old with the mutation how present angiokeratoma; his clinical outcome will add knowledge about the renal evolution, and a challenge to prevent CKD and ESRD, among other things, because it´s difficult to establish genotype/phenotype variations in just 2 members of the same family.
Conclusion
The genotype/phenotype variation of the c.835C>T mutation (p.Gln279Ter. Q279X) in exon 6 of the GLA gene may express an important renal variation with a wide range of clinical manifestations that cannot be predicted, therefore, an early nephrological evaluation and periodic follow-up of these family group are necessary.
Acknowledgements
We would like to thanks to the Nephropathology Section of the Anatomopathology Institute of the Central University of Venezuela (Caracas-Venezuela), for the renal biopsies precessing, and to Laboratory of Biology and Molecular Pathology Grabiela Serebrinsky (Buenos Aires-Argentina) for the molecular study and mutation determination.
References
- 1. Ishii S, Taguchi A, Okino N, Ito M, Maruyama H. Determination of globotriaosylceramide analogs in the organs of a mouse model of Fabry disease. J Biol Chem 2020; 295(17): 5577-5587. doi: 10.1074/jbc.RA120.012665
» https://doi.org/10.1074/jbc.RA120.012665 - 2. Svarstad E, Marti HP. The Changing Landscape of Fabry Disease. Clin J Am Soc Nephrol 2020;15(4):569-576. doi: 10.2215/cjn.09480819
» https://doi.org/10.2215/cjn.09480819 - 3. Acevedo O, Gago M F, Miltenberger- Miltenyi G, Robles A, Costa M, Pereira O, et al. Natural history of the late-onset phenotype of Fabry disease due to the p.F113L mutation. Mol Genet Metab Rep 2020;22:100565. doi: 10.1016/j.ymgmr.2020.100565
» https://doi.org/10.1016/j.ymgmr.2020.100565 - 4. Ortiz A, Oliveira J, Waldek S, Warnock D, Cianciaruso B, Wanner C. Nephropathy in males and females with Fabry disease: Cross-Sectional description of patients before treatment with enzyme replacement therapy. Nephrol Dial Transplant 2008;23(5):1600-1607. doi: 10.1093/ndt/gfm848
» https://doi.org/10.1093/ndt/gfm848 - 5. Ortiz A, Cianciaruso B, Cizmarik M, Germain D, Mignani R, Oliveira P et al. End-stage renal disease in patients with Fabry disease: natural history data from the Fabry Registry. Nephrol Dial Transplant 2010;25(3):769-775. doi: 10.1093/ndt/gfp554
» https://doi.org/10.1093/ndt/gfp554 - 6. Gal A, Beck M, Winchester R. Clinical utility gene card for: Fabry Disease. Eur J Human Genet 2012;20:e1-e3. doi: 10.1038/ejhg.2011.178
» https://doi.org/10.1038/ejhg.2011.178 - 7. Germain D, Shabbeer J, Cotigny S, Desnick R. Fabry Disease: Twenty novel α-Galactosidase a mutations and genotype-phenotype correlations in classical and variant phenotypes. Mol Med 2002;8:306-312. doi: 10.1007/bf03402156
» https://doi.org/10.1007/bf03402156 - 8. Chamoles N, Blanco M, Gaggioli D. Fabry disease: enzymatic diagnosis in dried blood spots on filter paper. Clin Chim Acta 2001;308(1-2):195-196. doi: 10.1016/s0009-8981(01)00478-8
» https://doi.org/10.1016/s0009-8981(01)00478-8 - 9. Civallero G, Michelin K, de Mari J, Viapiana M, Burin M, Coelho JC, et al. Twelve different assays on dried-blood spot paper samples for detection of patients with selected inherited lysosomal storage disease. Cli Chim Acta 2006; 372(1-2):98-102. doi: 10.1016/j.cca.2006.03.029
» https://doi.org/10.1016/j.cca.2006.03.029 - 10. Oliveira J, Ferreira S. Multiple phenotypic domains of Fabry disease and their relevance for establishing genotype-phenotype correlations. Appl Clin Genet 2019;12:35-50. doi: 10.2147/TACG.S146022
» https://doi.org/10.2147/TACG.S146022 - 11. Gorostidi M, Santamaría R, Alcázar R, Fernández-Fresnedo G, Galcerán J, Marian Goicoechea M. Documento de la Sociedad Española de Nefrología sobre las guías KDIGO para la evaluación y el tratamiento de la enfermedad renal crónica. Nefrología 2014;34(3):302-316. doi: 10.3265/nefrologia.pre2014.feb.12464
» https://doi.org/10.3265/nefrologia.pre2014.feb.12464 - 12. Ortiz A. Etiopatogénesis de las enfermedades glomerulares. In: Avendaño LH, Aljama García M, Arias Rodríguez M, Caramelo Díaz C, Egido de los Ríos JE, Lamas Peláez S (Editores). Nefrología Clínica Editorial Médica Panamericana; 2003: 255-267.
- 13. Herrera J, Vidau P. La Enfermedad de Fabry en pacientes en hemodiálisis: valoración del tratamiento enzimático sustitutivo. Dial Trasp 2012;33(3):71-74. doi: 10.1016/j.dialis.2011.11.002
» https://doi.org/10.1016/j.dialis.2011.11.002 - 14. Villalobos J, Febres Celestina, Colina Virginia Leticia. Comportamiento de los signos vitales durante la infusión de Agalsidasa B en el tratamiento de la Enfermedad de Fabry. VITAE Academia Biomédica Digital 2013;53. http://www.bioline.org.br/abstract?id=va13006⟨=en
» http://www.bioline.org.br/abstract?id=va13006⟨=en - 15. Koulansios K, Styhanou K, Pateinakis P, Zamanakou M, Loules G, Manou E, et al. Fabry disease due to D313Y and novel GLA mutations. BMJ Open 2017;7:e017098. doi: 10.1136/bmj.jopen2017-017098
» https://doi.org/10.1136/bmj.jopen2017-017098 - 16. Filoni C, CaciottiA, Carraresi L, Cavecchi C, Parini R, Antuzzi D. Functional studies of new GLA gene mutations leading to conformational Fabry disease. Biochim Biophys Acta 2010;1802(2):247-252. doi: 10.1016/j.bbadis.2009.11.003
» https://doi.org/10.1016/j.bbadis.2009.11.003 - 17. De Baere E, Beysen D, Oley C, Lorenz B, Cocquet J, De Sutter P, et al. FOXL2 and BPES: Mutational Hotspots, Phenotypic Variability, and Revision of the Genotype-Phenotype Correlation. Am J Hum Genet 2003;72(2): 478-487. doi: 10.1086/346118
» https://doi.org/10.1086/346118 - 18. Hilgert N, Huentelman M, Thorburn A, Fransen E, Dieltjens N, Mueller-Malesinska M. Phenotypic variability of patients homozygous for the GJB2 mutation 35delG cannot be explained by the influence of one major modifier gene. Eur J Hum Genet 2009;17:517-524. doi: 10.1038/ejhg.2008.201
» https://doi.org/10.1038/ejhg.2008.201 - 19. Ars E, Torra R. Rare diseases, rare presentations: recognizing atypical inherited kidney disease phenotypes in the age of genomics. Clin Kidney J 2017;10(5):586-593. doi: 10.1093/ckj/sfx051
» https://doi.org/10.1093/ckj/sfx051
-
Limitations of the Study
There is no lyso Gb3 determination in any of both patients that contribute to explain the renal disease outcome. It’s necessary to determine the presence of modifiers genes by molecular characterization of both females, mother and sister (Figure 4).
Publication Dates
-
Publication in this collection
11 Nov 2020 -
Date of issue
2020
History
-
Received
07 June 2020 -
Reviewed
23 Sept 2020 -
Accepted
01 Oct 2020




