










In the early 1990s, the concept that the inflammatory process is causally involved in plaque formation began to be recognised and referred to as ‘the inflammatory hypothesis of atherosclerosis’.1,2 Different investigations found evidence that inflammatory biomarkers are associated with the prognosis and severity of acute coronary syndrome (ACS) and other clinical manifestations of atherosclerosis.3,4 The contributions of Maseri, Libby, Ridker and Crea – among many others – have led to a better understanding of the importance of the inflammatory component in the pathogenesis of this condition.5,6 In fact, the idea that atherosclerosis carries characteristics of an inflammatory disease has been suspected since the 19th century, based on pathological observations made by Rudolf Virchow and others. However, it is only in recent years that chronic inflammation has been recognised as a contributing factor in the development, progression and complications of atherosclerosis, with new evidence supporting the inflammatory nature of the disease.7,8
While the purpose of an inflammatory process is the resolution of injuries, pathogens or infections by initiating an appropriate response, chronic inflammation represents a deviation from this natural biological response. In contrast to acute inflammatory events, which are usually self-limiting, atherosclerosis has been shown to be an unresolved chronic inflammatory condition that lacks the typical resolution phase.9
Greater attention has been focused on the relationship between inflammation and vascular calcification.10 Coronary artery calcification (CAC), which is concomitant with the development of advanced atherosclerosis, shows a close association with the total atherosclerotic plaque burden and an increased risk of cardiovascular (CV) events and mortality.11 CAC pathologically begins as microcalcifications (0.5–15.0 μm) and grows into larger calcium fragments that are observed to occur concurrently with the progression of plaques. Recent studies suggest that massive dense calcifications are usually associated with stable plaques, whereas, microcalcifications are related to vulnerability.12 Increasing evidence now supports the concept that arterial calcification is an inflammatory disease, an active process associated with macrophage burden, which is stimulated by inflammatory pathways and exacerbated in certain clinical conditions, including diabetes.13,14
Cholesterol and Inflammation
In atherosclerosis, inflammation begins and evolves in response to the accumulation of cholesterol in the intima of large and medium-sized arteries. New discoveries about innate immunity have improved the understanding of the events that initiate and drive inflammation, changing several concepts about the pathogenesis of inflammatory disorders and showing that innate and adaptive immune responses play a key role throughout the initiation, progression and clinical consequences of atherosclerotic disease. In the initial stages, the endothelial cells are activated and inflammatory cells are recruited in the vascular wall in response to the accumulation of cholesterol-rich lipoproteins in the innermost part of the arteries, giving rise to a wide variety of macrophages derived from monocytes, T lymphocytes, mast cells and dendritic cells among others.15–17 The role of T-cell-mediated adaptive immunity in the pathogenesis of atherosclerosis is an increasing focus of study. Activated T lymphocytes, primarily T helper 1 cells (Th1) accumulate early and abundantly in atherosclerotic lesions. The Th1 cells recruited to the lesion recognise oxidised low-density lipoprotein (ox-LDL) as an antigen and produce pro-inflammatory mediators such as interferon-gamma (IFN-γ) and tumour necrosis factor (TNF).18,19 IFN-γ is the main pro-atherogenic cytokine and it promotes local expression of adhesion molecules, cytokines and chemokines such as CXCL9, CXCL10 and CXCL11 and their main receptor CXCR3 by macrophages and endothelial cells. Chemokine signalling via CXCR3 facilitates the recruitment of active Th1 cells (Figure 1).20 The Th1 subset of CD4+ T cells is the most abundant T cell population in human atherosclerotic plaques with CXCR3 being required for the generation of Th1 cells.21. Strong evidence supports that CXCR3 and its ligands play a key role in atherosclerosis. Although this has not yet been fully established, it would seem they have both beneficial and deleterious actions depending upon their timing and level of activation. CXCL10, which is involved in sustaining inflammation through Th1 recruitment, appears to have a role in preventing excessive fibrosis.21
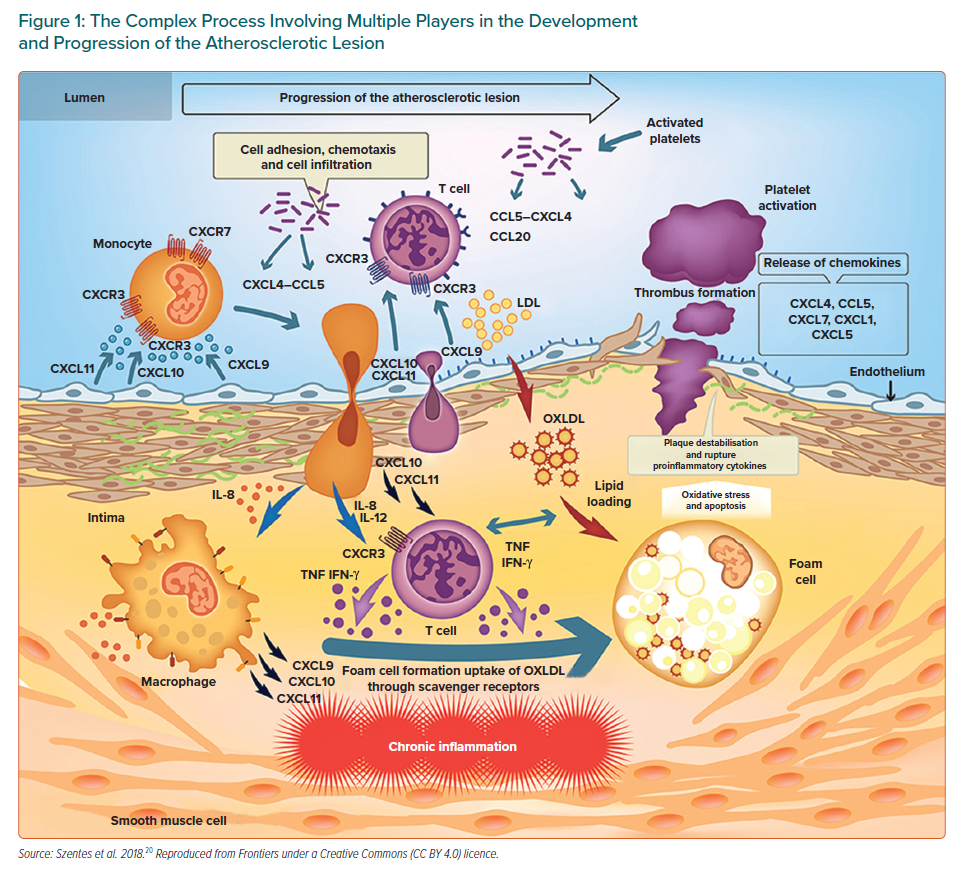
While all of the above can contribute to some extent to the formation and progression of atherosclerosis, macrophage retention within the arterial wall is fundamental in atherosclerosis, with macrophages being the major inflammatory cells involved in its progression. Structural alterations, in particular the exposure of proteoglycans, facilitate the retention of LDL particles in the intima, where they undergo oxidative modifications promoted by reactive oxygen species (ROS) and inflammatory cells.22,23 As a result, these lipoproteins become more pro-inflammatory and contribute to endothelial activation. Facilitated by adhesion molecules, the different types of leukocytes adhere to the activated endothelium covering the retained lipids and produce pro-inflammatory cytokines. Macrophages derived from monocytes take up the ox-LDL through scavenger receptors, transforming from the permanent accumulation of lipids into cells known as foams that secrete pro-inflammatory molecules and play an important role in collagen and matrix breakdown leading to plaque rupture.24,25 It has been reported that macrophages themselves can proliferate within atherosclerotic lesions. Related to this, both resident and recruited macrophages are thought to be mediated through the action of interleukin 4 (IL-4) which was sufficient to drive the accumulation of macrophages through self-renewal.26
Two different subtypes of macrophages – M1 and M2 – have been described, with M1 being induced (activated) by Th1 and cytokines such as IFN-γ and involved with pro-inflammatory activities. In contrast, M2 macrophages are stimulated by Th2 and cytokines including IL-4 or IL-13 and produce anti-inflammatory cytokines such as IL-10 and are able to counterbalance inflammatory responses to M1 macrophages by promoting the resolution of inflammation.27 Regulatory T cells that express transforming growth factor-beta (TGF-b) also tend to mitigate inflammation.8 Humoural immunity and B cells also participate in atherosclerosis with B1 lymphocytes appearing to be protective against atherosclerosis, whereas B2 lymphocytes can aggravate this process.28
Scavenger receptors class B type 1 (SR-B1) in endothelial cells have been postulated to mediate LDL delivery into arteries and its accumulation by macrophages, thereby promoting atherosclerosis.29 The ox-LDL exerts its action through several receptors, with the most important being the innate immune scavenger receptor lectin-like ox-LDL receptor 1 (LOX-1).30 An exacerbation of endothelial dysfunction has also been reported due to increased production of vasoconstrictors, an increase in ROS and a decrease in endothelial nitric oxide.31 Ox-LDL plays a key role in atherogenesis through LOX-1.32 This influences multiple cell types such as endothelial cells, smooth muscle cells (SMCs), fibroblasts, macrophages and platelets contributing to endothelial dysfunction, apoptosis, migration and the differentiation of monocytes and macrophages, proliferation and migration of SMCs and plaque instability, some of the mentioned critical factors in atherosclerosis.33
In synthesis, cholesterol and inflammation are interconnected, because the accumulation of cellular cholesterol promotes inflammatory responses. In addition, the activation of immune cells promotes cholesterol deposition by affecting the flow of cellular cholesterol.33 Therefore, atherosclerosis is characterised by quantitative and qualitative abnormalities of lipoproteins and an inadequate inflammatory response.15 The involvement of the inflammatory process is notably evident in acute coronary disease in which unstable plaques prone to rupture are characterised by significant infiltration of different inflammatory cells, a large and friable lipid core and a thin fibrous layer.16,34
Crystalline cholesterol, which is present and abundant in atherosclerotic lesions, has been identified as being the predominant endogenous danger signal that initiates an inflammatory response through the stimulation of the nucleotide-binding oligomerisation domain, NLR family pyrin domain containing 3 (NLRP3) and known as the inflammasome.35–37 As a result of the retention and oxidation of lipoproteins within the vessel wall, the accumulation of cholesterol can lead to the formation of cholesterol crystals, which are then absorbed by macrophages, causing an inflammatory reaction through the activation of the NLRP3 inflammasome and triggering a cascade of amplification of the immune responses.38 Therefore, cholesterol crystals can be an initiating or exacerbating factor in the atherosclerotic process, contributing to the rupture of foam cells and the expansion of the lipid-rich necrotic core in vulnerable plaques.39
In summary, the NLRP3 inflammasome is a fundamental component of the innate immune system which mediates the activation of caspase-1 and the secretion of pro-inflammatory cytokines IL-1β and IL-18 in response to microbial infection and cell damage, as well as being associated with several inflammatory disorders, including but not limited to diabetes and atherosclerosis.40
Imaging technologies, including CT, intravascular ultrasound (IVUS) and MRI, are critical for confirming the presence and extent of atherosclerosis, with MRI permitting the characterisation of plaque composition, such as lipid core, fibrosis, calcification and intraplaque haemorrhage.41 When MRI is combined with positron emission tomography (PET), some functional and molecular insights are provided into the underlying biological processes. For quantifying vascular inflammation, 18-fludeoxyglucose PET (18-F-FDG PET) has been validated for determining macrophage infiltration to plaques, the relationship with disease activity, response to treatment and it can be predictive of future events.42 In addition, 18 F-fluoride PET (18-F-NaF PET) can identify culprit and ruptured plaques in patients with MI and symptomatic carotid disease. Moreover, histological characterisation has demonstrated that 18 F-fluoride activity localises to regions of plaque rupture with evidence observed of increased inflammation, calcification activity, necrosis and cell death.43 The latter method, capable of detecting microcalcifications can be very useful in patients with early atherosclerosis.44
Inflammation as a Treatment Target
The pharmacological modulation of inflammation aimed at reducing CV events is difficult, since many agents that can modulate the inflammatory response have been shown to be ineffective or have had off-target negative effects. We will now focus more extensively on the results of Phase III clinical trials for different treatment options.
Statins
The idea of addressing inflammation as a way of reducing mortality and morbidity from coronary artery disease (CAD) has received strong support based on the JUPITER study, where treatment with rosuvastatin reduced LDL-C levels by 50% and high-sensitivity C-reactive protein (hs-CRP) by 37% after a median follow-up of 1.9 years. A concurrent and significant reduction in the combined primary endpoint of MI, stroke, arterial revascularisation, hospitalisation for unstable angina or death from CV causes, was verified in favour of rosuvastatin (HR 0.56; 95% CI [0.46–0.69]; p<0.00001).45 Likewise, it was observed that in those individuals who attained low levels of both LDL-C (<70 mg/dl) and hs-CRP (<1 mg/l), the relative risk reduction for CV events was 79%, (95% CI [0.09–0.52]; p<0.0001).46 Similarly, the IMPROVE-IT study analysed the relationship between the achievement of this dual target – LDL-C and hs-CRP – and the primary endpoint – CV death, major coronary event or stroke – for patients randomly assigned to simvastatin monotherapy or a combination of simvastatin and ezetimibe. In the 15,179 patients studied, simvastatin plus ezetimibe significantly increased the likelihood of achieving the predefined targets of LDL-C <70 mg/dl and hs-CRP <2 mg/l with those who achieved this dual target (39%) had lower primary event rates than those who did not (38.9% versus 28.0%, adjusted HR 0.73; 95% CI [0.66-0.81]; p<0.001).47 Nevertheless, a fundamental question remains as to whether or not inflammation still plays a role in patients who reach very low LDL-C levels, for example when they are treated with proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, which unlike statins have been shown not to modify hs-CRP values.48
In the FOURIER study, Bohula et al. explored whether the association of inflammation and risk of CV events persists even at very low levels of LDL-C. In patients with an LDL-C <20 mg/dl 1 month after randomisation, there was still a risk gradient under basal hs-CRP of <1, 1–3 and >3 mg/l with an event rate of 9.0%, 10.8% and 13.1%, respectively. This supports the concept of an inflammatory risk that is independent of LDL-C levels.49
Imaging-based studies support an association between statin and calcification progression, which is one of the ways by which statins prevent CV events, although the mechanism responsible for this effect is not completely understood.10 In a pooled analysis of eight randomised trials of serial IVUS, statins were shown to promote coronary calcium independent of plaque volume regression.50 Related to this, plaques with microcalcification (spotty calcium) found in the active stage of atherosclerosis inflammation have been shown to respond favourably to statin therapy confirming that the ability of statins to affect atheroma burden depends in part on the type of calcification.14 Yet, their modest effect on the absolute burden of atherosclerotic disease suggests that any protective effects on the arterial wall may result, in part, from compositional changes rather than a pure reduction in lesion size or the degree of stenosis.
In the REVERSAL study, patients were randomly assigned to receive a moderate lipid-lowering regimen (pravastatin 40 mg) or an intensive one (atorvastatin 80 mg), and the primary efficacy parameter was calculated as the percentage change in atheroma volume assessed by IVUS. A progression of coronary atherosclerosis occurred in the pravastatin group (2.7%; 95% CI [0.2–4.7]; p=0.001) compared with baseline whereas progression in the atorvastatin group did not occur (−0.4%; 95% CI [− 2.4– 1.5]; p=0.98). The baseline LDL-C level (mean = 150.2 mg/dl in both treatment groups) was reduced to 110 mg/dl in the pravastatin group and 79 mg/dl in the atorvastatin group (p<0.001). Interestingly, hs-CRP decreased by 5.2% with pravastatin and 36.4% with atorvastatin (p<0.001).51
Similarly, in the SATURN study, a significant lowering of the atherogenic lipoprotein levels (LDL-C), inflammatory status (hs-CRP) and elevations of anti-atherogenic lipoproteins (HDL-C) were associated with overall atheroma regression, mediated largely by a reduction in the fibro-fatty tissue component.52
In addition to reducing LDL-C, statins have also been shown to diminish inflammation independently of cholesterol, but this does not provide the proof of the principle of inflammatory causation in atherosclerosis. Therefore, the only way to corroborate this hypothesis was to attack inflammation without modifying the lipid levels and testing pure anti-inflammatory therapies.
Canakinumab: the First Proof of Concept
CRP is an excellent marker of systemic inflammation and it is a useful parameter for evaluating the effectiveness of anti-inflammatory treatments. However, since it does not cause atherothrombosis, it does not qualify as a primary target for therapeutic intervention.53 On the other hand, IL-1β is critically involved in atherosclerosis and induces the production of IL-6 and subsequently CRP. Therefore, the IL-1 signalling pathway is a valuable treatment target.54 This background and several studies have supported the rationale for targeting IL-β specifically without affecting IL-1α, which may be involved in host defense.
The CANTOS trial was designed to test whether reducing inflammation by neutralising IL-1β with canakinumab (a fully human monoclonal antibody) in patients with previous MI and elevated plasma hs-CRP levels (≥2 mg/l) would reduce the risk of recurrent CV events beyond standard secondary prevention therapies. This trial compared three doses of canakinumab – 50 mg, 150 mg and 300 mg administered subcutaneously every 3 months – with placebo. A total of 10,061 subjects were included, with a mean age of 61 years and with a median follow-up of 3.7 years. Although no changes in LDL-C were observed, the use of canakinumab resulted in large, dose-dependent reductions in CRP and IL-6. The primary endpoint, a combination of non-fatal MI, non-fatal stroke or CV death was reduced by 15% in the 150 mg group (HR 0.85; 95% CI [0.74–0.98]; p=0.021); and by 14% in the 300 mg group, (HR 0.86; 95% CI [0.75–0.99]; p=0.031). However, there was no significant difference in all-cause mortality for all doses of canakinumab versus placebo (HR 0.94, 95% CI [0.83–1.06]; p=0.31). A potentially limiting fact for the use of this drug in coronary heart disease is that canakinumab was associated with a higher incidence of fatal infection which, although small in proportion, was statistically significant.55
It is important to note that the reduction in CV events was stronger in those who were identified as cytokine responders as they achieved lower levels of hs-CRP after initiation of the drug. Patients treated with canakinumab who reached hs-CRP concentrations on treatment of <2 mg/l showed a 25% reduction in major adverse CV events (MACE; p=0.0001) and a 31% decrease in both CV mortality (adjusted HR 0.69, 95% CI [0.56–0.85]; p=0.0004) and all-cause mortality (adjusted HR 0.69, CI [0.58–0.81]; p<0·0001) with the latter being partially explained by concomitant reductions in deaths from lung cancer. However, no significant reduction in these endpoints was observed among those treated with canakinumab who had hs-CRP concentrations of ≥2 mg/l.56 The calculated number needed to treat (NNT) of patients to avoid one MI, stroke, coronary revascularisation or death from any cause in the entire CANTOS cohort was calculated to be 24. However, among patients who reached hsCRP <2 mg/l with canakinumab, the estimated 5-year NNT figure dropped to 16. In contrast, the NNT increased to 57 for patients who reached hsCRP >2 mg/l on treatment.56
Compared with those allocated to placebo, patients treated with canakinumab who reached lower levels of IL-6 after the first dose (below the study median value of 1.65 ng/l), experienced a 32% reduction in MACE, (adjusted HR 0.68, 95% CI [0.56–0.82]; p<0.0001), a 52% reduction in CV mortality (adjusted HR 0.48, 95% CI [0.34–0.68]; p<0.0001) and a 48% reduction in all-cause mortality (adjusted HR 0.52, 95% CI [0.40–0.68]; p<0.0001) thereby providing evidence that modulation of the IL-6 signalling pathway is associated with reduced CV event rates, independent of lipid lowering.57 Interestingly, canakinumab was shown to reduce MACE in chronic kidney disease (CKD) patients and was particularly effective in those who achieved a level of hsCRP <2 mg/l after the first dose with CV and all-cause mortality being reduced.58
Finally, subclinical inflammation-mediated in part by IL-1β participates in peripheral insulin resistance and impaired pancreatic insulin secretion. Although IL-1β inhibition with canakinumab had similar effects on major CV events in patients with or without diabetes in the CANTOS study, treatment over a median period of 3.7 years did not reduce incident diabetes.59
Methotrexate
The commonly used methotrexate (MTX) has shown favourable results in attenuating systemic inflammation and decreasing CV events.60–62 At low doses, it is also an effective, safe and well-tolerated anti-inflammatory agent used in patients with rheumatoid arthritis or psoriasis.63 The CIRT trial was designed to evaluate the results of low dose MTX (LD-MTX) on CV events in patients with chronic atherosclerosis and with diabetes or metabolic syndrome.64
It included 4,786 participants, most of whom had previous coronary revascularisation and were treated with standard preventive therapy prior to randomisation. These patients had a mean basal LDL-C of 68 mg/dl and a mean basal hs-CRP of 1.6 mg/l. However, the study was stopped early due to a lack of benefit of LD-MTX in preventing MACE in patients with CAD and either type 2 diabetes or metabolic syndrome. Thus, no benefit was observed in CV results with the use of LD-MTX (HR 0.96, 95% CI [0.79–1.16]; p=0.67) and no difference in all-cause mortality was observed between the groups. It is important to note that, in contrast to canakinumab, LD-MTX did not reduce IL-6, CRP or IL-1β levels and there was no reduction in CV events compared with placebo.64
The study populations in CANTOS and CIRT were similar but with a key difference. In the CANTOS trial all participants had a basal CRP ≥2 mg/l, they qualified as a population with high inflammatory risk (with a median basal hs-CRP of 4.2 mg/l), while in CIRT the median basal hs-CRP level was 1.6 mg/l. Another crucial difference was the inflammatory pathway affected by the pharmacological intervention. Canakinumab used in the CANTOS trial specifically inhibits IL-1β, resulting in a significant reduction in CRP, IL-6 and IL-1β levels. In contrast, LD-MTX used in CIRT had no effect on these biomarkers or inflammatory mediators, which may explain the lack of clinical benefit in these stable populations with atherosclerotic CAD.65
Colchicine
Colchicine, a broad-spectrum anti-inflammatory agent previously used to treat inflammatory disorders such as gout and recurrent pericarditis, is also a treatment option for atherosclerosis. Colchicine is a widely available, inexpensive and generally well-tolerated medication. Among several anti-inflammatory mechanisms, colchicine appears to block cholesterol crystal-induced activation of the NLRP3 inflammasome, which decreases the secretion of the pro-inflammatory cytokines IL-1β and IL-18 leading to downstream reductions in interleukin-6 and CRP, thus providing a reason to test this classical drug in patients with CAD.66–68 Colchicine also reduces the mobility and deformability of neutrophils and decreases their adhesion to endothelial cells and atherosclerotic lesions, features that can improve plaque morphology.69,70
The LoDoCo trial tested colchicine in a cohort of 532 patients with stable coronary disease receiving aspirin and/or clopidogrel and statins. The treatment with 0.5 mg/day of colchicine significantly reduced the prevalence of CV events (4.5%) compared to placebo (16.0%), although a small percentage of patients showed intestinal intolerance toward colchicine.71
Similarly, in the COLCOT study, treatment with colchicine of 0.5 mg per day compared to placebo over a two-year period in 4,745 patients after MI, resulted in a 23% relative reduction in the primary endpoint of the study, including MI, stroke, resuscitated cardiac arrest, hospitalisation for angina leading to urgent revascularisation and CV death (HR 0.77; 95% CI [0.61–0.96]; p=0.02). Despite the benefit obtained from colchicine being significant only for the components of coronary revascularisation and the endpoint stroke, all CV outcomes were positively affected. However, it should be noted that colchicine has been associated with increased cases of pneumonia and gastrointestinal disturbances.72
Finally, evidence from the recent LoDoCo2 trial has shown that the anti-inflammatory effects of colchicine reduce the risk of CV events in patients with chronic coronary disease. The study, randomised 5,522 patients with chronic stable coronary disease and compared a daily dosage of 0.5 mg colchicine against placebo. After an average follow-up of 2.4 years, the primary endpoint (CV death, non-fatal MI, non-fatal stroke and coronary revascularisation) decreased by 31% (HR 0.69; 95% CI [0.57–0.83]; p<0.001) in those treated with colchicine.73
A recent meta-analysis examined four randomised clinical trials, including 11,594 patients (colchicine n=5,774; placebo/no colchicine n=5,820) in two studies in stable CAD – LoDoCo and LoDoCo2 – and two in ACS – COLCOT and the Australian COPS Randomized Clinical Trial.74 Compared with placebo or no drug, colchicine was associated with a statistically significant reduction in the incidence of the primary composite endpoint (pooled HR 0.68; 95% CI [0.54-0.81]; I2 = 37.7%).75
Convincing evidence now supports the use of colchicine for secondary prevention in patients with recent MI or chronic CAD that has continued at residual CV risk despite good blood pressure control and an adequate reduction of atherogenic lipid.76 The features of this drug may contribute in different ways to its atheroprotective effects but many of these still need to be elucidated.77 In addition, further randomised and controlled studies will be needed to determine the long-term tolerability and efficacy of low-dose colchicine for secondary prevention in patients with CAD before its widespread use can be recommended.78 Indeed, two relevant randomised clinical trials are still running: CLEAR SYNERGY (NCT03048825) in ACS patients and CONVINCE (NCT02898610) for secondary prevention after stroke, that is expected to have the results shortly.
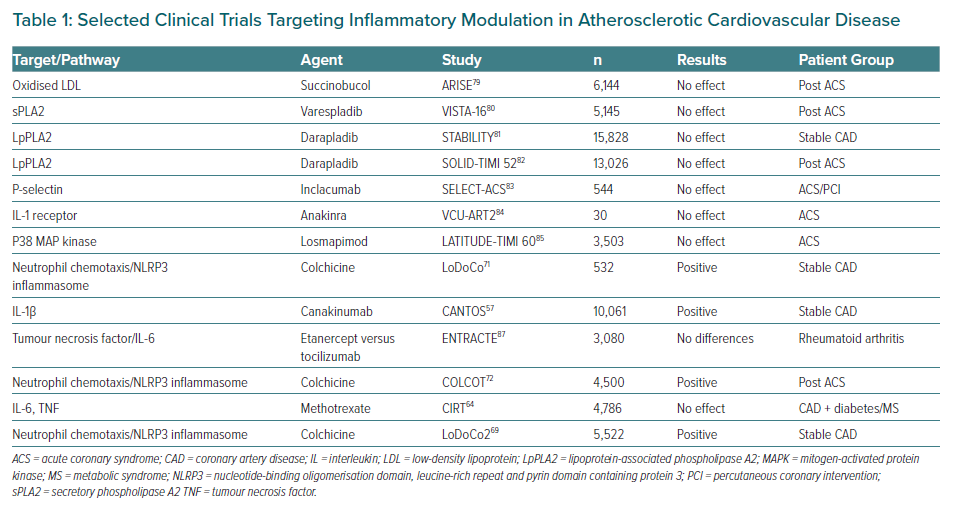
Multiple pathways have been identified as potential objectives for the prevention and treatment of CV diseases. In Table 1, several trials of agents that modulate a specific pathway are summarised including some that have been extensively analysed. Most clinical trials targeting specific downstream targets, such as those with oxidation of LDL, secretory phospholipase A2 (sPLA2) and lipoprotein associated phospholipase A2 (LpPLA2), P-selectin, the IL-1b inhibitor anakinra, losmapimod to inhibit p38 MAP kinase and methotrexate failed to meet their primary endpoints with respect to a decrease in CV events or a selected surrogate endpoint.64,79–85 Nevertheless, a spectrum of possibilities has now opened up for exploring new anti-inflammatory therapies to target inflammation in the prevention of atherosclerosis, although it is clear that more research is still needed on the role of anti-inflammatory and immunomodulatory interventions in atherosclerotic CV disease to achieve a better understanding of the complex players involved in the inflammatory process.86 Finally, the development of new potentially more promising drug compounds is highly desirable.
Conclusion
Inflammation plays a key role in all steps of the atherosclerotic process, from the initial stage, when leukocytes are recruited at the sites of subendothelial cholesterol accumulation to the late events of plaque rupture and thrombosis. Chronic inflammation of the arterial wall is promoted by the innate and adaptive immune responses and is sustained by the complex mechanism involving pro-inflammatory cytokines.
In the CANTOS study, designed specifically to demonstrate whether purely decreasing inflammation correlated with reducing clinical events, the expected proof of concept, the quarterly administration of the anti-IL-1β monoclonal antibody canakinumab showed clear benefits. Most recently this concept was endorsed by the use of colchicine in COLCOT and LoDoCo2 studies, but further confirmation and careful evaluation of the balance between benefits and adverse events is still needed before international guidelines can endorse the use of colchicine for the secondary prevention of CV diseases.