

Abstract
In recent years, the glutamatergic system has been implicated in the development and treatment of psychiatric disorders. Glutamate signaling is processed by different receptors, including metabotropic glutamate receptors (mGluRs), which in turn interact with the scaffolding protein Homer1 to modulate downstream Ca2+ signaling. Stress is a major risk factor for the incidence of psychiatric diseases, yet acute stress episodes may have diverging effects on individuals. Cognitive impairments have often been shown to occur after episodes of stress, however the specific role of mGluR5/Homer1 signaling in the interaction of stress and cognition has not yet been elucidated. In this study we show that a single episode of social defeat stress is sufficient to specifically induce cognitive impairments in mice 8 h after the stressor without affecting the animals' locomotion or anxiety levels. We also demonstrate that Homer1b/c levels as well as mGluR5/Homer1b/c interactions in the dorsal hippocampus are reduced up to 8 h after stress. Blockade of mGluR5 during the occurrence of social stress was able to rescue the cognitive impairments. In addition, a specific overexpression of Homer1b/c in the dorsal hippocampus also reversed the behavioral phenotype, indicating that both mGluR5 and Homer1b/c play a crucial role in the mediation of the stress effects. In summary, we could demonstrate that stress induces a cognitive deficit that is likely mediated by mGluR5/Homer1 signaling in the hippocampus. These findings help to reveal the underlying effects of cognitive impairments in patients suffering from stress-related psychiatric disorders.
Introduction
Stressful life events are commonly accepted as risk factors for the development of psychiatric disorders (de Kloet et al., 2005). Social and work stress in particular are prevalent in Western societies, and psychopathologies emanating from such stressors result in high economic losses (Tennant, 2001). Stress paradigms in animal models produce a variety of behavioral, physiological, and neuroendocrine changes that are related to clinical symptoms of psychiatric disorders (Nestler and Hyman, 2010). One major indication of these disorders is cognitive impairment, and numerous studies have investigated the interactions of stress and cognitive dysfunction at different stages in life in both animal models and humans (Lupien et al., 2009). Early life stress, chronic stress, and chronically elevated circulating glucocorticoids (GCs) have been shown to induce cognitive impairments (Landfield et al., 1981; Borcel et al., 2008; Wang et al., 2011a,b). In contrast, acute effects of stress and GCs critically depend on the timing and magnitude of the stressor or GC exposure and the stage of memory formation, consolidation, or retrieval (Sandi, 2011). In this context, glutamatergic pathways and their role in memory formation have received growing attention in the last years, with a particular focus on glutamate receptor signaling (Popoli et al., 2012).
Homer1 is a scaffolding protein located in the postsynaptic density (PSD) and interacts with various binding partners, most prominently group I metabotropic glutamate receptors (mGluRs) and inositol trisphosphate receptors (Brakeman et al., 1997; Tu et al., 1998). The best described Homer1 isoforms are on the one hand Homer1b/c, consisting of a conserved amino-terminal target-binding domain and a coiled-coil structure that allows for multimerization (Xiao et al., 1998). On the other hand, Homer1a, a short form that is missing the coiled-coil structure, has been shown to act as a dominant negative for long Homer1 isoforms at the mGluR and IP3 binding sites. The constitutively expressed Homer1b/c has been shown to mediate ligand-dependent signaling of mGluRs that may lead to downstream translational activation of protein kinase pathways (Ronesi and Huber, 2008; Ronesi et al., 2012) but is also involved in cell surface expression of mGluRs (Ango et al., 2002). In contrast, Homer1a induces ligand-independent activation of mGluRs, thus interfering with intracellular CA2+ release (Ango et al., 2001). In humans, a role of Homer1 has been suggested in the emergence of major depressive disorders (Rietschel et al., 2010), and it was also found to be implicated in memory formation and cognition (Lominac et al., 2005; Szumlinski et al., 2005).
In the current study, we hypothesized that mGluR5/Homer1 signaling plays a crucial role in mediating effects of stressful life events. To test this, we first devised an experimental setup that specifically produces a learning impairment in response to an acute stress exposure. Next, we investigated both mRNA and protein levels of Homer1 in response to the stressor. Additionally, we tested whether pharmacological modulation of the glucocorticoid receptor (GR) or mGluR5/Homer1 signaling or Homer1 overexpression can attenuate the stress-induced cognitive deficits.
Materials and Methods
Animals.
For all experiments, male C57BL/6N mice (Charles River Laboratories) at the age of 12 weeks were used. The mice were held under standard conditions (12 light:12 dark light cycle, lights on at 8:00 A.M., temperature at 23 ± 2°C) and were single housed and acclimated to the room for 2 weeks before the beginning of the experiments. Food (Altromin 1324, Altromin) and tap water were available ad libitum. Male CD1 mice (16–18 weeks of age) served as resident mice, which were held under the conditions described above. They were allowed to habituate to the social defeat cage for 2 weeks before the experiment. All experiments were carried out in the animal facilities of the Max Planck Institute of Psychiatry in Munich, Germany. The experiments were carried out in accordance with the European Communities' Council Directive 2010/63/EU. All efforts were made to minimize animal suffering during the experiments. The protocols were approved by the Committee for the Care and Use of Laboratory animals of the Government of Upper Bavaria, Germany.
Experimental design.
For all experiments, a separate batch of animals was used (n = 8–12 mice per group). The defeat procedure was performed between 8:00 A.M. and 12:00 P.M. Sampling and testing was performed after the stated amount of time. For in situ hybridization and Western blot, brains were removed, frozen in isopentane at −40°C, and stored at −80°C until further processing. For immunohistochemistry, animals were deeply anesthetized with ketamine/Rompun and perfused intracardially with 4% paraformaldehyde. Brains were removed, postfixed overnight in 4% paraformaldehyde following overnight incubation in 30% sucrose solution at 4°C, and then stored at −80°C. Blood samples were kept on ice and later centrifuged at 8000 rpm at 4°C for 15 min. Plasma was transferred to new, labeled tubes and stored at −20°C until determination of corticosterone by radioimmunoassay (sensitivity 12.5 ng/ml; MP Biomedicals).
Social defeat stress procedure.
Animals were exposed to an aggressive CD1 resident mouse with short attack latency for 5 min, and then returned to their home cage until testing or sacrifice. During the defeat procedure, fighting was not interfered with unless a severe injury occurred. In this case, the defeat was stopped and the experimental animal was excluded from further analysis. Control animals were allowed to explore an empty novel cage similar to the resident cage for 5 min as well.
In situ hybridization and immunohistochemistry.
For in situ hybridization, frozen brains were coronally sectioned in a cryostat microtome at 18 μm and kept at −80°C. In situ hybridization using a 35S UTP-labeled ribonucleotide probe for Homer1b/c (Forward primer: AACACTGGGAGGCTGAGCTA; Reverse primer: TACTGCGGAAAGCCTCTTGT) and Homer1a (Forward primer: TGGTTGCTCAAGTTGACTGAA; Reverse primer: CCAGTAATGCCACGGTACG) was performed as described previously (Schmidt et al., 2007). For fluorescence immunohistochemistry, serial coronal sections were cut at 30 μm thickness. Double-labeling immunofluorescence (rabbit anti-Homer1: 1:1000, Synaptic Systems; goat anti-GFP: 1:500, Abcam) was performed on free-floating sections (n = 3 per mouse) as described previously (Wang et al., 2011a).
Coimmunoprecipitation.
For coimmunoprecipitation (CoIP), a separate batch of experimental animals was killed 8 h after the defeat procedure, and the dorsal hippocampus was dissected. Membrane fractions were isolated using the Calbiochem Proteo-Extract Kit (EMD Biosciences) as described previously (Wagner et al., 2012). The protein concentration was determined, and 1.2 mg of lysate was incubated with 2.5 μg mGluR5 antibody (Millipore) overnight at 4°C. Twenty microliters of BSA-blocked Protein G Dynabeads (Invitrogen, catalog no. 100-03D) were added to the lysate–antibody mix followed by 3 h of incubation at 4°C. The beads were washed three times with PBS, and protein-antibody complexes were eluted with 100 μg/ml mGluR5-peptide solution (Millipore) in CoIP buffer for 30 min at 4°C. Fifteen micrograms of the cell lysates and 10 μl of the immunoprecipitates were further processed by Western blot analysis.
Western blot.
Purification of synaptosomal fraction of hippocampal tissue (Filiou et al., 2010) and Western blot analysis (Wang et al., 2011a) were performed as described previously. Antibodies used were rabbit anti-Homer1 (1:1000, Synaptic Systems), rabbit-anti mGluR5 (1:1000, Millipore), and goat anti-actin (1:2000, Santa Cruz Biotechnology) for primary as well as horseradish peroxidase-conjugated secondary antibodies (1:2000; Dako).
Behavioral testing.
All behavioral tests were recorded using a video-tracking system (Anymaze 4.20; Stoelting). The following behavioral tests were performed: spatial object recognition (sOR), Y-maze, elevated plus maze (EPM), and female urine sniffing test (FUST). The testing procedures were performed as described below.
Spatial object recognition.
The spatial object recognition task was performed in an open field apparatus (50 × 50 × 50 cm) under low illumination (15 lux) as described previously (Schmidt et al., 2011). Prominent spatial cues were provided. Mice were habituated to the testing environment for 10 min on two consecutive days before testing. During the acquisition trials, mice were presented with two identical aluminum cubes (5 × 5 × 5 cm) and allowed to freely explore the objects two times for 10 min separated by a 15 min intertrial interval (ITI). During the 5 min retrieval trial, 30 min following the last acquisition trial, mice were presented with a nondisplaced object and a relocated one. The percentage of time exploring the displaced and the nondisplaced objects was calculated, with a higher preference for the novel object being rated as intact spatial recognition memory. Animals that did not explore any of the two objects during the retrieval phase were excluded from the analysis.
Y-maze.
The Y-maze was performed as described previously (Schmidt et al., 2011). Briefly, the apparatus is made of gray polyvinyl chloride (PVC) and consisted of three evenly illuminated arms (30 × 10 × 15 cm, 15 lux) with an angle of 120° between each arm. The apparatus was surrounded by various spatial cues. To ensure that the mice had sufficient spatial cues available without having to stretch up and look outside of the maze, we also introduced intramaze cues (triangles, bars, and plus signs) that served the same purpose as the external cues. The Y-maze test comprises two trials separated by an ITI to assess spatial recognition memory. During the first trial (the acquisition phase), the mouse was allowed to explore only two of the three arms for 10 min while the third arm was blocked. After 30 min ITI, the second trial (the retrieval phase) was conducted during which all three arms were accessible for 5 min. The percentage of distance traveled in the novel arm compared with the known arms was scored with a significantly higher percentage than chance level (33.3%) and rated as successful spatial memory.
Elevated plus maze.
The EPM was conducted to display changes in anxiety-related behavior. The device consisted of a plus-shaped platform with two opposing open arms (30 × 5 × 0.5 cm) and two opposing enclosed arms (30 × 5 × 15 cm) made of gray PVC, which were connected by a central area (5 × 5 cm). The whole device was elevated 50 cm above the floor. The illumination was 20 lux in the open arms and <10 lux in the closed arms. Testing duration was 5 min, and mice were placed into the center zone facing one of the enclosed arms at the start of the test. The time spent in the open arms compared to the total arm time was analyzed.
Female urine sniffing test.
The FUST was performed as described previously (Malkesman et al., 2010; Wagner et al., 2012). Briefly, 1 h before the test mice were habituated to a sterile cotton swab inserted into their home cage. In the first stage of the test, mice were exposed to a cotton swab dipped in water for 3 min and sniffing time was scored. After an ITI of 45 min, mice were exposed to a cotton swab dipped in urine from estrous females of the same mouse strain. Again, total sniffing time was scored. The test was performed in a dark environment (<3 lux). Animals that escaped from the apparatus in any trial were excluded from the analysis.
Drugs.
The selective GR agonist dexamethasone (DEX) (Ratiopharm) or vehicle (0.9% NaCl) was administered subcutaneously (10 mg/kg body weight). Animals were then placed back in their home cages until sacrifice or behavioral testing.
For blockade of GR signaling, the specific GR antagonist RU486 (Sigma) was administered 5 min before the defeat procedure. Animals received a subcutaneous injection of either RU486 (100 mg/kg body weight) or vehicle (polyethylene glycol).
For blockade of mGluR5 signaling, the specific mGluR5 antagonist 3-((2-methyl-4-thiazolyl)ethynyl)pyridin (MTEP) (Tocris Bioscience) was solubilized in 0.9% NaCl (Anderson et al., 2003; Busse et al., 2004). Animals received a subcutaneous injection of either vehicle or MTEP (15 mg/kg body weight) 5 min before the defeat procedure.
Drug dosage was chosen according to previous reports (Anderson et al., 2003; Schmidt et al., 2011).
Viral overexpression of Homer1.
Viral overexpression was performed as described previously (Schmidt et al., 2011). We used an adeno-associated bicistronic AAV1/2 vector (GeneDetect) containing the CAG-Homer1-IRES-EGFP-WPRE-BGH-polyA expression cassette (containing coding sequence of Homer1 NCBI CCDS ID CCDS36745). For the control group, we used the same vector construct expressing only EGFP. Virus production, amplification, and purification were performed by GeneDetect. Mice were anesthetized with isoflurane, and 0.5 μl of either AAV-Homer1 or AAV-EGFP (titers: 1.2 × 1012 genomic particles/ml) were bilaterally injected in the dorsal hippocampus at 0.06 μl/min by glass capillaries with tip resistance of 2–4 MΩ in a stereotactic apparatus. The following coordinates were used: 1.9 mm posterior to bregma, 1.3 mm lateral from midline, and 1.3/1.8 mm below the surface of the skull, targeting the CA1 and dentate gyrus (DG) region of the dorsal hippocampus. After surgery, mice were treated for 5 d with Metacam via drinking water. Behavioral testing started 4 weeks after virus injection. Successful overexpression of Homer1 was verified by immunofluorescence. Animals that were not infected bilaterally in both the CA1 and DG region were excluded from the analysis (n = 4). Quantification of the overexpression was achieved by in situ hybridization using the riboprobe described above.
Statistical analysis.
The data presented are shown as means ± SEM and were analyzed by the commercially available software SPSS 16.0. Student's t test was employed for comparison of means. Two-way analysis of variance was used to investigate effects of locomotion and object interaction in experiments including two conditions (control/defeat and vehicle/drug treatment). A nominal level of significance, p < 0.05, was accepted.
Results
A single social defeat induces specific spatial memory impairments after an intermediate period of recovery
We subjected mice to a single defeat session and investigated the corticosterone response as a measure of hypothalamic–pituitary–adrenal axis activity after four distinct time points (Fig. 1A). Circulating corticosterone was significantly increased 1 h T(9.614) = −8.623, p < 0.001), 4 h (T(9.898) = −3.273, p < 0.01), and 8 h (T(18) = −3.753, p < 0.01), but not 24 h after the defeat session. As the defeat stress showed a significant neuroendocrine impact 8 h after the stress, we tested whether stressed animals show a behavioral phenotype. Indeed, 8 h after a defeat session, experimental mice were not able to distinguish a displaced object in the sOR test (control: T(22) = 7.721, p < 0.001; defeat: T(22) = −0.234, p = 0.817) (Fig. 1B). We also replicated this cognitive impairment in the Y-maze test, where stressed animals did not discern between a novel and known arms (Fig. 1C). To investigate for potential biases, we also investigated the total distance traveled in the apparatus in both tests, but did not find a difference (sOR: control: 8.70 ± 1.26 m; defeat: 7.21 ± 1.19 m, p = 0.398; Y-maze: control: 12.01 ± 0.85 m; defeat: 12.75 ± 2.04 m, p = 0.724). Additionally, the total interaction time with both objects in the sOR test did not differ between control and defeated animals (control: 3.79 ± 0.61 s; defeat: 3.19 ± 0.75 s, p = 0.542). These memory deficits were not visible in either test when animals were tested 24 h after the defeat session (control: displaced object: 78.61% ± 2.62; nondisplaced object: 21.39% ± 2.62; T(18) = 15.419, p < 0.001; defeat: displaced object: 76.72% ± 3.42; nondisplaced object: 23.27% ± 3.42; T(18) = 11.032, p < 0.001).

Neuroendocrine and behavioral effects of a single defeat session. A, Defeated mice show significantly increased corticosterone levels up to 8 h after onset of the stressor. B, In the spatial object recognition test, animals did not discriminate between the displaced and the nondisplaced object when stressed 8 h before. C, In the Y-maze, we found the same memory impairment as when animals were defeated 8 h before. D, Both control and defeated mice spent equal time sniffing in the female urine sniffing test 8 h after the defeat, indicating that there is no anhedonic behavior induced by a single defeat session. E, In the elevated plus maze, defeated animals show the same anxiety-related phenotype as their control littermates when tested 8 h after the stress. *p < 0.05; data are expressed as mean ± SEM.
To further narrow down the behavioral phenotype, we also performed an EPM test (Fig. 1D) as well as a FUST (Fig. 1E) 8 h after the defeat but did not find any difference between control and defeated mice (EPM, time on the open arm: T(22) = 0.830, p = 0.415; FUST, urine sniffing time: T(22) = 0.284, p = 0.779), indicating that there is no underlying locomotion phenotype, anxiety phenotype, or anhedonic phenotype in these animals.
A single defeat immediately reduces Homer1b/c levels in the dorsal hippocampus and decreases mGluR5/Homer1b/c coupling
Brains from the same animals that were killed to investigate corticosterone levels were prepared for in situ hybridization, and Homer1 mRNA levels were investigated in the dorsal and ventral hippocampi. We detected a reduction in Homer1b/c mRNA 4 h (T(18) = 3.734, p < 0.01) and 8 h (T(18) = 2.317, p < 0.05) after the stressor in the CA1 (Fig. 2A,B) and DG region of the dorsal hippocampus (DG: 4 h: control 22.20 ± 1.39, defeat 16.98 ± 1.03, T(18) = 3.019, p < 0.01; 8 h: control 17.47 ± 0.55, defeat 15.47 ± 0.72, T(17) = 2.214, p < 0.05). After 24 h, no significant effect was visible. Also, we did not find a change in mRNA levels in the ventral hippocampus, indicating a high specificity of this regulation. Additionally, we investigated short-form Homer1a mRNA levels in the CA1 region of the dorsal hippocampus after the same time points but did not see a significant regulation (Fig. 2C,D). In a separate cohort of animals that were defeated and killed 8 h after the stressor, we extracted the highly enriched synaptosomal fraction of dorsal hippocampus tissue and measured Homer1b/c levels by Western blot. Here, we found a significant decrease of Homer1 protein levels in the synapse when animals were defeated previously (T(13) = 2.436, p < 0.05). Another cohort of animals was defeated, killed 8 h after the stressor, and dorsohippocampal membrane fractions were processed for CoIP. While total protein levels remained unchanged in response to stress, coupling of mGluR5/Homer1b/c was significantly reduced (T(21) = 2.867, p < 0.01) (Fig. 2E,F).

Homer1 mRNA level alterations in response to stress. A, After 4 and 8 h, Homer1b/c mRNA levels in the CA1 region are reduced. This reduction normalizes after 24 h. B, Representative autoradiographs of Homer1b/c mRNA levels in the hippocampus. C, Homer1a levels are not significantly altered in response to a single social defeat. One hour after onset of the stressor, an increase in Hoemr1a mRNA failed to reach significance (p = 0.077). D, Representative autoradiographs of Homer1a mRNA levels in the hippocampus. E, Interactions of mGluR5 and Homer1b/c are decreased 8 h after defeat stress. F, Representative Western blot of the mGluR5/Homer1 immunoprecipitation. For the technical control, a pooled lysate was incubated without primary antibodies. *p < 0.05, data are expressed as mean ± SEM.
GR signaling is not sufficient to elicit memory impairments, while blockade of mGluR5 signaling reverses stress effects
To investigate whether activation of the GR in the hippocampus is already sufficient to induce changes in Homer1b/c levels that subsequently lead to memory impairments, we injected a batch of experimental animals with the GR agonist DEX to mimic the corticosterone response to a severe stressor. Next, we analyzed the expression profile of Homer1b/c in response to the DEX injection but did not find a regulation in any of the investigated time points (Fig. 3A). Furthermore, a DEX injection was not able to induce spatial memory deficits 8 h after the administration (Fig. 3B). In the Y-maze test, both vehicle- and DEX-injected animals were able to discriminate between the new and the known arms (vehicle: T(20) = −10.266, p < 0.001; DEX: T(20) = −5.178, p < 0.001). Total locomotion was not affected by DEX treatment.
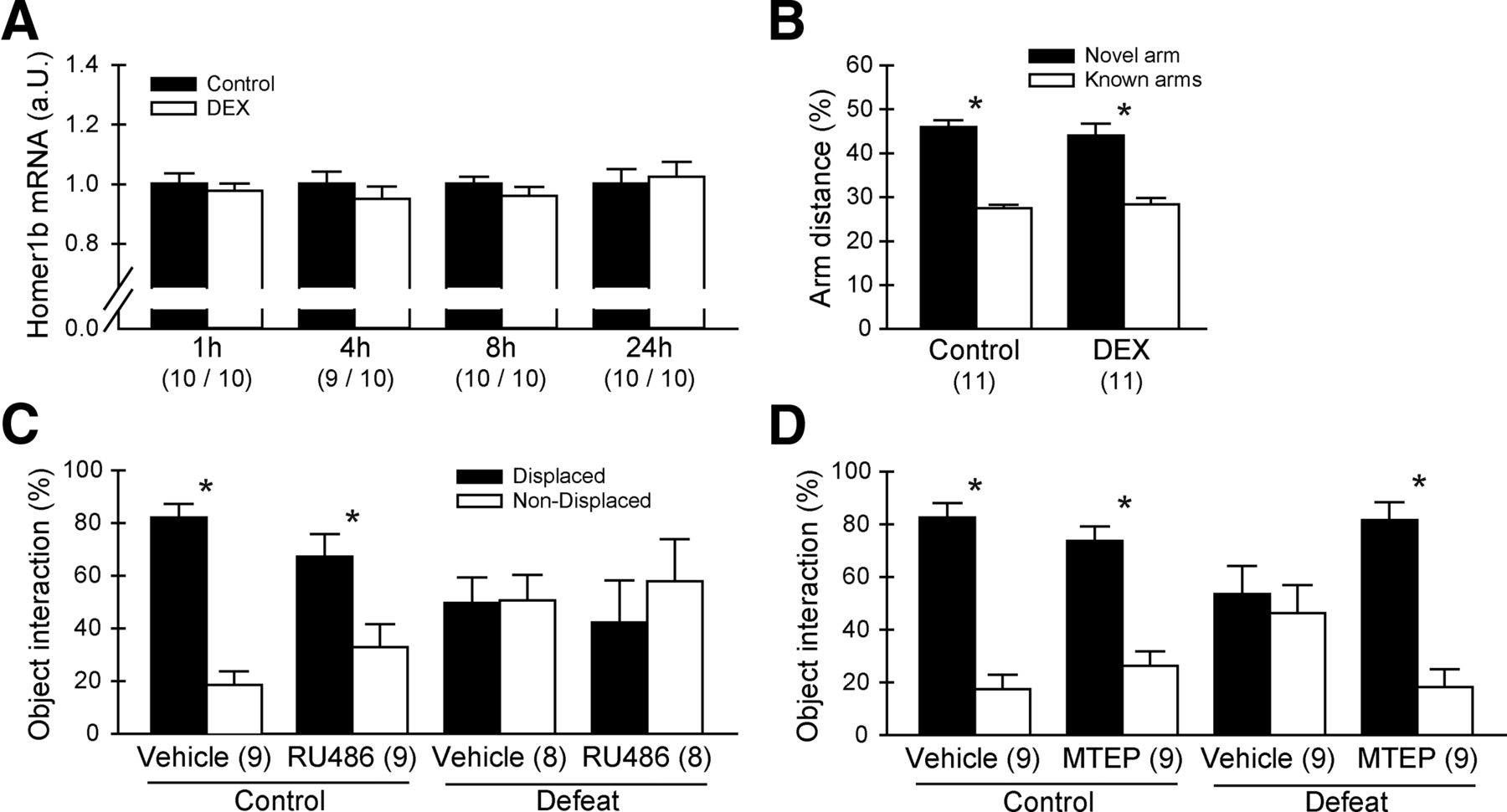
Manipulation of mGluR5/Homer1 but not glucocorticoid receptor signaling rescues cognitive impairments. A, A single injection of DEX (dexamethasone) that mimics a corticosterone response to a stressor was not able to induce changes in Homer1b/c mRNA levels. B, Eight hours after a single injection of DEX, experimental mice are not impaired in the Y-maze memory performance task. C, Mice that received a RU486 injection before the stressor are still affected by the defeat stress and cannot discriminate between the displaced and the nondisplaced object. D, Mice that received a MTEP injection before the stressor show functional learning behavior independent of the condition. *p < 0.05, data are expressed as mean ± SEM.
We then investigated whether GR or mGluR5/Homer1 signaling during the sensation of the defeat stress is necessary to induce the memory deficits that are occurring 8 h after the stress. We injected either RU486, a specific GR antagonist, or the specific mGluR5 inverse agonist before the defeat and tested the learning behavior in a subsequent sOR test 8 h after the onset of the stressor. Blockade of GR signaling by RU486 was not able to prevent the stress-induced memory impairments in the sOR task (Fig. 3C). Here, both unstressed control groups discriminated between the two objects (vehicle: T(16) = 8.747, p < 0.001; RU486: T(16) = 2.796, p < 0.05), while the defeated animals did not learn the novel position of the object regardless of treatment (vehicle: T(14) = −0.067, p = 0.948; RU486: T(14) = −0.699, p = 0.495). Total locomotion and total interaction time with both objects did not differ between the experimental groups.
When animals were administered with MTEP treatment before the defeat, their learning behavior normalized in a subsequent test 8 h after the onset of the stressor (Fig. 3D). In the sOR, both control groups showed normal learning behavior with a significant preference for the displaced object (vehicle: T(16) = 8.367, p < 0.001; MTEP: T(16) = 6.065, p < 0.001). In the defeated group, only the MTEP-treated animals showed a learning phenotype (vehicle: T(16) = 0.479, p = 0.638; MTEP: T(16) = 6.600, p < 0.001). We also replicated this pharmacologically induced rescue of the behavioral phenotype in the Y-maze test (control vehicle: novel arm 40.90% ± 3.95, known arms 29.55% ± 1.97, T(22) = 2.570, p < 0.05; control MTEP: novel arm 39.75% ± 2.44, known arms 30.12% ± 1.22, T(22) = 3.519, p < 0.01; defeat vehicle: novel arm 37.59% ± 4.39, known arms 31.21% ± 2.19, T(22) = 1.298, p = 0.208; defeat MTEP: novel arm 42.86% ± 4.51, known arms 28.57% ± 2.26, T(22) = 2.832, p < 0.01). In both experiments we did not find significant differences in locomotion or total object interaction time between any of the groups.
Overexpression of Homer1b/c rescues spatial memory impairments induced by defeat stress
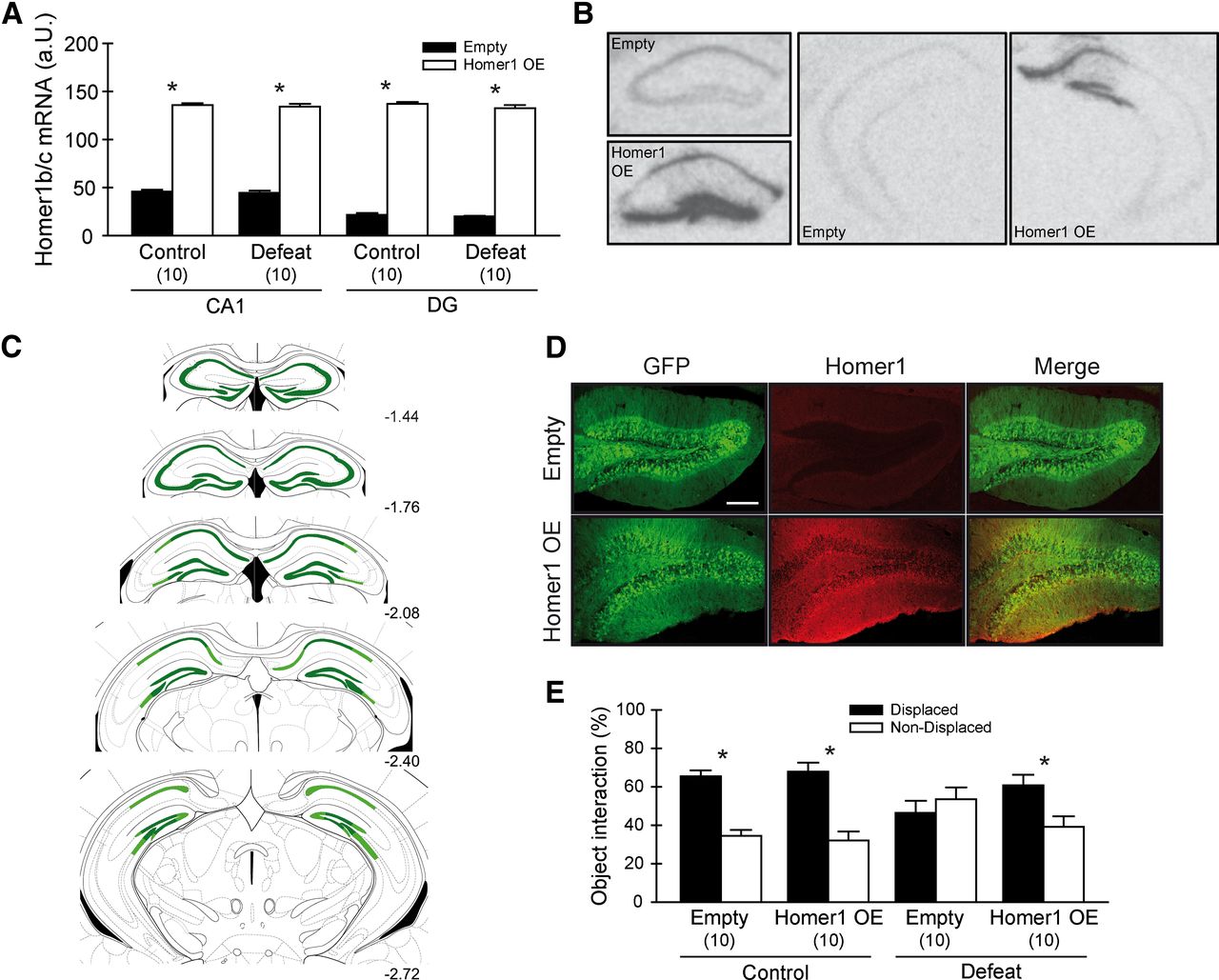
We examined whether we could rescue the stress-induced cognitive decline, which correlated with reduced Homer1 levels in the dorsal hippocampus, by overexpressing Homer1b/c in the dorsal hippocampus by AAV injection. We achieved a stable overexpression in the CA1 and DG regions of the dorsal hippocampus that was quantified by in situ hybridization (Fig. 4A,B). Viral infection spread (Fig. 4C) and verification of the infection sites were additionally controlled by immunofluorescence (Fig. 4D). Animals were tested in the sOR test, and while defeated animals infected with a control virus showed cognitive impairments (T(18) = 0.802, p = 0.433), animals that were overexpressing Homer1b/c were able to discriminate the objects' locations (T(18) = −4.215, p < 0.001) (Fig. 4E). Both control groups were able to discriminate between the displaced and nondisplaced objects (empty: T(18) = −5.381, p < 0.001; Homer1 overexpression: T(18) = −5.411, p < 0.001). Again, no locomotion or total object interaction differences were present when comparing the experimental groups.

Overexpression of Homer1b/c in the dorsal hippocampus rescues memory impairments induced by defeat stress. A, Homer1b/c mRNA levels in the dorsal hippocampus. Infection with the viral construct induced a robust increase. B, Representative autoradiographs of Homer1b/c mRNA levels in the dorsal and ventral hippocampus of control and Homer1 OE animals. C, Schematic representation of the extent of viral infection in the hippocampus from −1.44 to −2.72 mm posterior to bregma. Dark green, Strongest overexpression; light green, weaker overexpression. D, Visualization of Homer1b/c expression in the hippocampal DG region 5 weeks after injection of control (top panels) or Homer1b/c-expressing virus (bottom panels) (scale, 200 μm). E, Overexpression of Homer1b/c in the dorsal hippocampus prevents learning deficits induced by acute defeat stress. *p < 0.05, data are expressed as mean ± SEM.
Discussion
In our study, we show that a single social defeat leads to temporally specific cognitive impairment after a recovery of 8 h. In parallel, hippocampal Homer1b/c levels mRNA and protein levels, as well as mGluR5/Homer1b/c interaction, are decreased in response to the stressor. To further investigate the relationship between the behavioral and molecular phenotype, we first blocked mGluR5 signaling by applying MTEP, a specific inverse agonist of mGluR5, before the defeat stress and could thereby rescue the stress-induced phenotype. Furthermore, a specific Homer1b/c overexpression in the dorsal hippocampus by viral transfection was also able to reverse the stress-induced effects on cognition. These results give substantial evidence that postsynaptic signaling via mGluR5 and Homer1b/c is strongly involved in mediating cognitive deficits induced by acute stress.
The impact of stress on cognition and memory formation is well described but strongly dependent on the chosen experimental setup (Schwabe et al., 2010). An acute stress session has been reported to interfere with the induction of long-term potentiation (LTP), but it facilitates long-term depression (LTD) (Joëls and Krugers, 2007). In animal models, acute stressors disrupt the performance in memory tasks when the test is immediately after the sensation of stress (Howland and Cazakoff, 2010), most likely due to elevated GCs during memory encoding (Sandi, 2011). An acute stress session can also induce long-term effects such as an increase in anxiety-related behavior that develops 7 days after the stress session (Kinn Rød et al., 2012). In this study, we were able to induce a specific cognitive deficit by social defeat after an intermediate time of recovery (8 h) without interfering with other behavioral traits such as anxiety or locomotion. A possible confounding factor in this behavioral setup is neophobia, which can be induced by stress as well (Bats et al., 2001). However, all parameters that we measured for control, such as the locomotion in the behavioral tests as well the interaction times with the objects to observe, do not indicate a possible corruption of the data. Furthermore, both the FUST and the EPM test did not show significant effects in anxiety-related behavior and interaction with novel stimuli.
In our molecular analysis, we show that Homer1b/c levels are reduced in response to stress in a time window that coincides with the observed memory deficits. Various studies have identified Homer1 to be involved in memory processes in the prefrontal cortex (Lominac et al., 2005; Jaubert et al., 2007). Furthermore, Homer1 has recently been associated with stress-induced behavioral changes in the context of fear conditioning (Tronson et al., 2010). There, the intrinsic activity of mGluR5 and its subsequent signaling via Homer1 multimers in the hippocampus have been shown to mediate the enhancing effects of stress on contextual fear. We now confirm that acute stress has a profound effect on metabotrobe glutamate signaling pathways in the hippocampus. This is demonstrated by a significant reduction in both Homer1b/c mRNA levels as well as in decreased Homer1b/c protein levels in the PSD and Homer1/mGluR5 interactions 8 h after the defeat stress.
We did not observe a significant induction of Homer1a, an immediate early gene triggered by synaptic activity (Bottai et al., 2002), which is in contrast to previous studies that report increased Homer1a mRNA in response to various stimuli (Sala et al., 2003; Hu et al., 2010; Mahan et al., 2012). Possibly, the induction of Homer1a due to the defeat stress is already diminishing at the 1 h time point, and early time points of investigation might have shown a significant increase in Homer1a mRNA levels. Previous studies have already reported a significant role of Homer1 in memory processes, such as memory impairments of Homer1 knockout mice (Jaubert et al., 2007) that could be rescued by Homer1c overexpression in the hippocampus of these animals (Gerstein et al., 2012). Homer1a, in turn, has been shown to be essentially involved in the processing of fear (Brouillette et al., 2007) and working memory (Celikel et al., 2007). Our findings are contributing further to understanding the role of Homer1 in memory formation, suggesting that Homer1b/c signaling is immediately altered by stress and subsequently changes hippocampal memory processing.
Interestingly, the observed behavioral and molecular effects do not seem to be dependent on GR signaling. This is on the one hand shown by the absence of memory impairments as well as changes in Homer1b/c mRNA levels when the synthetic GR agonist DEX is administered to the animals instead of exposing them to a defeat stress. On the other hand, antagonizing GR by RU486 before stress was not able to recover the memory deficits. Corticosterone has frequently been shown to both positively and negatively alter memory formation, depending on the timing of corticosterone release and learning (Sandi, 2011; Popoli et al., 2012). A very recent study elucidated on the role of GR signaling in the dorsal and ventral hippocampi in memory retrieval, showing that stress impairs hippocampus-dependent memory up to 105 min after the stressor (Dorey et al., 2012). The present results further complement these findings, suggesting that it is not GR signaling but mGluR5/Homer1-mediated processes that impair memory formation in a more intermediate time window.
Inverse agonists of mGluR5 such as MPEP and MTEP have received growing attention for their potential in the treatment of neurologic disorders and were extensively tested in various behavioral parameters (Simonyi et al., 2010). With regard to learning paradigms, an immediate treatment before the test predominantly led to cognitive impairments (Steckler et al., 2005). When MTEP was applied before the stressor, we could show that stress-induced learning deficits were abolished. (Anderson et al., (2003) could convincingly show that MTEP is rapidly metabolized in mice, suggesting that 8 h postadministration, mGluR5 blockade by residual MTEP is unlikely and therefore does not interfere with the behavioral tests performed. Given the anxiolytic and antidepressant-like properties that have been reported for mGluR5 antagonists (Palucha and Pilc, 2007), one can speculate that blockade of the mGluR5 during and directly after the sensation of stress severely alters the perception and processing of the stressor and therefore its long-term consequences. The concept that environmental stimuli can trigger changes in the ability of synapses to express plasticity via intracellular signaling cascades has been coined “metaplasticity” (Abraham, 2008). It has previously been shown that prior activation of group I mGluRs can induce metaplasticity, thereby altering the ability of neurons in the CA1 or DG region of the hippocampus to express LTP or LTD (Rush et al., 2002; Gisabella et al., 2003). Our current data provide further evidence for this concept, showing that stress perception and subsequent molecular signaling pathways, in this case driven by mGluR5/Homer1 interactions, are critical for the future capacity of an individual to retain novel spatial information. This is particularly intriguing, since acute stress has been convincingly shown to enhance fear memory via mGluR5 signaling, with Homer1a having been shown to be necessary for the augmenting effects of stress on fear memory (Inoue et al., 2009; Tronson et al., 2010). Previous evidence also suggests that stress impairs spatial memory, which is in line with the results presented here (Sandi, 2011). The reduction of Homer1b/c in response to acute defeat stress, accompanied by increased dissociation from mGluR5, indicates that this pathway exerts detrimental effects on spatial memory performance, possibly shifting the hippocampal memory formation system toward a more sensitive fear response in an intermediate time frame.
Taken together, our study introduces the mGluR5/Homer1b/c signaling pathway as a major mediator of stress-induced spatial memory deficits. We also provide evidence that mGluR5 signaling plays a critical role during the perception of stress and most likely induces metaplasticity changes that lead to an altered Homer1b/c signaling. These findings underline the importance of glutamatergic pathways in the development of psychopathologies that are related to stress.
Footnotes
This study was partially supported by National Alliance for Research on Schizophrenia and Depression Grant No. 17322. We thank Kathrin Hafner, Daniela Harbich, and Bianca Schmid for their excellent technical assistance as well as Christine Kohl for proof reading the manuscript.
The authors declare no competing financial interests.
- Correspondence should be addressed to Mathias V. Schmidt, Max Planck Institute of Psychiatry, Neurobiology of Stress Research Group, Kraepelinstrasse 2-10, 80804 Munich, Germany. mschmidt{at}mpipsykl.mpg.de





{kind=link}
{kind=link}
{kind=link}
{kind=link}