

Abstract
Preventing and reversing opioid dependence continues to be a clinical challenge and underlying mechanisms of opioid actions remain elusive. We report that matrix metalloproteinase-9 (MMP-9) in the spinal cord contributes to development of physical dependence on morphine. Chronic morphine exposure and naloxone-precipitated withdrawal increase activity of spinal MMP-9. Spinal inhibition or targeted mutation of MMP-9 suppresses behavioral signs of morphine withdrawal and the associated neurochemical alterations. The increased MMP-9 activity is mainly distributed in the superficial dorsal horn and colocalized primarily with neurons and small numbers of astrocytes and microglia. Morphine exposure and withdrawal increase phosphorylation of NR1 and NR2B receptors, ERK1/2, calmodulin-dependent kinase II, and cAMP response element binding proteins; and such phosphorylation is suppressed by either spinal inhibition or targeted mutation of MMP-9. Further, spinal administration of exogenous MMP-9 induces morphine withdrawal-like behavioral signs and mechanical allodynia, activates NR1 and NR2 receptors, and downregulates integrin-β1, while a function-neutralizing antibody against integrin-β1 suppresses MMP-9-induced phosphorylation of NR1 and NR2B. Morphine withdrawal-induced MMP-9 activity is also reduced by an nNOS inhibitor. Thus, we hypothesize that spinal MMP-9 may contribute to the development of morphine dependence primarily through neuronal activation and interaction with NR1 and NR2B receptors via integrin-β1 and NO pathways. The other gelatinase, MMP-2, is not involved in morphine dependence. Inhibiting spinal MMP-9 or MMP-2 reduces chronic and/or acute morphine tolerance. This study suggests a novel therapeutic approach for preventing, minimizing, or reversing opioid dependence and tolerance.
Introduction
Opioid drugs have been used and abused for their analgesic and rewarding properties. Mechanisms of opiate dependence and tolerance are complex and involve factors at levels of the drug receptor, the cell, and neural networks. Roles of diverse neurotransmitter and receptor systems and intracellular signaling proteins in opioid actions have been demonstrated. A glutamate/NMDA receptor (NMDAR)/NO cascade is the most intensively studied system (Kolesnikov et al., 1993; Inoue et al., 2003; Zachariou et al., 2003; Bailey and Connor, 2005; Pasternak and Kolesnikov, 2005; Muscoli et al., 2007; Pasternak, 2007; Liu et al., 2009a). Adaptive changes following chronic opioid exposure that might underlie physical dependence by altering neuronal excitability and synaptic transmission include a rebound increase in cAMP levels and in expression of certain types of adenylyl cyclase, guanylyl cyclase, PKA, PKG, and cAMP response element binding protein (CREB) (Nestler, 2001; Barrot et al., 2002; Shaw-Lutchman et al., 2002). The regions important in opioid actions include the spinal cord and supraspinal levels (Jhamandas et al., 1996; Bailey and Connor, 2005; Muscoli et al., 2007; Han et al., 2008; Liu et al., 2009a). Despite decades of investigation, the specific cellular and molecular mechanisms underlying opioid actions remain elusive. Opiate tolerance and dependence and many side effects during repeated use have limited opioid use in the clinic. Thus, preventing and reversing opioid dependence and tolerance is still a clinical challenge.
Matrix metalloproteinases (MMPs), which belong to the metzincin clan of the metalloproteinase superfamily and are expressed in both neurons and glia, are widely implicated in inflammation and tissue remodeling through the cleavage of extracellular matrix (ECM) proteins, cytokines, and chemokines. Studies have shown that MMPs can regulate diverse biological processes including inflammation and responses to injury of peripheral nerve, spinal cord, and brain (Wang et al., 2000; Yong et al., 2001; Overall and López-Otín, 2002; Parks et al., 2004; Yong, 2005; Zhao et al., 2006; Chattopadhyay et al., 2007; Ethell and Ethell, 2007). In particular, the two gelatinases, MMP-9 and MMP-2, distinguished by three cysteine-rich fibronectin type II repeats in the catalytic domain, have recently been shown to play important roles in neuropathic pain (Kawasaki et al., 2008). We hypothesized that MMP-9 and/or MMP-2 might play an important role in chronic opioid action. We here provide the first evidence that spinal MMP-9, but not MMP-2, plays a critical role in development of morphine dependence. Mechanistically, MMP-9 may act primarily through neuronal activation and interact with NMDAR subunits NR1 and NR2B via integrin-β1 and NO pathways.
Materials and Methods
Experimental animals.
Adult male and female CD-1 mice (Charles River Laboratories) and MMP-9 mutant (Mmp9−/−) and wild-type (WT) FVB mice (The Jackson Laboratory) weighing 24–28 g at 8–10 weeks of age were used. Female mice were used for the pain threshold and morphine analgesia/tolerance tests using the hot plate apparatus because pain was predicted to be evoked by contact of the testicle with the hot plate; male mice were used for all other experiments. A cutoff time of 30 s was set to avoid tissue damage. As described by The Jackson Laboratory, a targeting vector containing a neomycin resistance gene driven by the mouse phosphoglycerate kinase promoter was used to disrupt most of exon 1 and all of intron 2 of the MMP-9 gene. The construct was electroporated into 129S-derived ZW4 embryonic stem cells. Correctly targeted embryonic stem cells were injected into C57BL/6J blastocysts. The resulting chimeric male animals were mated with Black Swiss females. Progeny animals were mated to Black Swiss mice for an unknown number of generations before being mated with FVB animals. The mice were maintained in FVB background for more than five generations. The experimental protocol was approved by the Parker Research Institutional Animal Care and Use Committee. Adult Kunming (KM) mice were also used to confirm the behavioral studies in different mouse strains (Fig. 1D). KM mice were provided by the Experimental Animal Center at Xuzhou Medical College (XMC). The experiments in KM mice were performed in the Jiangsu Province Key Laboratory of Anesthesiology at XMC.
Animal anesthesia, drugs, and administration.
Mice were anesthetized with pentobarbital (50 mg/kg, i.p.) for preparing the spinal cord tissue for gelatin zymography, Western blotting, and immunobiochemistry. We purchased MMP-9 inhibitor Inhibitor-I (MMP9i), MMP-2 inhibitor Inhibitor-III (MMP2i), and MMP-9 from Calbiochem; an nNOS inhibitor, 7-nitroindazole (7NI), morphine, and naloxone from Sigma-Aldrich; and a function-neutralizing integrin-β1 antibody (β1ab) from Millipore. Each of these drugs was dissolved in PBS or DMSO and diluted in PBS (final concentration of DMSO for intrathecal administration was 1%). MMP-9 (0.4 pmol), MMP9i (5 μg), MMP2i (5 μg), 7NI (50 nmol), and β1ab (2 μg) and the vehicle controls PBS and DMSO were injected intrathecally (each in 10 μl), respectively, under brief inhalational anesthesia or ∼5 min after each morphine injection, by means of lumbar puncture at the intervertebral space of L4–5 and L5–6 for multiple injections, using a stainless steel needle (30 gauge) attached to a 25 μl Hamilton syringe.
Morphine withdrawal.
Mice were repeatedly injected with morphine in seven escalating doses every 8 h (20, 40, 60, 80, 100, 100, and 100 mg/kg, i.p.). Two hours after the last morphine injection, mice were injected with naloxone (1 mg/kg, s.c.), and withdrawal symptoms were monitored for 30 min after naloxone administration. For testing the morphine withdrawal-like behavioral signs following intrathecal MMP-9, the withdrawal symptoms were monitored for 30 min 2 h after naloxone administration. In addition to measuring individual withdrawal signs, an overall opiate withdrawal score was calculated as (no. of backward walking steps × 0.1) + (diarrhea × 2) + (no. of jumps × 0.1) + (paw tremor × 0.1) + ptosis + tremor + (% weight loss × 5) + no. of wet-dog shakes (Zachariou et al., 2003; Liu et al., 2009a).
Pain threshold and morphine analgesia tests.
To test effects of MMP-9 and MMP-2 inhibitors on the pain threshold and the initial analgesic response to morphine, mice were placed on a 55°C hot plate apparatus, and the latency to lick a paw was measured. Data were calculated as percentage maximal possible effect (MPE%), which was calculated by the following formula: 100 × [(drug response time − basal response time)/(30 s − basal response time)] = MPE%. Morphine (10 mg/kg, s.c.), MMP9i (5 μg, i.t.), MMP2i (5 μg, i.t.), and the control vehicles PBS and DMSO (1%, i.t.) were administered 30 min before testing. The protocol was the same as that previously described (Zachariou et al., 2003; Liu et al., 2009a).
Mechanical allodynia test.
Mechanical allodynia was indicated by a significant decrease in the threshold of paw withdrawal in response to mechanical indentation of the plantar surface of each hindpaw with a sharp, cylindrical probe with a uniform tip diameter of ∼0.2 mm provided by an Electrovonfrey (Almemo 2390-5, Anesthesiometer IITC). The probe was applied to four designated loci distributed over the plantar surface of the hindpaw. The minimal force (in grams) that induced paw withdrawal was read off the display. Threshold of mechanical withdrawal in each mouse was calculated by averaging the four readings, and the force was converted into millinewtons.
Morphine tolerance tests.
To evaluate the physical tolerance to morphine, a profound decrease in analgesic effect, each female mouse was placed on a 55°C hot plate apparatus, and the latency to lick a paw was measured following subcutaneous morphine injection. For testing acute tolerance, the latency to lick a paw was measured at 0.5, 1, 1.5, and 2 h after a single dose of morphine at 10 mg/kg, administered 24 h after a morphine treatment at 100 mg/kg (protocol abbreviated as Mor100-10). To mimic clinical long-term use of morphine at high doses, we further tested morphine tolerance using a model measuring the analgesic effect of a single dose of morphine at 10 mg/kg each day followed by repetitive morphine at 50 mg/kg 2 h afterward, for 7 consecutive days (protocol abbreviated as Mor10-50). Chronic tolerance was also tested following repetitive treatment of morphine at 10 mg/kg given daily for 7 d, and the analgesic effect was measured 30 min after each injection (protocol abbreviated as Mor10).
Gelatin zymography.
The protocol was similar to that previously described (Gasche et al., 1999; Kawasaki et al., 2008). The animals were anesthetized deeply and transcardially perfused with PBS, and then a length of spinal cord containing segments L1–L6 was rapidly dissected and homogenized in a lysis buffer containing proteinase inhibitors. Aliquots (10 μl) of the homogenates were saved for total protein measurement (BCA kit). The homogenates were centrifuged at 12,000 RPM for 5 min. The supernatants were recovered and incubated for 60 min with gelatin-Sepharose 4B (Pharmacia Biotech) with constant shaking. After incubation, the samples were centrifuged at 500 RPM for 2 min. The pellets were resuspended in 80 μl of elution buffer for 30 min. The entire sample was loaded onto a 8% SDS gel containing 1 mg/ml gelatin. The gel was washed for 3–4 h to remove SDS and allow renaturation of MMPs. The gel was then left for 48 h in an incubation buffer to allow MMPs to degrade the gelatin in their immediate vicinity. Finally, zones of gelatin degradation representing proteolytic activity were identified by staining the gel with Coomassie Blue and destaining with methanol and acetic acid in water (3:1:6).
Western blotting.
To identify temporal expression of the phosphorylated protein levels of NR1, NR2B, ERK, calmodulin-dependent kinase II (CaMKII), CREB, glial fibrillary acidic protein (GFAP), and GAPDH, whole-cell protein extract lysates were used. The protocol was similar to that described previously (Bundesen et al., 2003; Liu et al., 2009a). Under anesthesia and immediately after perfusion with PBS, the mice spinal cord tissue at segments L1–L6 was rapidly removed and homogenized. After transfer to nitrocellulose filters, the filters were blocked with 5% bovine serum albumin (BSA) and then incubated overnight at 4°C with the primary antibodies [pNR1 (Ser897), 1:800 from Millipore; pNR2B (Tyr1472), 1:300 from Millipore Bioscience Research Reagents; pERK1/2 (Thr202/Tyr204), 1:500, and pCaMKII (Thr286), 1:1000 from Cell Signaling Technology; GAPDH, 1:1000 from Sigma; GFAP and pCREB (Ser133) from Santa Cruz Biotechnology] and anti-integrin β1 (MAB1987Z, 1:500, Millipore). The filters were developed using ECL reagents (PerkinElmer) with secondary antibodies from Millipore Bioscience Research Reagents. Data were analyzed with a Molecular Imager (Gel DocTM XR, 170–8170) and the associated software Quantity One-4.6.5 (Bio-Rad Laboratories).
Immunohistochemistry.
Under deep anesthesia, mice were transcardially perfused with PBS followed by 4% paraformaldehyde with 1.5% picric acid in 0.16 m PB (pH 7.2–7.4, 4°C), and then the L4 and/or L5 lumbar segment was dissected out and postfixed in the same fixative overnight. The embedded blocks were sectioned (30 μm thick) and processed for immunofluorescence. Sections from each group (five mice in each group) were incubated with rabbit anti-c-Fos polyclonal antibody (1:100, sc-52, Santa Cruz Biotechnology), rabbit anti-calcitonin gene-related peptide (CGRP) polyclonal antibody (1:1000, Millipore), goat anti-MMP-9 (1:500, M9570, Sigma) and rabbit anti-MMP-9 (1:500, AB19016, Millipore) polyclonal antibodies, mouse monoclonal anti-neuronal nuclear protein (NeuN) for identifying neurons (1:100, Alexa Fluor-488 conjugated, MAB377X, Millipore Bioscience Research Reagents), rabbit polyclonal anti-GFAP (1:500, ab7260, Abcam), and rabbit polyclonal anti-IBA (1:100, 019-19741, Wako Pure Chemical Industries), respectively. Rabbit IgG (1:200, Vector Laboratories) was used as an isotype control. For double immunofluorescence staining, the free-floating sections were incubated in PBS containing 10% donkey serum and 1% BSA for 2 h, incubated at 4°C in primary antibody, then washed three times in 50 mm Tris-HCl (pH = 7.4) PBS, and incubated in the secondary antibody either for 2 h at room temperature or overnight at 4°C. After washing three times in PBS, sections were reincubated in blocking serum for 1 h. Morphologic details examined with a confocal microscope (Leica TCS SP2). Images were randomly coded and transferred to a computer for further analysis. Fos-immunoreactive neurons were counted in a blind fashion. The number of Fos-like-immunoreactive neurons in the dorsal horn (DH) of the spinal cord (laminae I–VII) was determined by averaging the counts made in 20 spinal cord sections for each group. To obtain quantitative measurements of CGRP immunofluorescence, 15–20 fields covering the entire DH in each group were evaluated and photographed at the same exposure time to generate the raw data. Fluorescence intensities of the different groups were analyzed using MicroSuite image analysis software (Olympus America). The average green fluorescence intensity of each pixel was normalized to the background intensity in the same image.
Statistics.
SPSS Rel 15 was used to conduct all the statistical analyses. Alteration of expression of the proteins detected and the behavioral responses to morphine withdrawal were tested with one-way ANOVA and the differences in latency over time among groups were tested with two-way ANOVA with repeated measures, respectively, followed by Bonferroni post hoc tests. All data are presented as mean ± SEM. Statistical results are considered significant if p < 0.05.
Results
Inhibition of spinal MMP-9, but not MMP-2, or targeted mutation of MMP-9 suppresses behavioral and neurochemical signs of naloxone-precipitated morphine withdrawal
Spinal coadministration of an MMP-9 inhibitor, MMP9i (5 μg/10 μl, i.t.), but not an MMP-2 inhibitor, MMP2i (5 μg/10 μl, i.t.), with morphine (i.p., seven escalating doses every 8 h) significantly attenuated behavioral signs of morphine withdrawal in CD-1 mice. The overall score and individual behavioral signs were reduced significantly compared with the control groups (Fig. 1A). Neither MMP9i nor MMP2i altered the pain threshold (Fig. 1B) or the initial morphine-induced analgesia (Fig. 1C) in naive mice. Similar effects were observed in another mouse strain, the KM mice (Fig. 1D). These pharmacological data were further confirmed in mice lacking MMP-9 (Mmp9−/−). The overall score significantly decreased and the individual behavioral signs observed were significantly suppressed in Mmp9−/− mice compared with those in WT FVB mice with the same genetic background (Fig. 2). We noticed that WT FVB mice exhibited significantly lower overall scores and individual signs than did WT CD-1 (Fig. 1A) or WT KM mice (Fig. 1D) (overall scores: CD-1 vs KM = 76.0 ± 3.54% vs 68.2 ± 3.36%, p > 0.05; CD-1 vs FVB (48.6 ± 1.98%), p < 0.01; KM vs FVB, p < 0.01). We further tested pharmacological effects on WT FVB and the Mmp9−/− mice. MMP9i significantly attenuated behavioral signs of morphine withdrawal in WT FVB mice, but did not produce further inhibition in Mmp9−/− mice (Fig. 2). These results demonstrate an important role of spinal MMP-9, but not MMP-2, in the development of morphine physical dependence.

Inhibition of spinal MMP-9, but not MMP-2, attenuates behavioral signs of naloxone-precipitated morphine withdrawal. A, Effects of MMP-9 inhibitor MMP9i and MMP-2 inhibitor MMP2i on behavioral signs. In addition to receiving morphine (intraperitoneally) and naloxone (subcutaneously), the different groups received one of the following treatments (intrathecally): PBS and DMSO (1%) for vehicle controls, MMP9i, and MMP2i (5 μg each, 7 doses accompanied morphine). *p < 0.05 and **p < 0.01 compared to vehicle control. #p < 0.05 and ##p < 0.01 compared to group of MMP9i. Number of mice in each group: PBS = 12, DMSO = 14, MMP9i = 12, MMP2i = 16. B, Effects of MMP9i and MMP2i (intrathecal) on the pain threshold in naive mice (n = 14 each group). C, Effects of MMP9i and MMP2i (intrathecal) on the initial analgesic response to morphine (Mor, intraperitoneal) (n = 14 each group). PBS and DMSO were used as intrathecal controls in B and C. D, Effects of MMP9i and MMP2i on behavioral signs of morphine withdrawal in another strain of animals, KM mice. Sixteen mice were included in each group. **p < 0.01 compared to vehicle controls; ##p < 0.01 compared to group of MMP9i.

Spinal inhibition and targeted mutation of MMP-9 attenuate behavioral signs of naloxone-precipitated morphine withdrawal. A, B, Overall withdrawal scores (A) were calculated from the individual signs (B). *p < 0.05, **p < 0.01 compared to the control (Ctrl) group in WT or MMP-9 mutation (Mmp9−/−) group. Number of mice in each group: WT Ctrl = 20, WT MMP9i = 10, Mmp9−/− Ctrl = 15, Mmp9−/− MMP9i = 10.
In addition to the behavioral signs, morphine exposure and withdrawal caused significant neurochemical alterations, induction of c-Fos (Fig. 3A), increased CGRP-like immunoreactivity (Fig. 3B), and spinal astrocyte activation (Fig. 4) manifested as upregulation of expression of GFAP (an astrocyte marker) and increases in both astrocyte number and size in the DH. Repetitive administration of MMP9i, but not MMP2i, prevented or suppressed these neurochemical changes.

Effects of MMP-9 inhibitor MMP9i and MMP-2 inhibitor MMP2i on induction of c-Fos and expression of CGRP in the DH following morphine (Mor) treatment and/or naloxone (Nlx)-precipitated withdrawal. A, MMP9i, but not MMP2i, inhibits induction of c-Fos in the DH following Nlx-precipitated withdrawal. Confocal image shows examples of induction of c-Fos in the DH. Bottom, Data summary. B, MMP9i, but not MMP2i, prevents accumulation of CGRP in the superficial DH following Mor treatment. Confocal image shows examples of expression of CGRP in the DH. Bottom, Data summary. Five spinal cord segments were included in each group. **p < 0.01 compared to group of sham; ##p < 0.01 compared to group of Mor; @p < 0.05 compared to group of (Mor+Nlx); &p < 0.05 compared to group of Mor+MMP9i+Nlx (A) or Mor+MMP9i (B). Magnification: 200×.

MMP-9 inhibitor MMP9i, but not MMP-2 inhibitor MMP2i, suppresses morphine (Mor) exposure-induced activation of astrocytes in the spinal cord. A, Western blot analysis shows expression of GFAP (a marker for astrocytes) (top: examples; bottom: data summary). *p < 0.05 compared to group of sham; **p < 0.01 compared to group of sham; #p < 0.05 compared to group of Mor and MMP2i. Four spinal cord segments were included in each group. B, Examples of confocal images showing GFAP activation in the DH. Magnification: 200× (top), 400× (bottom).
Activity and distribution of spinal MMP-9 and MMP-2 following morphine exposure and withdrawal
Given that blocking spinal activity of MMP-9, but not MMP-2, or targeted mutation of MMP-9 can prevent development of behavioral and neurochemical signs associated with morphine exposure and withdrawal, we examined the activity of MMP-9 and MMP-2 using gelatin zymography in the spinal cord under the same conditions. MMP-9 activity was significantly increased following morphine exposure and further increased after morphine withdrawal. Activity peaked 30 min after withdrawal, whereas MMP-2 (pro- and active) activity was not changed (Fig. 5A). The increased immunostaining for MMP-9 was located mainly in the superficial layers and inner parts of laminae III–VI of the DH (Fig. 5B).

Upregulation of MMP-9, but not MMP-2, in the spinal cord following morphine (Mor) exposure and naloxone (Nlx)-precipitated withdrawal. A, Gelatin zymography shows activity of MMP-9 and MMP-2 (Pro-MMP-2 and active MMP-2). Intensity of MMP-9 and pro-MMP-2 bands, expressed as multiples of values in the sham control, is shown on the right. **p < 0.01 compared to sham. Six segments of the spinal cord were used in each group for MMP-9 and MMP-2, respectively. B, Confocal image of immunostaining for MMP-9 showing increased activity of MMP-9 in the DH. Magnification: 100×.
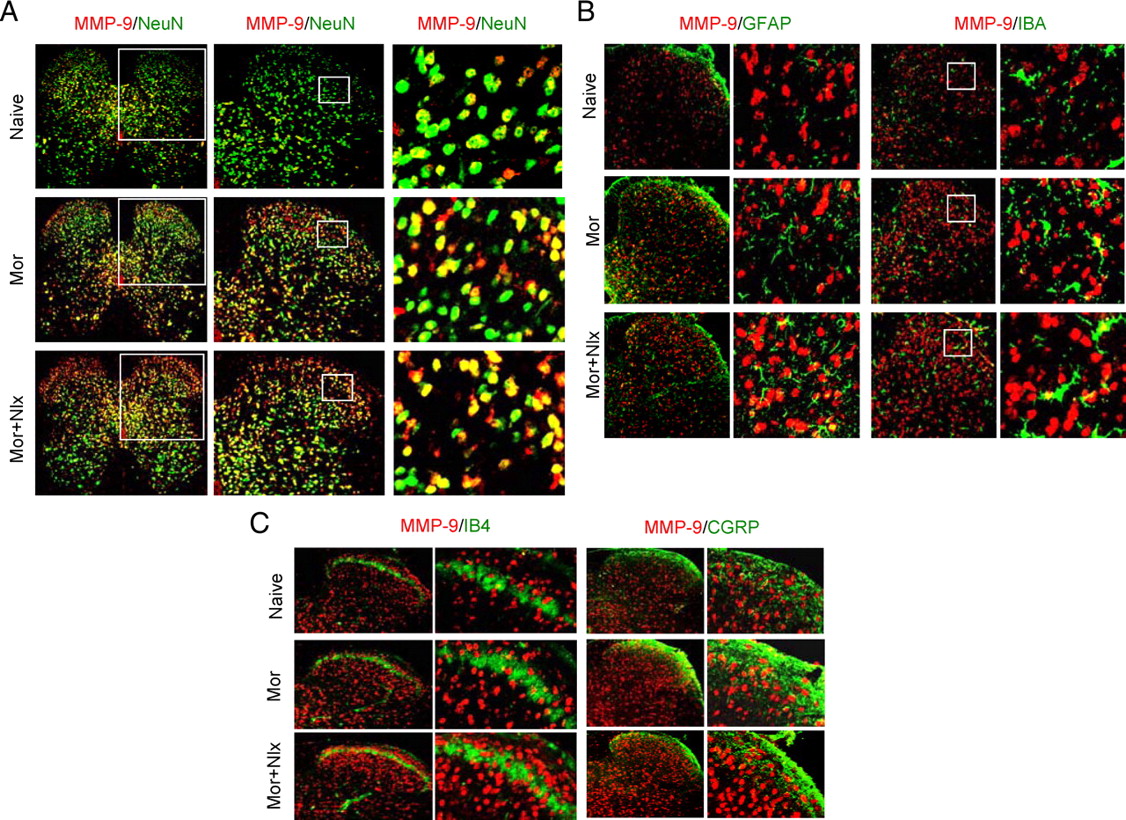
We further asked (1) with which neurons, astrocytes, and/or microglia within the DH MMP-9 immunoreactivity was colocalized; (2) whether the MMP-9 was associated with IB4-positive nociceptor terminals; and c) whether the MMP-9 colocalized with CGRP afferents following morphine treatment. Confocal images showed that, in naive mice, the MMP-9 immunoreactivity was distributed throughout the DH and ventral horn and colocalized primarily with neurons (as indicated by NeuN, a neuronal marker) (Fig. 6A, top). The increased MMP-9 immunoreactivity after morphine exposure (Fig. 6A, middle) and naloxone-precipitated morphine withdrawal (Fig. 6A, bottom) was mainly distributed in the superficial layers and inner parts of laminae III–VI of the DH and colocalized primarily with neurons, while only a small amount of colocalization of MMP-9 with astrocytes (GFAP) (Fig. 6B, left two panels) and microglia (IBA, a microglia marker) (Fig. 6B, right two panels). However, our results failed to show any colocalization of MMP-9 immunoreactivity with either IB4-positive terminals (Fig. 6C, left two panels) or CGRP-positive nociceptive primary afferents (Fig. 6C, right two panels). These results indicate that morphine treatment primarily upregulates neuronal MMP-9 within the DH.

Distribution and colocalization of MMP-9 with neurons, astrocytes, and microglial cells, but not the primary afferents in the dorsal horn after morphine (Mor) exposure and naloxone (Nlx)-precipitated withdrawal. A, Distribution of MMP-9 (red) and its colocalization with the neurons (NeuN, green, a marker of neurons). Magnification: 100× (left); 200× (middle); 400× (right). B, A small amount of colocalization of MMP-9 (red) with astrocytes (GFAP, green, left two panels) or microglia (IBA, green, right two panels). C, MMP-9 (red) was not colocalized with IB4 (green, left two panels) or CGRP (green, right two panels) primary afferents. Magnification in B and C: 200× (the first and the third panels); 400× (the second and the fourth panels).
Inhibition of spinal MMP-9 or targeted mutation of MMP-9 suppresses upregulation of phosphorylation of NR1, NR2B, ERK, CaMKII, and CREB associated with morphine exposure and withdrawal
NMDARs have a well established role in opiate-related neural plasticity. Chronic morphine treatment causes increases in Ca2+ levels and alterations of ERK (Ren et al., 2004), CaMKII (Lu et al., 2000; Liang et al., 2004), and CREB (Nestler, 2001). Our Western blotting analysis showed that morphine exposure and withdrawal-induced increases of levels of phosphorylation of NR1, NR2B, ERK1/2, CaMKII, and CREB protein were significantly reduced by MMP-9 inhibitor MMP9i (5 μg, i.t.) (Fig. 7A,B). These findings indicate that MMP-9 may contribute to morphine dependence, probably via interaction with NR1 and NR2B receptors and activation of subsequent signaling pathways. These alterations were further confirmed in Mmp9−/− mice; morphine withdrawal-induced phosphorylation of NR1 and NR2B was significantly less in Mmp9−/− mice than in WT FVB mice (Fig. 7C).

Inhibition of spinal MMP-9, but not MMP-2 or targeted mutation of MMP-9 suppresses increased phosphorylation of NR1, NR2B, ERK, CaMKII, and CREB in the spinal cord associated with morphine (Mor) exposure and naloxone (Nlx)-precipitated withdrawal. A, B, Expression of phosphorylation of the molecules after treatment of Mor (A) and Mor+Nlx (B) with MMP-9 inhibitor MMP9i. Top, Examples of Western blot; bottom, data summary (n = 5 each group). **p < 0.01 compared to naive; #p < 0.05 and ##p < 0.01 compared to Mor. C, Expression of phosphorylation of pNR1 and pNR2B after treatment of Mor or (Mor+Nlx) in Mmp9−/− mouse spinal cord (n = 5 each group). **p < 0.01 compared to naive; #p < 0.05 and ##p < 0.01 compared to WT. *p < 0.05, **p < 0.01 compared to naive. Bottom, Expression of NR1 and NR2B were not statistically different between naive and MMP9−/− groups; thus, naive was used as control.
Spinal administration of exogenous MMP-9 induces morphine withdrawal-like behavioral signs and increases levels of phosphorylation of NR1, NR2B, and CREB
To determine whether exogenous MMP-9 is sufficient to mimic the morphine withdrawal-like behavioral signs and the morphine treatment-activated and MMP-9-regulated NMDARs and subsequent signaling, we examined alterations of behavior and levels of phosphorylation of NR1, NR2B, and CREB in the spinal cord following spinal administration of MMP-9 in naive mice. A single dose of MMP-9 (0.4 pmol, i.t.) produced behavioral signs that were similar to the naloxone-precipitated morphine withdrawal (Fig. 8A). The overall scores increased to 35.4 ± 2.51 after MMP-9 treatment, which was significantly higher (p < 0.01) than the 19.6 ± 3.33 in the control treated with PBS. Of the nine monitored behavioral signs, scores for diarrhea, paw tremor, ptosis, tremor, and wet dog shakes increased significantly, while the others did not. In a separate group of mice, spinal administration of MMP-9 (0.4 pmol) produced significant mechanical allodynia tested in the hindpaws. The mechanical hypersensitivity developed within 1 h, peaked at 2 h, and lasted for at least 6 h (Fig. 8B). Meanwhile, the same treatment of MMP-9 significantly increased phosphorylation of NR1 and NR2B receptors and CREB (Fig. 8C). These results indicate that spinal MMP-9 can induce morphine withdrawal-like behavioral signs and activate NR1 and NR2B receptors and the downstream pathways.

Spinal administration of exogenous MMP-9 causes morphine withdrawal-like behavioral signs, mechanical allodynia, and increases in phosphorylation of NR1, NR2B, and CREB in the spinal cord. A, Morphine withdrawal-like behavioral signs induced by MMP-9 (0.4 pmol, i.t.). PBS was injected as vehicle control. Ten mice were included in each group. B, Mechanical allodynia induced by MMP-9 (0.4 pmol, i.t.). Data presented are averages of both hindpaws in both hindpaws. C, Increased phosphorylation of NR1, NR2B, and CREB in the spinal cord after MMP-9 treatment. Left, Examples; right, data summary. Four spinal cords were included in each group. *p < 0.05, **p < 0.01 compared to PBS (A, B) or sham (C).
A function-neutralizing antibody against integrin-β1 prevents MMP-9-induced phosphorylation of NR1, NR2B, and CREB
We investigated a potential role of integrin-β1, which mediates MMP-9 regulation of synaptic plasticity and long-term potentiation in the spinal cord (Nagy et al., 2004), in mediating effects of MMP-9 on NR1 and NR2B receptors. First, our results showed that expression of integrin-β1 in the spinal cord was markedly downregulated by morphine exposure and withdrawal (Fig. 9A). Expression of integrin-β1 was also significantly downregulated by spinal administration of exogenous MMP-9 (0.4 pmol) (Fig. 9B). Further, β1ab (2 μg, i.t.) significantly suppressed MMP-9-induced phosphorylation of NR1 and NR2B (Fig. 9C). These results suggest that integrin-β1 is involved in MMP-9-induced phosphorylation of NR1 and NR2B receptors during morphine treatment.

Interactions among MMP-9, NR1, and NR2B receptors and integrin-β1 in the spinal cord. A, Morphine exposure and naloxone precipitated-withdrawal-induced downregulation of integrin-β1. B, Spinal administration of exogenous MMP-9 (0.4 pmol) induced downregulation of integrin-β1. C, β1ab (2 μg, i.t.) reduced MMP-9 (2 h)-induced upregulation of NR1 and NR2B. Four spinal cords were assayed in each group. *p < 0.05, **p < 0.01 compared to the corresponding group of sham (A–C); #p < 0.05, ##p < 0.01 compared to MMP-9 (C).
Spinal inhibition of nNOS suppresses MMP-9 activity after morphine withdrawal
NO has been demonstrated to play an important role in opioid dependence (Pasternak, 2007) and directly activates MMP-9 in a model of cerebral ischemia (Gu et al., 2002). We used a selective nNOS inhibitor 7NI to reduce NO signaling and found that morphine withdrawal-induced MMP-9 activity in the spinal cord was significantly suppressed by spinal administration of 7NI (50 nmol, i.t., one dose before naloxone administration) (Fig. 10). This result supports a role of NO regulation of spinal MMP-9 in the development of morphine dependence.

Spinal administration of an nNOS inhibitor 7NI attenuates the increased activity of MMP-9 after naloxone (Nlx)-precipitated morphine (Mor) withdrawal. Activity of MMP-2 was altered by neither Mor nor 7NI treatment. Top, Gelatin zymography showing activity of MMP-9 and MMP-2 in the spinal cord (n = 6 each group). Bottom, Data summary. *p < 0.05 and **p < 0.01 compared to naive. #p < 0.05 compared to the corresponding Mor group.
Effects of inhibiting spinal MMP-9 and MMP-2 on morphine tolerance
Physical tolerance, a profound decrease in analgesic effect, is another major limitation to long-term use of morphine, in addition to physical dependence. We examined whether analgesic effects of morphine could be rescued by inhibiting spinal MMP-9 or MMP-2. MMP9i and MMP2i (each 5 μg, i.t., 30 min before the dose of morphine at 100 mg/kg), respectively, significantly reduced the decrease in analgesic effect of morphine at 10 mg/kg, which was administered 24 h after a treatment of morphine at 100 mg/kg (protocol Mor100-10) (Fig. 11A). To mimic long-term, clinical uses of morphine at high doses, we tested the analgesic effect of morphine at 10 mg/kg followed by repetitive morphine at 50 mg/kg 2 h afterward, for 7 consecutive days (protocol Mor10-50). MMP9i, but not MMP2i, rescued the analgesic effect of morphine during the test days 3 through 7 (Fig. 11B). However, neither MMP9i nor MMP2i significantly changed the tolerance following repetitive injection of morphine at 10 mg/kg given daily for 7 consecutive days (protocol Mor10), although MMP9i showed a tendency to reduce tolerance during days 5–7 (Fig. 11C). Further, Western blot analysis showed that expression of MMP-9 was significantly increased following morphine treatment using protocol Mor10-50, but not that using protocol Mor10. Expression of MMP-2 was not changed by morphine treatment using either protocol Mor10-50 or Mor10 (Fig. 11D).

Effects of spinal inhibition of MMP-9 and MMP-2 on morphine tolerance. A, Acute tolerance measured after morphine (Mor) at 10 mg/kg (s.c.) alone (Mor10) and 24 h after Mor at 100 mg/kg (protocol Mor100-10). MMP9i and MMP2i were administered (intrathecally), respectively, 30 min before the dose of Mor at 100 mg/kg. *p < 0.05, **p < 0.01 compared to the control group Mor10. #p < 0.05, ##p < 0.01 compared to the model group Mor100-10. Ten mice were included in each group. B, Tolerance measured after Mor at 10 mg/kg (s.c.) followed by repetitive treatment of Mor at 50 mg/kg 2 h afterward each day for 7 consecutive days (protocol Mor10-50). MMP9i and MMP2i were administered, respectively, immediately after Mor (50 mg/kg) injection. *p < 0.05, **p < 0.01 compared to the group of Mor10-50. Number of mice: Mor10-50 = 10, Mor10-50+MMP9i = 15, Mor10-50+MMP2i = 15. C, Tolerance following repetitive treatment of Mor at 10 mg/kg given daily for 7 d (protocol Mor10) with the analgesic effect measured 30 min after each injection. MMP9i or MMP2i was administered immediately after Mor injection. Fifteen mice were included in each group. D, Upregulation of MMP-9, but not MMP-2, in the spinal cord following treatment of Mor10-50, but not Mor10. **p < 0.01 compared to the group of sham and Mor10.
Discussion
Our study reveals a critical role for gelatinase MMP-9 in development of physical dependence on morphine. Spinal neuronal MMP-9 can induce activation of NR1 and NR2B receptors and the downstream pathways in addition to the behavioral and neurochemical signs of morphine dependence and withdrawal. These actions of MMP-9 are regulated by integrin-β1 and NO in the spinal cord. It provides a novel mechanism underlying morphine physical dependence and suggests that inhibition of MMP-9 signaling may be of potential clinical benefit in preventing, minimizing, or reversing morphine-induced physical dependence and other side effects, thereby facilitating the clinical utility of opioid drugs.
MMP-9 and MMP-2 are expressed in the brain and spinal cord, and are often increased in response to inflammation, injury, or neurological diseases (Yong et al., 2001; Kawasaki et al., 2008). Analysis of the cellular distribution of MMP-9 and MMP-2 has revealed that MMP-9, but not MMP-2, increases both in neurons and astrocytes in the rat hippocampus following intraperitoneal kainate administration (Szklarczyk et al., 2002) and in DRG neurons after spinal nerve ligation (Kawasaki et al., 2008), while MMP-2 activity increases in astrocytes in the DH (Kawasaki et al., 2008). The present study demonstrates that morphine exposure and withdrawal increase spinal activity of MMP-9, but not MMP-2. The increased MMP-9 activity occurs mainly in the superficial layers of the DH, an essential area for generation and processing of pain signals, and colocalizes primarily with neurons and, to a lesser extent, with astrocytes and microglia. In the spinal nerve injury model, MMP-9 was found colocalized with DRG neurons and CGRP primary afferents, suggesting that the spinal MMP-9 derives primarily from axonal transport from DRG neurons (Kawasaki et al., 2008). However, in the present study MMP-9 was colocalized with neither IB4 nor CGRP primary afferents within the DH after morphine treatment; the upregulated MMP-9 following morphine treatment was found primarily in neurons in the DH and in some astrocytes and microglia. In contrast, our data demonstrate that spinal MMP-2 activity is not important for the development of morphine dependence, similar to its unimportance for kainate-induced excitotoxicity in the hippocampus (Szklarczyk et al., 2002). MMP-2 is, however, important for the maintenance of neuropathic pain caused by nerve injury (Kawasaki et al., 2008). Together, MMP-9 and MMP-2 activity may contribute differently to different pathological conditions. Patterns of expression of MMP-9 and MMP-2 in the hippocampus and the DH are similar to each other under conditions of morphine treatment and kainate-induced excitotoxicity (Szklarczyk et al., 2002). In the case of nerve injury, the initial spinal MMP-9 derives from axonal transport in DRG neurons (Kawasaki et al., 2008), while following morphine treatment in the spinal cord or kainate treatment in the hippocampus the initial MMP-9 may originate primarily locally, within central neurons (Szklarczyk et al., 2002).
NMDARs have a well established role in opiate-related neural plasticity. NMDAR activation results in Ca2+ influx through the NMDAR ion-channel complex (Mao and Mayer, 2001; Nestler, 2001; Inoue et al., 2003; Bajo et al., 2006). Subsequent activation of various Ca2+-dependent signaling enzymes, such as CaMKII, can phosphorylate CREB, leading to increases in c-Fos mRNA and c-Fos protein levels (Mao and Mayer, 2001; Nestler, 2001; Liu et al., 2009a,b). Gene expression is thought to play an important role in many forms of plasticity, including morphine dependence. Here we provide evidence that morphine exposure and withdrawal or spinal administration of exogenous MMP-9 increases the phosphorylation of NMDAR subunits, NR1 and NR2B, as well as CaMKII, ERK, and CREB, and induces c-Fos expression. These effects are prevented or suppressed by spinal inhibition or targeted mutation of MMP-9. Therefore, we hypothesize that MMP-9 in the DH may contribute to morphine dependence probably via interaction with the NR1 and NR2B receptors and therefore result in subsequent activation of Ca2+-dependent signaling pathways.
MMPs can influence dendritic spine development and hence synaptic stability in hippocampal neuron cultures, and can induce synapse modeling by interacting with NMDAR activity (Bilousova et al., 2006; Ethell and Ethell, 2007). MMP-9 may regulate synaptic plasticity and long-term potentiation through integrins (Nagy et al., 2004), possibly through the cleavage of laminin or other ECM components to expose otherwise inaccessible arginine-glycine-aspartic acid sites that can then induce integrin signaling (Ethell and Ethell, 2007). Integrin signaling has been shown to modulate NMDAR currents, control the structural remodeling of dendritic spines/synapses, and play an important role in hippocampal long-term potentiation (Bahr et al., 1997; Chavis and Westbrook, 2001; Lin et al., 2003; Bernard-Trifilo et al., 2005; Moeller et al., 2006; Shi and Ethell, 2006; Michaluk et al., 2009). Here we provide evidence that spinal administration of exogenous MMP-9 downregulates expression of integrin-β1 while activating NR1 and NR2B receptors, in addition to producing morphine withdrawal-like behavioral signs and mechanical allodynia. Moreover, a function-neutralizing antibody against integrin-β1 can suppress MMP-9-induced phosphorylation of NR1 and NR2B receptors. Interestingly, integrin-β1 is also downregulated after morphine treatment. These findings support a role for integrin-β1 in mediating MMP-9 modulation of NMDARs in morphine dependence in the spinal cord. Another signaling system with which MMP proteolysis may interact and therefore modulate NMDARs is the ephrin–Eph receptor pathway. EphrinB2 and EphB2 receptors are cleaved by MMPs in vitro and in cultured primary hippocampal neurons (Hattori et al., 2000; Georgakopoulos et al., 2006; Lin et al., 2008). We recently implicated a role for spinal ephrinB–EphB in morphine dependence probably via modulation of NR2B (Liu et al., 2009a).
NO can S-nitrosylate a wide range of proteins, including NR1 and NR2 (Jaffrey et al., 2001). In addition, exposure of NO to superoxide generates peroxynitrite, which can nitrosylate tyrosine residues (Muscoli et al., 2007; Pasternak, 2007). In a stroke model of cerebral ischemia, colocalization of MMP-9 and nNOS was found in ischemic cortical neurons, where MMP-9 could be S-nitrosylated and activated by NO (Gu et al., 2002). The present study shows that an nNOS inhibitor suppresses morphine exposure/withdrawal-induced upregulation of MMP-9. These findings indicate that free radicals may regulate MMP-9 activity during morphine treatment and cerebral ischemia. In addition, MMPs in the nucleus accumbens may be involved in methamphetamine-induced behavioral sensitization and reward by regulating dopamine release and receptor signaling (Mizoguchi et al., 2007a,b), and ECM remodeling in the hippocampus may be a persisting effect of chronic abuse that contributes to the compulsive and relapsing nature of cocaine addiction (Mash et al., 2007). The first demonstration of an opioid-mediated regulation of metalloproteinase activity was reported in cultured rat mesangial cells, in which morphine decreased 72 kDa metalloproteinase activity (Sagar et al., 1994).
In addition to physical dependence, another major limitation to the long-term use of opiates is the development of physiological tolerance. We here show that the acute morphine tolerance can be markedly improved by inhibiting spinal MMP-9 or MMP-2; the chronic tolerance following repetitive treatment of morphine at higher doses (10 mg/kg + 50 mg/kg), but not that at 10 mg/kg only, can be markedly improved by inhibiting MMP-9, but not MMP-2. Further, lack of MMP-9 and MMP-2 activity in response to repetitive morphine at 10 mg/kg may explain, at least partly, the failure of spinal inhibition of MMP-9 or MMP-2 in rescuing the analgesic effect of morphine. These results suggest a complexity of mechanisms underlying morphine tolerance. Such a complexity and potentially different roles of MMP-9 in morphine dependence and tolerance need to be further investigated. Possible pathways by which MMP-9 in the spinal cord may contribute to morphine dependence are illustrated in Figure 12.

Model of spinal MMP-9 contributions to the development of physical dependence on morphine. Flowchart illustrates a functional link among MMP-9 activation, MMP-9-dependent cellular responses, interactions between MMP-9 and integrins, NMDAR subunits NR1 and NR2B, ephrinB–EphB receptors, other intracellular signals, and behavioral manifestations of morphine dependence.
Footnotes
-
This project was supported by grants from the Parker Research Foundation (PCCRF-BSR0804/0905), National Natural Science Foundation of China (NSFC-30628027), and National Institutes of Health of the United States (NIH-1R43AT004933-01). We thank Dr. E.T. Walters for his comment and editing the manuscript, Dr. Z.J. Huang for technical assistance in data analysis, Dr. R.L. Rupert for advice and stimulating discussion, Dr. P. Brandon for proofreading, and M. Dominguez, Y.-P. Wu, and P.C. Xu for technical support and assistance in laboratory management. W.-T.L., Y.H., Y.-P.L., A.A.S., and B.B. designed and performed the experiments; X.-J.S. conceived the study, designed and supervised the overall project and the experiments, analyzed data, and wrote the manuscript. This work should be attributed equally to the institutions.
- Correspondence should be addressed to Xue-Jun Song, Department of Neurobiology, Parker University Research Institute, 2500 Walnut Hill Lane, Dallas, TX 75229. song{at}parkercc.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}