1. 引言
在肿瘤的表观遗传学研究中,组蛋白去乙酰化酶(histone deacetylases, HDACs)的主要功能是催化组蛋白的去乙酰化,抑制其转录活性,组蛋白的乙酰化修饰对肿瘤的发生起重要作用。如果正常的细胞体出现核内组蛋白乙酰化与去乙酰化失衡,正常细胞周期与细胞代谢就会发生改变从而诱发肿瘤 [1]。
大多数HDACs活性都具有Zn2+依赖性,组蛋白去乙酰化酶抑制剂(histone deacetylase inhibitor, HDACIs)可与HDACs活性位点的Zn2+离子结合,干扰HDACs的活性 [2],增加细胞内组蛋白的乙酰化程度,抑制肿瘤细胞的增殖,诱导细胞分化和(或)凋亡。因此,HDACIs的结构研究对于设计新型抗癌药物的合成具有非常重要的意义。
本文主要利用Topomer CoMFA方法研究对香豆素基苯甲酰胺作为组蛋白去乙酰化酶抑制剂的HCT116细胞系和A2780细胞系活性进行研究,分析其药物结构与生物活性之间的关系,建立3D-QSAR模型,根据主成分分析和偏最小二乘的表征手法对所建立的模型进行验证,然后在ZINC数据库中进行构象搜索,设计出更具活性的新型组蛋白去乙酰化酶抑制剂,最后结合分子对接,对新设计的化合物进行进一步分析,研究化学结构与生物活性之间的相互影响关系。
2. 实验材料和方法
2.1. 分子构建与数据来源
本文分子构建中化合物所包含的小分子结构是利用SYBYL2.0-X软件中的sketch molecule绘图工具进行构建。全部分子加载Gasteger-Huckel电荷,采用标准的Tripos分子力场和Powell能量梯度算法对所有小分子进行能量优化,最大迭代次数1000次,能量收敛限定设为0.005 Kcal∙mol−1,其余参数均采用系统默认值 [3] [4]。能量最小化使得化合物都处于能量最低的稳定状态。
本文对Abdizadeh [5] 组报道的21种以香豆素为基础的苯扎酰胺类新一类抗癌药物和HDACs抑制剂进行了QSAR研究,研究其降低半数感染细胞有效浓度(IC50单位mol∙L−1)。为了计算方便,把这些IC50值转换为相应的pIC50(-lg IC50)值用来表征其生物活性。21种以香豆素为基础的苯扎酰胺类HDAC抑制剂化学结构与活性数据见表1。将数据集随机分为两个部分,分别为用于生成QSAR模型的训练集(14个化合物,占2/3)和用于模型外部验证的测试集(7个化合物,占1/3)。

Table 1. Compounds structure and activity value
表1. 化合物的结构及pIC50值
*为测试集。
2.2. 三维定量构效关系
定量构效关系(Quantitative Structure-Activity Relationship, QSAR)是一种探索分子结构与其生物活性关系的常用研究手段。通常使用数学模型来研究分子结构和分子生物活性之间的定量关系,以分子结构参数为自变量,分子生物活性为因变量来建立出活性与结构之间的定量关系表达式 [6]。三维定量构效关系(3D-Quantitative Structure-Activity Relationship, 3D-QSAR)是根据化合物和生物大分子的三维结构进行QSAR的研究。与传统的QSAR方法相比,3D-QSAR增添了生物活性分子的三维构象的性质,在QSAR中引入了与生物活性分子三维结构信息有关的参数作为变元,因此能更加精确地反映生物活性分子与受体作用的真实情况,更深刻地说明药物与受体之间的相互作用机制,为新药的研发设计提供一定的理论基础。
2.3. 比较分子力场分析法
比较分子力场(Comparative molecular field analysis, CoMFA)是研究药物–受体三维定量构效关系(3D-QSAR)最常用的方法之一。CoMFA能够充分考虑分子的三维结构信息,通过分子结构表征立体场和静电场,再用偏最小二乘法(Partial least squares method, PLS)获取这些三维特征信息与化合物活性之间的联系 [7]。通过构建分子结构,获取分子的最低能量构象,选择公共骨架进行分子叠合,根据探针原子计算化合物周围的分子力场,分析药物结构与活性的关系。其中,分子叠合结果对模型预测的精准性有很大的影响 [8]。因此要想获取更精确的结果往往需要尝试多种方式进行分子叠合,并且需要花费大量的时间。
2.4. 易位体比较分子力场
易位体比较分子力场(Topomer CoMFA)是在2004年,由Cramer等人 [9] 提出,是一种可以预测化合物生物活性或性质的新型3D-QSAR方法。不同于CoMFA,Topomer CoMFA是同时结合了Topomer与CoMFA技术。它弥补了传统CoMFA方法中的手动叠合分子的缺点,它结合了3D-QSAR构建技术和基于配体的虚拟筛选技术,并且可在短时间内构建较为可靠的3D-QSAR模型,具有便捷、快速、准确的特点 [10]。另外,Topamer CoMFA还具有重复性高的优势。利用Topomer CoMFA可快速建立预测模型并进行分析与评价,为同类小分子抑制剂的结构优化提供理论依据 [11]。
2.5. 虚拟筛选
本文中的分子设计采用的是虚拟筛选的方法,使用SYBYL2.0-X中Topomer Search技术进行分子虚拟筛选。此工具是在数据库中基于R基团的搜索技术进行搜索,可以找到训练集中没有出现过的更高活性的化学结构片段,进而将找到的高贡献值的片段通过重新排列组合的方式设计出更高活性的化合物 [12]。本文中,将具有最高活性的11号化合物作为模板分子进行切割以获得分子的结构片段,用获得的模型预测其对活性的贡献值大小 [13]。采用Topomer search技术在ZINC数据库中对基于R基团的虚拟筛选,用距离作为打分的依据,去评判搜索化合物小片段与数据库化合物片段之间的相似性 [14]。Topomer的最大距离设置为185,在数据库中筛选出来的化合物片段Topomer距离要尽量接近185,且片段的贡献值超过模板分子R基团的贡献值,筛选出来符合条件的片段,最后将筛选出来的小分子片段重新进行组合排列去替换模板分子中相应的R基团片段进行分子设计 [15]。
2.6. 分子对接
分子对接(molecular docking)是通过受体的特征以及受体和药物分子之间的相互作用方式来进行药物设计的方法。其主要研究化合物与所选蛋白质活性位点残基之间的相互作用,比较它们之间的亲和性,并根据停靠能量最低来评价最佳构象。通过小分子配体和受体之间的几何、能量匹配与识别,预测其结合模式和亲合力,找到两者之间最佳的结合模式 [16]。是计算机辅助药物研究领域的一项重要技术。
本文中采用的是SYBYL-X2.0 软件中的Surflex-Dock模块进行分子对接。本次对接所使用的蛋白酶晶体来源于PDB数据库(Protein Data Bank),晶体结构的ID为:3JSX。在进行分子对接前对3JSX大分子蛋白进行预处理,将需要的小分子配体从大分子复合物中提取出来,不需要的小分子配体及所有的水分子删除,给蛋白加氢、添加Castelger电荷。根据小分子配体确定对接的活性位点,设置对接格子的格点为40 × 40 × 40,格点间距为0.375Å。然后采用拉马克遗传算法对分子对接的配体构象进行搜索,每个配体分子进行50次独立对接操作,能量评估的最大值设置250万。
先将活性最高的11号化合物作为模板与蛋白晶体进行对接,之后再将设计的分子全部与之对接,最后观察所有化合物的打分函数Total-Score、Crash和Polar的值 [17] [18]。Total-Score 的打分函数值表示从大分子蛋白中提取出来的小分子配体与受体的亲和力,其值越高亲和力越高;Crash表示从大分子蛋白中提取出来的配体与受体之间的不适度,其值越接近零越好。Polar为极性函数得分,当结合位点在分子表面时,其值越大越好;在分子内部时,其值越小越好 [19]。
2.7. Topomer CoMFA模型的建立
不同于CoMFA和CoMSIA,Topomer CoMFA不需要采用基于骨架的三维结构叠合。通常Topomer CoMFA的操作流程大致可分为以下两个步骤:
第一、根据训练集分子结构片段建立Topomer的三维模型。通过化合物的结构特点确认化合物结构的断裂方式,Topomer CoMFA可以自动识别结构片段的化学结构和电荷排布,将整个配体分子切割成两个或两个以上的小片段,没有被切割的化合物可以进行手动切割,然后小片段会自行调整,自动生成Topomer模型和小片段的三维构像。
第二、采用偏最小二乘法分析(PLS) [20]。先通过抽一法(Leave-One-Out)进行交叉验证的CoMFA分析,形成一个预测模型。在生成模型的过程中,CoMFA方法可处理大量数据客观分析和自动匹配相应的化合物结构,以使Topomer CoMFA形成的预测模型更有效 [11]。然后根据交叉验证得到的最佳主成分数,进行非交叉验证回归,建立回归模型表达式。
采用LOO交叉验证方法作为内部验证,通过公式计算得到交叉验证系数q2,非交叉验证系数r2。相关公式与说明如下 [21] [22] [23] [24]:
式中
和
分别为预测值和实验值,
和
分别为训练集的实验值和预测值的平均活性值。
是预测残差平方和(PRESS)。
良好的内部验证只能说明化合物训练集的q2较高,但并不表明所建立模型的预测能力较高,因此外部验证是必要的,通过计算测试集的化合物的生物活性来进一步验证3D-QSAR模型的预测能力 [22]。在交叉验证之后,通常采用常规相关系数r2进行非交叉验证分析,计算出的估计标准误差(SEE)和F值。
通常,交叉验证系数q2越大,相关系数越大,SEE越小,表示相关性越好,方程的预测能力越强,q2的标准误差(q2 stderr)和r2的标准误差(r2 stderr)越小说明模型质量越高 [11]。通常,当交叉验证系数q2大于0.5且非交叉验证系数r2大于0.6时,说明由该方法所建立的模型是比较理想的Topomer CoMFA模型 [25]。
2.8. 分子片段划分
以活性最高的化合物11作为模板,将所有化合物按照图1所示的分割方式分割,将分子划分为三个部分,未自动分割的分子进行手动切割,切割后的化合物被自动划分为R1 (红色)、R2 (蓝色)和公共骨架(绿色)。

Figure 1. Template molecule segmentation mode (left) and fragment schematic diagram (right)
图1. 模板分子分割方式(左)和片段示意图(右)
3. 结果与讨论
3.1. Topomer CoMFA模型分析
采用Topomer CoMFA方法分别对以香豆素为基础的苯扎酰胺类化合物作为HDAC抑制剂的21种化合物的HCT116细胞系和A2780细胞系研究结果如下:
Table 2. Results of Topomer CoMFA model
表2. Topomer CoMFA模型结果
如表2所示,Topomer CoMFA模型HCT116细胞体系的统计学参数为最佳主成分值为3,交叉验证相关系数q2为0.517,非交叉验证系数r2为0.880;A2780细胞体系的统计学参数为最佳主成分值为2,交叉验证相关系数q2为0.572,非交叉验证系数r2为0.869。数据显示,两者的q2大于0.5,r2大于0.6,说明通过该方法建立的模型是比较理想的Topomer CoMFA模型,其统计结果具有很高的预测能力。同时预测值的SEE为0.039,q2 stderr为0.36,r2 stderr为0.18,进一步说明所建立的模型具有较高的可靠性。
图2和图3分别是HCT116细胞体系与A2780细胞体系的训练集和测试集化合物的实验值与预测值之间的线性回归相关图和柱状分析图。从图中可以看出,线性回归图中所有样本均匀地分布在45°线附近,柱状分析图中所有样本基本在同一水平高度。由图可知两种细胞系构建的QSAR模型是比较可靠的,具有较好的拟合力。
3.2. 3D-QSAR分析
图4是以活性最高的11号分子为模板建立的Topomer CoMFA模型的分子场三维等势图,根据切割方式,所有化合物的R1基团都是一样的,所以无法对R1基团进行比较分析,因此本文只对两种细胞系的R2基团进行比较分析。
图4(a)与图(b)分别为HCT116细胞系R2基团的立体场等势图和静电场等势图;A2780细胞系R2基团的立体场等势图和静电场等势图为图4(c)与(d)。在图4(a)和(c)中,绿色的部分表示提高取代基的体积可以增加活性;黄色的部分则相反,减少取代基的体积可以提高化合物活性;在图4(b)和(d)中,红色部分表示引入带负电子的基团可以提高化合物活性,而蓝色部分则表示引入带正电子的基团可以提高化合物活性
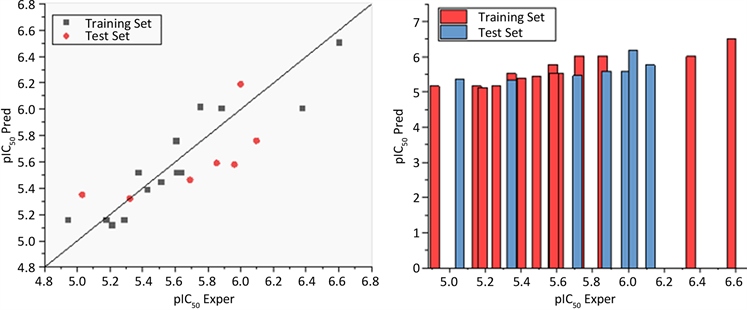
Figure 2. Linear regression diagram (left) and histogram diagram (right) of HCT116 cell system
图2. HCT116细胞体系的线性回归图(左)和柱状分析图(右)

Figure3. Linear regression diagram (left) and column analysis diagram (right) of A2780 cell system
图3. A2780细胞体系的线性回归图(左)和柱状分析图(右)
对于HCT116细胞系,在图4(a)中的R3、R4和R5取代基的位置上有绿色区域,若在此区域有大面积基团则活性相对较高。例如,化合物10、11、16、17、18和21 (pIC50值分别为5.88、6.60、5.75、6.38、6.00和6.10)相比化合物8 (pIC50值为5.60)都是增加了取代基的体积(分别用−C、−C、−Cl、−Br、−CO和−Cl取代了−H),体积增大,活性明显升高;同理,化合物R6位置有大面积黄色区域,需要在此位置引入小基团的基团以提高化合物的活性。例如,化合物12和15 (pIC50值分别为5.03和5.32)与化合8 (pIC50值为5.60)相比,在R6取代基的位置上用−F和−Cl取代了−H活性降低。
从图4(b)中可以看出R4、R5和R6取代基上有蓝色区域,因此在这些基团上减小取代基的电负性或者引入带正电荷的基团有利于提高活性。例如,R6取代基的位置上化合8、9和15 (pIC50值分别为5.60、5.85和5.32)分别在用−H、−C和−Cl取代化合物12 (pIC50值为5.030)的−F,电负性增强,活性明显升高;在R4取代基的位置上化合物14 (pIC50值为5.61)用-F取代化合物11、16、17和18 (pIC50值分别为6.60、5.75、6.38、6.00和6.10)相同位置上的−C、−Cl、−Br和−CO基团活性明显降低。另外,在图4(b)中R1、R2取代基的位置被红色区域包围,因此需要增加这些基团上取代基的电负性来提高活性。例如,化合物4、5和7 (pIC50值分别为5.51、5.64、和5.18)相比化合物1 (pIC50值为4.94)都是在取代基上引入了高电负性的基团,活性都有所提高。
同理,对于A2780细胞系也是如此。
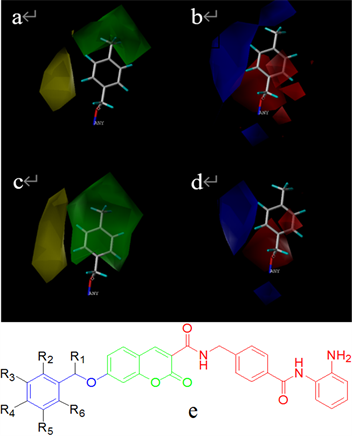
Figure 4. 3D equipotential diagram of Topomer CoMFA model based on template 11
图4. 基于模板11的Topomer CoMFA模型的三维等势图
因此,要想提高化合物的活性应该优先增大整个取代基的体积尤其是R4基团取代基的体积,并且减少R6基团取代基的体积。同时在R4、R5和R6取代基上减小取代基的电负性或者引入带正电荷的基团,在苯环的内、外侧和R1基团上增加取代基的电负性或者引入带负电荷的基团。
3.3. 分子设计结果
本文的分子设计是以3D-QSAR分析作为参考,基于化合物11的R2基团,采用Topomer Search技术在ZINC数据库中的Drug-like分子库中进行搜索 [25],以R2基团作为提问结构,在化合物数据库中进行筛选。在筛选出的这些分子结构片段中挑选出活性贡献值高于11号模板分子的结构片段作为新的R2基团。本文中所建立的Topomer CoMFA模型中HCT116细胞系的R2基团贡献值为1.36,挑选的时候选择Topomer距离越接近185且片段的贡献值超过模板分子R2基团的贡献值的R基团。选择出符合条件的16个集团,最终设计出16个新的化合物。按照训练集分子的构建方法,在SYBYL-X 2.0中构建出新分子的三维结构,然后优化、命名,用化合物11作为模板预测其活性,16个新设计的化合物结构和预测活性值(pIC50)见表3。
Table 3. Molecular design and predicted activity values of HCT116 cell lines and A2780 cell lines
表3. HCT116细胞系与A2780细胞系的分子设计和预测活性值
如表3所示,新设计出的16个分子的预测活性值(pIC50)都在6.60以上,比模板分子的活性值高。
3.4. 分子对接结果分析
将模板分子和新设计出的16个分子都进行分子对接,模板分子作为对照,以打分函数Total-Score、Crash和Polar的值作为评判标准,结果如表4所示。
Table 4. Molecular docking scoring function and number of hydrogen bonds
表4. 分子对接打分函数和氢键个数
对接分析图见图5,其中棒状化合物代表分子配体,球棍状化合物代表氨基酸残基,黄色虚线为氢键。从图5(a)可以看出,11号模板分子配体与3JSX蛋白晶体结构中的主要氨基酸残基共形成了3个氢键,与Tyr128、Tyr126、和Trp105形成氢键相互作用,Total-score,crash和polar分别为8.2690,−4.2223,1.7664。
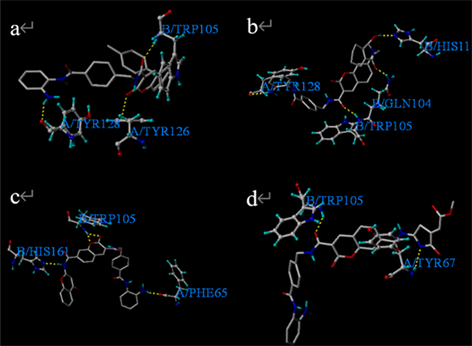
Figure 5. Molecular hydrogen bonding interaction diagram
图5. 分子对接氢键作用图
如图5(b)~(d)分别表示设计的新化合物1-01,1-14,1-02与3JSX蛋白晶体进行分子对接的活性位点,黄线色虚线表示氢键作用力。图5(b)所示,化合物1-01与蛋白酶晶体的His11,Tyr128,Gln104,和Trp105氨基酸残基形成氢键;图5(c)为化合物1-14与3JSX蛋白中的Trp105,His161,和Phe65氨基酸残基形成氢键。结果显示所设计的化合物对接结果是可靠。另外,从表4可以看出,新设计的化合物1-02、1-04和1-07均具有较高的预测活性,但总打分函数相对较低。特别是化合物1-02,在设计的16个分子中化合物1-02的预测活性值(pIC50)最高,但总打分函数最低。因此,新分子的设计不仅需要有较高的活性值,还需要分子与大蛋白之间的空间匹配和能量匹配。
4. 结论
本文利用Topomer CoMFA方法研究了香豆素基苯甲酰胺作为组蛋白去乙酰化酶抑制剂。HCT116细胞系和A2780细胞系的HDAC抑制剂活性进行了研究,建立了稳定且预测能力良好的3D-QSAR模型;结合两种细胞系通过Topomer search进行在ZINC数据库筛选出活性更好的分子片段,设计出了16个比模板分子活性更高的新化合物;最后,使用Surflex-dock方法将模板分子和新设计的分子与3JSX蛋白进行对接,对抑制剂和受体蛋白之间的结合机制进行研究。并最终在新设计的化合物中确定了13个新化合物,但这些化合物是否可以应用于实践中,还有待进一步的研究。本研究对于理解香豆素基苯甲酰胺作为组蛋白去乙酰化酶抑制剂的生物活性,寻找新型抗癌药物具有一定的指导作用。
基金项目
本课题得到国家自然科学基金(21475081)、陕西省自然科学基金(2019JM -237)、陕西科技大学研究生创新基金的资助。