





Abstract
By the action of chlorinating agent in the presence of alcohol, α-alkoxybenzocyclobutenone oximes are converted to stable benzonitrile oxides with an o-acetal moiety via facile ring-opening reaction.
In our studies on complex natural product synthesis,1 we have exploited stable benzonitrile oxide I2 as a versatile building block for assembling polycyclic structures (Figure 1). The nitrile oxide moiety in I is kinetically stabilized by two ortho substituents, but reactive enough for the 1,3-dipolar cycloaddition3 with cyclic enones, and the acetal moiety serves as a progenitor to an aldehyde for the stereoselective benzoin cyclizations (Figure 1, A).4
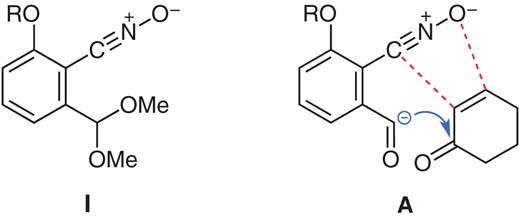
Stable benzonitrile oxide I.
We became interested in developing an alternative approach to nitrile oxide I from benzocyclobutenone oxime II,5 which is available from the [2+2] cycloaddition of benzyne6 and oximation (Scheme 1). Our expectation was the following. Given oxime II was chlorinated,7 the resulting α-chloronitroso compound IV would undergo a ring opening, thanks to the donor/acceptor effect as shown, thereby releasing the ring strain. The zwitterion V, thus generated, would undergo elimination of a chloride to generate nitrile oxide III. If the reaction was conducted in methanol, trapping of the oxonium species would give acetal I.8 Given the case, an additional hope was that an access to mixed acetals by employing propargyl alcohols as nucleophilic reaction partner, allowing a rapid molecular assembly by the intramolecular 1,3-dipolar cycloadditions of adduct VI.
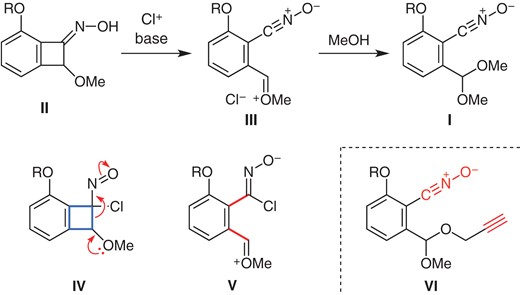
Access to stable benzonitrile oxide I.
In this communication, we report realization of this scenario, enabling a unique access to benzonitrile oxides I, and its application to tandem trapping and intramolecular cycloaddition reactions.
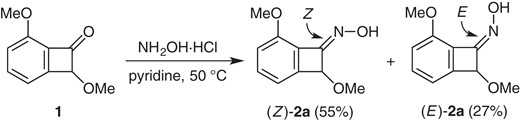
Equation 1 shows preparation of model substrates: The readily available benzocyclobutenone 16 was subjected to oxime formation. Chromatographic separation (SiO2) gave Z-oxime (Z)-2a and E-oxime (E)-2a in 55% yield and 27% yield, respectively.9
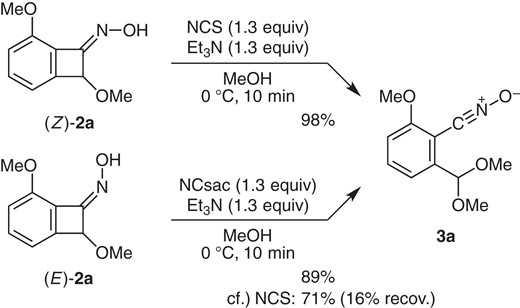
As the initial trial, oxime (Z)-2a was treated with various oxidants (Scheme 2). Among others,10 NCS gave the desired product 3a in excellent yield: When oxime (Z)-2a was treated with NCS (1.3 equiv) and Et3N (1.3 equiv) in MeOH (0 °C, 10 min), the starting material was smoothly consumed, giving nitrile oxide 3a in 98% yield.11 On the other hand, the isomeric oxime (E)-2a was less reactive, and under the same conditions as above, nitrile oxide 3a was obtained in 71% yield and 16% recovery of (E)-2a. However, use of N-chlorosaccharin (NCsac),12 a stronger chlorinating agent, enabled full conversion of (E)-2a to give 3a in 89% yield.
Oxidative ring opening.
Having found suitable conditions, application to several oxime substrates was examined with attention to the reactivity difference depending upon substituent(s) of the aromatic ring (Table 1).13
Substrate scopea
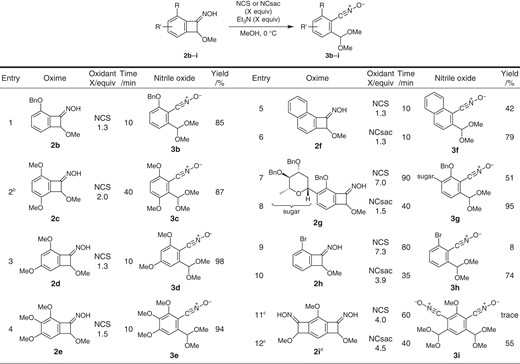
a0.3 mmol scale. bMeOH and CH2Cl2 (v/v = 2:1). cMeOH and CH2Cl2 (v/v = 1:1). dAn inseparable mixture of four stereo/geometrical isomers (1:1:1:1).
Substrate scopea
a0.3 mmol scale. bMeOH and CH2Cl2 (v/v = 2:1). cMeOH and CH2Cl2 (v/v = 1:1). dAn inseparable mixture of four stereo/geometrical isomers (1:1:1:1).
Entries 1–4 show the reactions of oximes 2b–2e having alkoxy group(s) on the benzene ring. Benzyloxy-substituted oxime 2b was cleanly converted by NCS (Et3N, MeOH, 0 °C, 10 min) to nitrile oxide 3b in 85% yield (Entry 1). We had a prior concern that aromatic chlorination may compete for the reactions of 2c–2e with an electron-rich aromatic ring. However, clean reactions occurred in all cases to give nitrile oxides 3c–3e in excellent yields (Entries 2–4). While NCS was not effective for the reactions of oximes 2f–2i, giving low yields of the corresponding nitrile oxides 3f–3i, the reactions were achieved by using NCsac (Entries 5–12). Entries 5–8 show the reactions of oximes 2f having a naphthalene ring and 2g having a sugar moiety, for which NCsac worked nicely to give nitrile oxides 3f and 3g in excellent yields (Entries 6 and 8). Entries 9 and 10 show the reactions of oxime 2h with an electron-deficient aromatic ring bearing an ortho-bromo group: The yield of 3h was poor when NCS was employed (8%, Entry 9), whereas NCsac gave much better yield (74%, Entry 10). Furthermore, the dual reactions of dioxime 2i were only possible with NCsac to give bis-nitrile oxide 3i (Entry 12).
As a potential application of the process, we examined the reaction in the presence of propargyl alcohol (4a), rather than methanol (Scheme 3). The hope was that the expected primary adduct 5 would undergo an internal 1,3-dipolar cycloaddition to give isoxazole 6a.
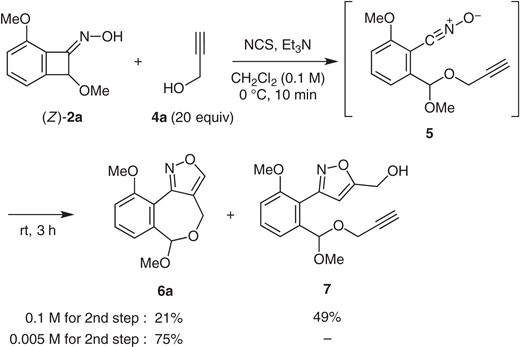
Tandem trapping/cycloaddition reaction.
As a preliminary attempt, oxime (Z)-2a was treated with NCS (1.3 equiv) and Et3N (1.3 equiv) in the presence of 4a (20 equiv).14 The reaction (0.1 M in CH2Cl2, 0 °C, 10 min, then room temp., 3 h) gave isoxazoles 6a (21%) and 7 (49%), derived respectively from the intra- and intermolecular cycloadditions.
Formation of 7 suggested that the intramolecular 1,3-dipolar cycloaddition within the mixed acetal 5 is not dramatically fast, so that the intermolecular reaction with the unreacted 4a competes.
To suppress this intermolecular reaction, a dilution protocol was devised. A mixture of oxime (Z)-2a and propargyl alcohol (4a) (20 equiv) was treated with NCS (1.3 equiv) and Et3N (1.3 equiv) in 0.1 M CH2Cl2 (0 °C). Formation of 5 was suggested by TLC monitoring. After a complete conversion of (Z)-2a (TLC, 10 min), the reaction was diluted with CH2Cl2 to 0.005 M, by which a clean conversion (room temp., 3 h) occurred to give the desired isoxazole 6a in 75% yield. None of the product 7 was identified.
Scheme 4 shows the reaction of oxime (Z)-2a and propargyl alcohol 4b (20 equiv) in the coexistence of phenylacetylene (20 equiv) by using the same protocol as above. It turned out that isoxazole 6b was obtained in 75% yield, and the unreacted phenylacetylene was recovered. Note that terminal alkynes are normally more reactive dipolarophiles in comparison with internal alkynes. Indeed, a reversed chemoselectivity was observed in the reaction of nitrile oxide 3a with 4b and phenylacetylene, that is, exclusive formation of isoxazole 8 (91% yield).
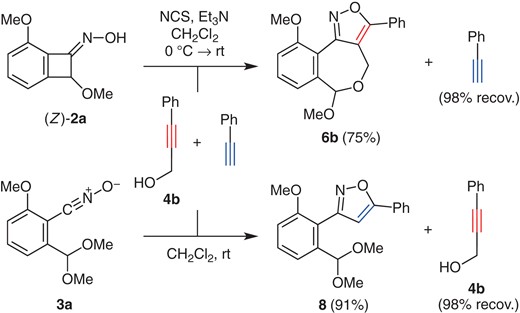
Chemoselective 1,3-dipolar cycloaddition.
In conclusion, an oxidative ring opening of benzocyclobutenone oximes allows facile access to stable benzonitrile oxides. The highly functionalized 1,3-dipole would be useful for complex polycyclic natural product syntheses. Further work along these lines is in progress.
Acknowledgment
This work was supported by JSPS KAKENHI (Grant Nos. JP23000006, JP16H06351, and JP26750363).
Supporting Information is available on http://dx.doi.org/10.1246/cl.170328.
References and Notes
a)
b)
c)
d)
a)
a)
b)
c)
b)
c)
b)
c)
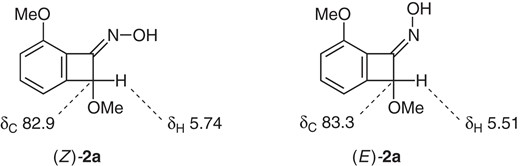
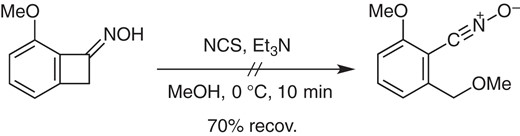
The electron donation from the α-methoxy group is crucial, as suggested by the unsuccessful reaction of an oxime without the methoxy group.
a)
b)
For oximes 2b–2d, 2f, and 2h, the respective Z-isomers were employed. Oximes 2e and 2g were obtained as virtually single isomers, whose E/Z geometries unassigned. Oxime 2i was obtained as an inseparable mixture of four stereo/geometrical isomers (1:1:1:1), which was, without separation, subjected to the reaction. See the Supporting Information.

K. Suzuki

H. Takikawa

S. Sato

R. Seki

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}