





Abstract
Androgen signaling, in particular overexpression of the androgen receptor (AR), is critical for the growth and progression of prostate cancer. Because the AR is amenable to targeting by small-molecule inhibitors, it remains the major druggable target for the advanced disease. Inflammation has also been implicated in the cancerous growth in the prostate. Here we show that 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), an endogenously produced antiinflammatory prostaglandin, targets the AR and acts as a potent AR inhibitor, rapidly repressing AR target genes, such as FKBP51 and TMPRSS2 in prostate cancer cells. However, exposure of prostate cancer cells to 15d-PGJ2 does not simply evoke a general inhibition of nuclear receptor activity or transcription because under the same conditions, peroxisome proliferator-activated receptor-γ is activated by 15d-PGJ2. Moreover, 15d-PGJ2 rapidly triggers modifications of AR by small ubiquitin-related modifier-2/3 (SUMO-2/3), which may modulate the repressing effect of 15d-PGJ2 on AR-dependent transcription. Chromatin immunoprecipitation assays indicate that the inhibitory effect of 15d-PGJ2 on FKBP51 and TMPRSS2 expression occurs in parallel with the inhibition of the AR binding to the regulatory regions of these genes. However, the DNA-binding activity is not the only AR function targeted by 15d-PGJ2 because the prostaglandin also blunted the androgen-dependent interaction between the AR amino and carboxy termini. In conclusion, our results identify 15d-PGJ2 as a potent and direct inhibitor of androgen signaling, suggesting novel possibilities in restricting the AR activity in prostate cancer cells.
Carcinogenesis is generally linked to chronic inflammation via increased production of reactive oxygen species (ROS) contributing to cancer initiation and progression (1). Elevated levels of ROS combined with defective detoxification processes lead to an imbalance in cellular redox state, causing DNA damage and misfolding of proteins (1). Oxidative stress also influences posttranslational modifications of crucial regulator proteins, such as transcription factors (TFs). In particular, there is often increased accumulation of small ubiquitin-related modifier (SUMO) conjugates (SUMOylation) in TFs as a consequence of cellular stress, including oxidative stress (2).
Prostate cancer is the most common malignancy of men in the Western countries. Androgen receptor (AR) activity is essential for prostate cancer development, growth, and progression (3, 4). Despite progress in unraveling the molecular etiology of prostate cancer, AR remains the major druggable target for the advanced disease (5). The proliferation of prostate cancer cells can initially be restricted by androgen deprivation therapy. However, for reasons that are still elusive, androgen deprivation therapy eventually fails and the cancer reverts to a castration-resistant prostate cancer (6, 7). Despite this apparent androgen independence, the AR remains a critical factor influencing the growth and survival of the majority of the castration-resistant prostate cancer tumors. Up to 50% patients with metastatic disease have an AR gene amplification, resulting in overexpression of the AR protein (8–10). The metastases often occur in bones. No durable treatment has been identified to combat this stage of disease.
The AR is a male sex steroid-activated TF, a member of nuclear receptor superfamily, that regulates gene expression in response to androgen exposure (11, 12). In addition to the hormone-occupied receptor, its cognate DNA-binding sites [androgen-responsive elements (AREs)] and RNA polymerase II, formation of an active AR transcription complex requires several AR-interacting coregulator proteins (13). Currently the clinically available antiandrogens, such as bicalutamide, act as competitive inhibitors for natural androgens, without supporting the formation of productive AR transcription complexes. Unfortunately, overexpression of AR may convert the bicalutamide (that in normal cells acts as a pure antagonist) into an agonist (14). The functions of many coregulator proteins are linked to posttranslational modifications, such as acetylation, methylation, or phosphorylation, which often target nucleosomal histones. These posttranslational modifications also directly modify the structure and the activity of AR (15, 16). The AR is also modified and regulated by SUMOylation (17, 18). Cyclopentenone prostaglandins, such as 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), are physiologically endogenous electrophilic compounds that are derived from the cyclooxygenase pathway (19). In addition, 15d-PGJ2-like compounds (deoxy-J2-like isoprostanes) can be formed nonenzymatically under oxidative conditions (20). 15d-PGJ2 is well characterized as a potent activator of the antiinflammatory and cytoprotective Keap1-nuclear factor erythroid 2-related factor 2 (Nrf2) system. Interestingly, it is also an endogenous ligand for the nuclear receptor peroxisome proliferator-activated receptor (PPAR)-γ (21–23). Many of the biological actions of 15d-PGJ2 are attributable to its ability to form covalent adducts with cysteine residues in target proteins (24–26). For example, 15d-PGJ2 covalently modifies estrogen receptor (ER)-α, inhibiting its activity (27). Because 15d-PGJ2 and related electrophilic signaling intermediates are formed during inflammation and other conditions associated with oxidative stress and the activity of SUMOylation pathway is regulated by a variety of stress conditions including oxidative and electrophilic stress (28, 29), we have investigated the effect 15d-PGJ2 on the activity of AR in prostate cancer cells. Our results show that 15d-PGJ2 acts as a potent and direct inhibitor of AR, providing a novel lead for targeting of the AR in prostate cancer cells.
Materials and Methods
Cell culture
Vertebral cancer of the prostate (VCaP) and COS-1 cells were from American Type Culture Collection (Manassas, VA), and maintained as previously described (18, 30). PC-3 human prostate cancer cells stably expressing wild-type AR (wtAR)- and SUMOylation-deficient ARK386,520R were generated by transfecting pcDNA-Flag-hAR or pcDNA-Flag-hARK386,520R (17) into PC-3 cells (from American Type Culture Collection) using TransIT-LT1-transfection reagent (Mirus Bio Corp., Madison, WI). Cells were grown in F-12 with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin under G418 selection (400 μg/ml), and G418-resistant single foci were isolated and expanded for analyses. Representative clones for further analyses were chosen on the basis of comparable AR levels as confirmed by Western blotting and quantitative real-time RT-PCR (RT-qPCR). In the experiments, the cells were grown in F-12 with 5% charcoal-stripped FBS.
Chemicals
15d-PGJ2-Δ12,14-prostaglandin J2 (18570), its biotinylated form (10141), and its biologically inactive analog CAY10410 (18590) were purchased from Cayman Chemicals (Ann Arbor, MI). The following AR ligands were used: R1881 (PerkinElmer Inc., Waltham, MA), 5α-dihydrotestosterone (Steraloids Inc., Newport, RI), and bicalutamide (Bidragon Pharmservice LCC, Burlingame, CA).
Immunoblotting and immunoprecipitation
VCaP cells seeded on six-well plates (800,000 cells/well) were grown as above and protein samples were prepared as described (31) and analyzed by immunoblotting with anti-AR (32), anti-PPARγ (sc-7196), anti-SUMO-1 (anti-GMP1; Invitrogen, Carlsbad, CA; 33–2400), anti-SUMO-2/-3 (MBL, M114-3; Woburn, MA) and antiubiquitin (Santa-Cruz Biotechnology, Santa Cruz, CA; sc-8017) antibodies. Horseradish peroxidase-conjugated secondary antibodies were from Invitrogen and chemiluminescence detection reagent from Pierce (Rockford, IL). For immunoprecipitations, VCaP cells were grown on 10-cm dishes and treated with or without R1881 for 2 h and with or without 10 μm 15d-PGJ2 for 1 h. Cells were harvested and processed as described elsewhere (31).
Biotin pull-down assay
VCaP cells were treated with androgen and 15d-PGJ2 or biotinylated 15d-PGJ2 as described above for the immunoprecipitation assays except that biotin bound proteins were affinity isolated with Streptavidin Plus UltraLink resin (Pierce). Bound complexes were analyzed by immunoblotting with anti-AR and anti-PPARγ antibodies. For in vitro analyses, AR with amino terminal 3× Flag tag was stably expressed in human embryonic kidney-293 cells (Flp-In system; Invitrogen) and immunoaffinity purified with EZview Red ANTI-FLAG M2 affinity gel (Sigma-Aldrich, St. Louis, MO) according to the manufacturer's instructions. The purified protein was eluted with 150 ng/μl 3× Flag peptide (Sigma-Aldrich) in Tris-buffered saline buffer containing 1:100 of protease inhibitor mixture (Sigma-Aldrich), 0.1 mm dithiothreitol and 10 nm R1881 and supplemented with 20% (vol/vol) glycerol. Silver staining of the 3× Flag-AR preparation showed that the preparation was essentially free of non-AR proteins. Purified AR (0.1 μm) was incubated for 2 h at 22 C with biotinylated 15d-PGJ2 (10 μm) in the presence or absence of 15d-PGJ2 (1 mm), and a reaction with 10 μm 15d-PGJ2 acted as an additional control. Reaction mixtures were subsequently diluted with a buffer containing 140 mm NaCl, 10 mm Tris-HCl (pH 7.5), 10% (vol/vol) glycerol, 0.5% Triton X-100, 1:50 of the protease inhibitor mixture, and 10 nm R1881. Proteins A and G agarose beads (Millipore, Bedford, MA) were used for preclearing and streptavidin beads from Pierce to pull down biotin-bound AR. Samples were analyzed by immunoblotting with anti-AR antibody.
Isolation of RNA and RT-qPCR
VCaP cells seeded on 12-well plates (300,000/well) were grown for 2 d, and medium containing 2.5% charcoal-stripped FBS in DMEM was changed on cells for 2 d before hormone treatments. Alternatively, serum-free medium (DMEM) was changed on the cells for the treatments as indicated. Total RNA was extracted using TriReagent (Invitrogen) and converted to cDNA using Transcriptor first-strand cDNA synthesis kit (Roche, Mannheim, Germany) according to the manufacturers' instructions. cDNA was used as a template in RT-qPCR carried out in an MX3000P real-time PCR System (Stratagene, La Jolla, CA) with Fast Start SYBR Green Master (Roche) and specific primers for FKBP51, TMPRSS2, CDKN1A, and GAPDH (primer sequences available upon request).
Reporter gene assays
COS-1 cells on 12-well plates were transfected using TransIT-LT1-transfection reagent (Mirus Bio Corp.) as described (18). The following constructs were used as reported elsewhere (18): pARE2-TATA-luc, pPB-luc, pSG5-hARΔLBD, pcDNA-Flag-hAR, and pcDNA-Flag-hARK386,520R. pM-LBD and pVP16-NTD (both 200 ng/well) have been described elsewhere (33) and pSG5-glucocorticoid receptor-interacting protein 1 (GRIP1; 200 ng/well) as reported elsewhere (34). pG5-luc (200 ng/well) and the pCMVβ (20 ng/well) were from Promega (Madison, WI) and CLONTECH (Palo Alto, CA), respectively. Cells were lysed in passive lysis buffer (Promega), and luciferase (luc) and β-galactosidase activities were measured as reported elsewhere (18).
Confocal microscopy
VCaP cells seeded onto glass coverslips in 24-well plates (160,000 cells/well) were grown and exposed to androgen and 15d-PGJ2 as indicated. Cells were fixed, permeabilized, and immunostained using anti-AR antibody and fluorescein isothiocyanate-labeled secondary antibody (Jackson ImmunoResearch, West Grove, PA). Confocal images were visualized in a Zeiss LSM700 laser scanning microscope (Jena, Germany).
Electrophoretic mobility shift assay
One hour before extraction, VCaP cells were exposed to R1881 (10 nm) in the presence of indicated concentrations of 15d-PGJ2 and extracted in 20 mm Tris-HCl (pH 7.5), 400 mm KCl, 15% (vol/vol) glycerol, 1 mm EDTA, 2 mm dithiothreitol, 10 nm R1881, and 1:100 diluted protease inhibitor cocktail (Sigma-Aldrich) and analyzed as described elsewhere (34, 35). Twelve micrograms of cell extract with 32P-labeled ds-oligomer corresponding to the intronic ARE of rat prostatein peptide C3(1) gene were used. The complexes were separated on 4% nondenaturing PAGE. The gels were dried, and a phosphoimager (FLA3000; Fuji, Tokyo, Japan) was used for visualization and quantification of the intensities of AR-ARE complexes.
Chromatin immunoprecipitation (ChIP)
VCaP cells were seeded at approximately 70% confluence onto 75-cm2 bottles and allowed to grow in steroid-depleted transfection medium for 72 h before ChIP. They were then exposed to vehicle (ethanol) or R1881 in the presence and absence of 15d-PGJ2 for 1 h and processed as described (30, 36). Quantitative PCR analyses were carried out with FastStart SYBR Green Master (Roche Diagnostics) and an MX3000P real-time PCR system (Stratagene). The results were calculated using the formula E-(ΔCt) × 10, where E (efficiency of target amplification) is a coefficient of DNA amplification for every one PCR cycle for a particular primer pair and ΔCt is Ct(ChIP-template)-Ct(Input). The results are presented as fold over the value IgG-precipitated sample.
Results
15d-PGJ2 triggers SUMOylation of AR in prostate cancer cells
We have recently reported that several types of cellular stress induce accumulation of AR-SUMO-2/3 conjugates in prostate cancer cells (37). To investigate whether prostaglandin 15d-PGJ2 influences AR signaling, we used VCaP cells that are derived from a bone metastasis of hormone-refractory cancer; these cells express amplified AR levels (36, 38). Interestingly, immunoblotting with anti-AR antibody revealed that even a short-term (1 h) exposure of the cells to low micromolar concentrations of 15d-PGJ2 (2.5–10 μm) resulted in a dose-dependent accumulation of slowly migrating conjugates in the agonist-bound AR. Immunoprecipitation and immunoblotting analyses proved that these conjugates corresponded to SUMO-2/3-modified AR but not SUMO-1 or ubiquitin-modified AR (Fig. 1, A and B). Treatment of wtAR and SUMOylation-deficient AR (ARK386,520R)-expressing PC-3 prostate cancer cell lines with 15d-PGJ2 confirmed that 15d-PGJ2 triggered SUMOylation to the consensus acceptor lysines K386 and K520 of AR (Fig. 1C). The effect of the prostaglandin on SUMOylation of AR appears to be transient because exposure of VCaP cells to 15d-PGJ2 for 8 h decreased the amount of AR-SUMO-2/3 conjugates, without markedly reducing the level of un-SUMOylated AR (Fig. 1D).
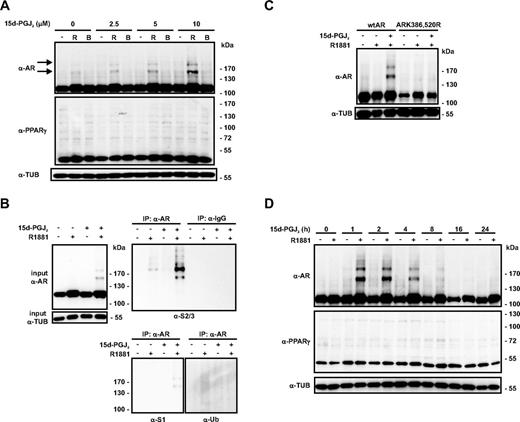
15d-PGJ2 induces formation of endogenous AR-SUMO-2/-3 conjugates in VCaP prostate cancer cells. A, VCaP cells were grown in the presence and absence of R1881 (R; 10 nm) or bicalutamide (B; 10 μm) for 2 h, and 15d-PGJ2 was added at the indicated final concentrations. After 1 h, cells were lysed in a denaturing buffer and the lysates were analyzed by immunoblotting with anti-AR, anti-PPARγ, and anti-α-tubulin (α-TUB) antibodies. The arrows depict the positions of AR-SUMO conjugates. B, VCaP cells were treated with or without R (10 nm) and 15d-PGJ2 (10 μm) as described above. AR was immunoprecipitated with anti-AR antibody, and the precipitates were analyzed by immunoblotting with anti-SUMO-1, anti-SUMO-2/3, and antiubiquitin antibodies. C, PC-3 prostate cancer cells stably expressing wild-type AR (wtAR) or SUMOylation-deficient receptor (ARK386,520R) were exposed to R1881 (R; 10 nm) for 2 h, and 15d-PGJ2 (10 μm final) was added for 1 h as indicated. Lysates were analyzed by immunoblotting with anti-AR and anti-TUB antibodies. D, VCaP cells were treated with 15d-PGJ2 (10 μm) in the presence or absence of androgen (R1881, 10 nm) for the indicated time periods, and immunoblotted with indicated antibodies.
Another electrophile, 9- and 10-nitro-octadec-9-enoic acid, which is also a reactive electrophile and shares some of the signaling features as 15d-PGJ2, e.g. the activation of the Keap1-Nrf2-signaling system (39, 40), had no effect on the SUMOylation of AR (Supplemental Fig. 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). Because 15d-PGJ2 is a ligand for PPARγ and the PPARγ has also been reported to be SUMOylated (41), we examined whether the endogenous PPARγ in VCaP cells undergoes SUMOylation in the same conditions under which the modification of AR can be triggered by the prostaglandin. However, no signs of PPARγ SUMOylation were detectable under these conditions (Fig. 1, A and D). Notably, 15d-PGJ2 did not markedly influence the overall level of SUMO-1, SUMO-2/3, or ubiquitin-conjugated proteins, whereas a more severe cell stress, heat shock (43 C, 1 h), resulted in a massive increase in the amount of SUMO-2/3- and ubiquitin-modified proteins in VCaP cells (Supplemental Fig. 1). Together these results indicate that the effect of 15d-PGJ2 on AR SUMOylation is not due to its general effect on protein SUMOylation in these prostate cancer cells.
15d-PGJ2 binds to AR
Because 15d-PGJ2 has been reported to form covalent adducts with thiol residues in specific signaling proteins (24, 25, 42), we next investigated whether 15d-PGJ2 can bind to the AR. To that end, VCaP cells were grown with or without R1881 for 2 h and treated with or without the biotinylated form of 15d-PGJ2 for 2 h. Cells were harvested, biotinylated proteins were affinity-isolated with streptavidin beads, and the eluates were analyzed by immunoblotting with anti-AR antibody. As shown in Fig. 2A, 15d-PGJ2 is capable of forming complexes with both apo-AR and holo-AR, including its SUMOylated forms. In contrast, we did not observe any PPARγ-15d-PGJ2 adduct formation as assessed in the biotin pull-down assays (Fig. 2B), which is in accordance with previous observations (25). Thus, the ability of 15d-PGJ2 to form protein adducts is not a general, nonspecific feature of the compound in the VCaP prostate cancer cells. Additionally, incubation of purified recombinant AR with biotinylated 15d-PGJ2in vitro evoked the formation of specific biotin-15d-PGJ2-AR adducts (Fig. 2C). These results indicated that 15d-PGJ2 is able to directly bind to the AR.
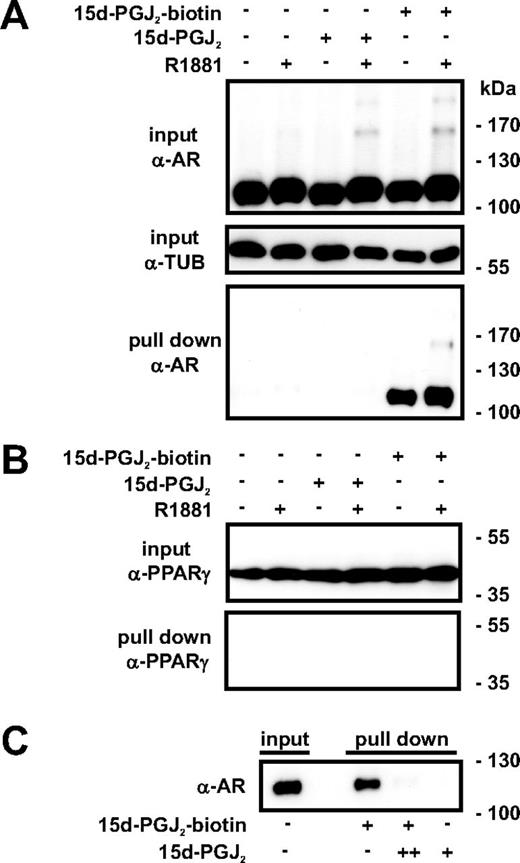
15d-PGJ2 binds to AR in intact cells and in vitro. A, VCaP cells were treated with or without R1881 (10 nm) for 2 h and subsequently exposed to 15d-PGJ2 or biotinylated 15d-PGJ2 (10 μm) for 2 h as indicated. Cells were lysed and the biotin-15d-PGJ2-bound proteins were affinity isolated with streptavidin beads. C, Purified recombinant AR was incubated for 2 h at 22 C with biotin-15d-PGJ2 (10 μm, +) in the presence or absence of nonbiotinylated 15d-PGJ2 (1000 μm, ++) or 15d-PGJ2 (10 μm) alone as indicated, and biotin-bound AR was pulled down with streptavidin beads. Formation of 15d-PGJ2 adducts was analyzed by immunoblotting with anti-AR antibody (A and C) and anti-PPARγ antibody (B).
15d-PGJ2 inhibits AR-dependent transcription
Because 15d-PGJ2 is capable of binding to the AR and triggering SUMOylation of the receptor, we tested its effect on the transcriptional activity of the AR. VCaP cells grown in the presence and absence of R1881 were exposed to increasing concentrations (2.5–10 μm) of 15d-PGJ2 for 6 h. The amounts of the AR target FKBP51 and TMPRSS2 mRNAs were measured by RT-qPCR (Fig. 3A). Interestingly, AR-dependent expression of both genes was dramatically and dose dependently repressed by 15d-PGJ2, with the IC50 value being approximately 2 μm. To rule out the possibility that 15d-PGJ2 exposure was not producing a general inhibition of transcription in VCaP cells, we analyzed the expression of PPARγ target CDKN1A mRNA, which was markedly up-regulated by 10 μm 15d-PGJ2, a concentration that totally repressed the androgen-dependent expression of both FKBP51 and TMPRSS2. Moreover, the inhibitory effect of 15d-PGJ2 on AR-dependent gene expression in VCaP cells was clearly enhanced (IC50 ∼1 μm), when the cells were treated with the prostaglandin in serum-free medium (Fig. 3B). Interestingly, 9,10-dihydro-15d-PGJ2 (CAY10410), a compound structurally similar to 15d-PGJ2, but lacking the cyclopentenone moiety, did not influence the AR activity in VCaP cells, suggesting that the cyclopentenone ring of 15d-PGJ2 had been responsible for the inhibition. We also tested the effect of 15d-PGJ2 in another AR-expressing prostate cancer cell line, LNCaP cells, and observed a similar inhibition of AR activity (data not shown), indicating that the effect of 15d-PGJ2 is not restricted to VCaP cells.
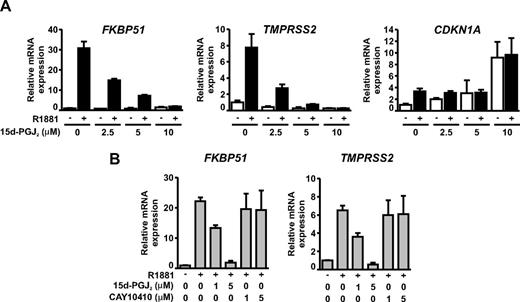
Transcriptional activity of AR is effectively and dose dependently inhibited by 15d-PGJ2 in VCaP prostate cancer cells. A, VCaP cells were treated with or without R1881 (10 nm) and increasing concentrations of 15d-PGJ2 for 6 h. Expression of endogenous AR target genes FKBP51 and TMPRSS2 and a PPARγ target gene CDKN1A were analyzed by RT-qPCR. The CDKN1A is also induced by R1881, but the androgen induction (compared with vehicle treated cells) is blunted as concentration of 15d-PGJ2 reaches the concentration activating PPARγ. Results were normalized to the amount of GAPDH mRNA. B, VCaP cells were treated as above, but serum was omitted from the growth medium and the effect of indicated concentrations of CAY10410 (a biologically inactive analog of 15d-PGJ2) on androgen-induced accumulation of FKBP51 and TMPRSS2 mRNAs was compared with that of 15d-PGJ2.
To study further the effect of 15d-PGJ2 on the AR-dependent transcription in a heterologous system, we transfected COS-1 cells with an expression vector encoding the AR along with a luciferase reporter driven by a minimal promoter harboring two AREs in front of a TATA box (pARE2-TATA-luc) or a luciferase reporter driven by a probasin gene promoter (pPB-luc, Fig. 4). AR-dependent transcription in COS-1 cells was inhibited in response to 15d-PGJ2 in a fashion comparable with that of the endogenous AR in VCaP cells. This result strongly supports the notion that the inhibition of AR-dependent transcription by 15d-PGJ2 in prostate cancer cells is due to a direct inactivation of the AR and not via cell-specific factors involved in AR transcription complexes.
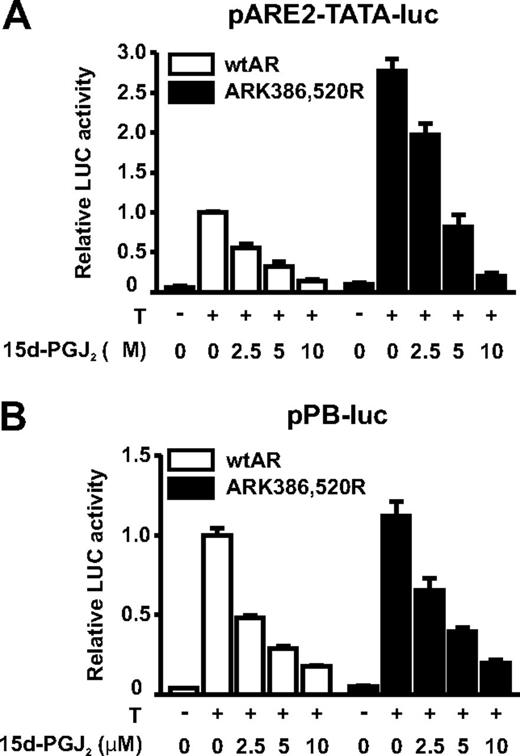
The AR is directly inhibited by 15d-PGJ2. COS-1 cells were cotransfected with a minimal pARE2-TATA-luc (A) or pPB-luc (B) reporter together with expression vectors encoding wtAR- or SUMOylation-deficient ARK386,520R. After 28 h, cells were exposed to testosterone (T; 100 nm) and increasing concentrations of 15d-PGJ2 as indicated. After 14 h, luciferase and β-galactosidase (a control assessing for transfection efficiency) activities were measured. Relative luciferase activity of wtAR in the presence of T and in the absence of 15d-PGJ2 is set as 1 and other values are expressed in relation to that value. The columns represent the mean ± sd values of a representative experiment with triplicate samples.
SUMOylation can contribute to the ability of 15d-PGJ2 to inhibit AR activity
To study the role of AR SUMO acceptor sites in the inhibition by 15d-PGJ2, we measured the inhibitory effect of the prostaglandin on the activity of SUMOylation-deficient AR mutant ARK386,520R vs. that of wtAR, first in reporter gene assays in COS-1 cells. Both receptor forms were inhibited by 15d-PGJ2, but the SUMOylation-deficient AR mutant was less sensitive than the wtAR at 2.5 μm 15d-PGJ2 (29 ± 5 vs. 45 ± 6% inhibition on pARE2-TATA-luc and 42 ± 7 vs. 52 ± 1% inhibition on pPB-luc) (Fig. 4). We next compared the effect of 15d-PGJ2 between PC-3 prostate cancer cells stably expressing wtAR or ARK386,520R. As shown in Fig. 5, the expressions of both FKBP51 and TMPRSS2 were significantly less inhibited by 2.5 μm 15d-PGJ2 in the ARK386,520R-expressing PC-3 cells in comparison with the wtAR-expressing cells (P < 0.01 and P < 0.001, respectively). However, at higher concentrations of 15d-PGJ2, the sensitivity differences between the wtAR and the SUMOylation mutant were blunted. Thus, these data suggest that the AR SUMOylation can contribute to the inhibitory effect of 15d-PGJ2 on the AR activity.
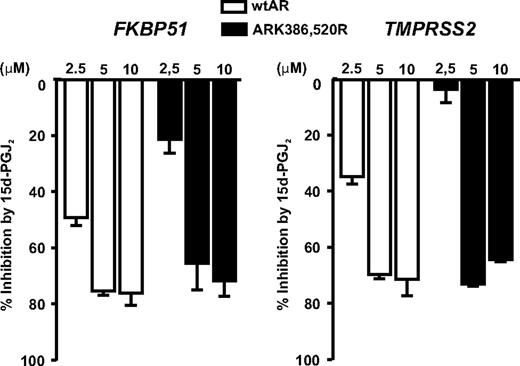
Repression of endogenous AR target genes by 15d-PGJ2 is modulated by AR SUMOylation. PC-3 prostate cancer cell lines stably expressing wtAR or ARK386,520R were exposed to R1881 (10 nm) and the indicated concentrations of 15d-PGJ2 for 12 h. The expression of FKBP51 and TMPRSS2 were analyzed by RT-qPCR, and the results are shown as inhibition percentage by 15d-PGJ2 (mRNA levels of 15d-PGJ2 treated cells compared with the mRNA level of cells grown in the presence of R1881 but in the absence of 15d-PGJ2 in each cell line).
15d-PGJ2 blunts the interaction of AR with chromatin in prostate cancer cells
In an attempt to clarify the function of AR when it is repressed by 15d-PGJ2, we first used immunofluorescence and confocal microscopy to examine whether the compound could impair the nuclear localization of holo-AR. As shown in Fig. 6A, this was not the case. Because the DNA-binding domain (DBD) of AR is rich in cysteine residues, we next tested whether the attack by 15d-PGJ2 would impair the DNA-binding function of AR in vitro by using EMSAs. To that end, androgen-exposed VCaP cells were treated with or without 15d-PGJ2, and cell extracts were prepared and incubated with a 32P-labeled consensus ARE DNA. As shown in Fig. 6B, 15d-PGJ2 reduced the binding of AR to the ARE consensus DNA in vitro. However, the inhibitory effect of the prostaglandin on the DNA-binding activity of the receptor was weaker than that seen on the AR-dependent transcription (c.f.Fig. 3) because 10 μm 15d-PGJ2 inhibited the DNA-binding by 67 ± 4%, whereas the AR-dependent transcription was virtually completely abolished (>90% inhibition) at the same prostaglandin concentration.
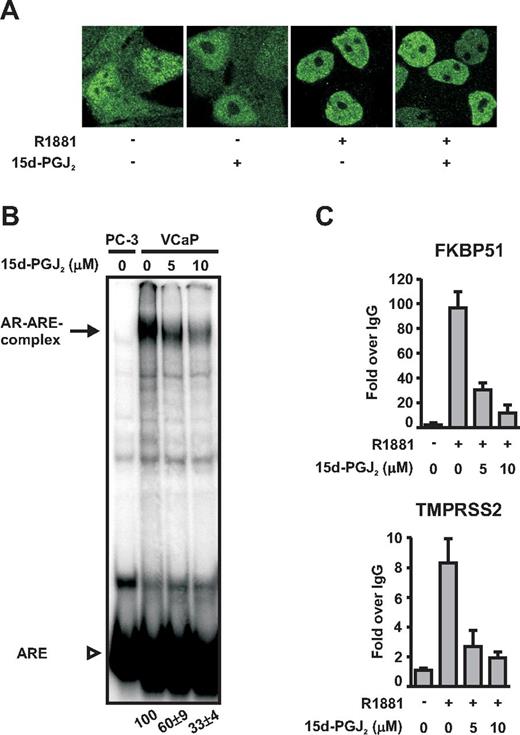
15d-PGJ2 inhibits binding of AR onto chromatin without altering subcellular distribution of the receptor. A, VCaP cells grown on glass coverslips were treated with or without R1881 (10 nm) and 15d-PGJ2 (5 μm for 1 h) as indicated and immunostained with anti-AR antibody and fluorescein isothiocyanate-labeled secondary antibody. Confocal images were collected by a Zeiss LSM 700 microscope. B, EMSAs were performed as described in Materials and Methods with extracts prepared from VCaP cells grown in the presence of R1881 (10 nm) and the indicated concentrations of 15d-PGJ2 for 1 h. An extract from AR-negative PC-3 prostate cancer cells was used as the negative control. The arrow depicts the position of AR-ARE complexes and the arrowhead depicts free ARE. Numbers below the lanes indicate the intensity of AR-ARE complexes in relation to the situation in the absence of 15d-PGJ2 (= 100). Quantification represents the mean ± sd of three experiments. C, For ChIP, VCaP cells were treated with R1881 (10 nm) and the indicated concentrations of 15d-PGJ2 for 1 h, and the samples were processed as described in Materials and Methods. Binding of AR onto FKBP51 and TMPRSS2 regulatory regions was monitored by using real-time quantitative PCR. ChIP assays with normal IgG monitored nonspecific binding. Results are shown as fold increases over the normal IgG-precipitated samples and represent the mean ± sd of three experiments.
We also investigated how 15d-PGJ2 could influence the interaction of androgen-bound AR with chromatin in intact cells by using real-time PCR-based quantitative ChIP assays. Androgen-treated VCaP cells were exposed to 15d-PGJ2 for 1 h and binding of AR onto FKBP51 and TMPRSS2 androgen-regulated enhancers was analyzed by quantitative ChIP assays. In line with the gene expression data, binding of the AR onto these regulatory gene regions in vivo was dramatically inhibited by 15d-PGJ2 exposure, with 10 μm 15d-PGJ2 inhibiting the AR-TMPRSS2 enhancer and the AR-FKBP51 enhancer interaction by 77 ± 3 and 88 ± 7%, respectively (Fig. 6C). Thus, the inhibitory effect of 15d-PGJ2 on the AR-chromatin interaction is more robust than with AR-naked DNA binding, suggesting that the compound does not merely impair the zinc finger-based the DNA-binding function of the AR.
15d-PGJ2 disrupts the N-terminal/C-terminal (N/C) interaction of AR
The androgen-dependent interaction between the N-terminal domain and the C-terminal domain of AR is essential for the transcriptional capacity of the receptor as well as its ability to interact with the chromatin but not for the binding of AR to AREs in vitro (34, 43, 44). Therefore, we performed mammalian two-hybrid assays in COS-1 cells cotransfected with expression vectors encoding the AR N-terminal domain (NTD) fused to the VP16 transactivation domain and the AR C-terminal domain [ligand-binding domain (LBD)] fused to the GAL4 DBD together with luciferase reporter regulated by GAL4-binding elements (pG5-Luc). Interestingly, the androgen-dependent AR N/C interaction was dramatically and dose dependently decreased by 15d-PGJ2, and a concentration as low as 2.5 μm 15d-PGJ2 could inhibit the interaction by 70 ± 4% (Fig. 7A). In contrast, the same concentration of 15d-PGJ2 had no effect on the transcriptional activity of the AR NTD harboring hormone-independent transcription activation function 1 (Fig. 7B). Moreover, this concentration of 15d-PGJ2 had no influence at all on the activity of a constitutively active AR form devoid of the LBD (ARΔLBD) (Fig. 7C).
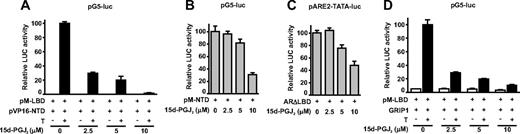
15d-PGJ2 disrupts androgen-dependent N/C interaction of the AR. A, AR N/C interaction was assessed by a mammalian two-hybrid assay. COS-1 cells were cotransfected with pG5-luc driven by Gal4 DNA-binding elements and vectors encoding Gal4 DBD fused to AR LBD (pM-LBD) and VP16 activation domain fused to AR NTD (pVP16-NTD). After 42 h, the cells were cultured for 6 h in the presence or absence of 100 nm testosterone (T) and increasing concentrations of 15d-PGJ2. Relative luciferase activity in the presence of T and the absence of 15d-PGJ2 was set as 100, and other values are expressed in relation to that value. The columns represent the mean ± sd values of a representative experiment with triplicate samples. B, Effect of 15d-PGJ2 on the transcriptional activity of AR NTD fused to Gal4 DBD (pM-NTD) as assessed by the activity derived from the cotransfected pG5-luc. C, Effect of 15d-PGJ2 on AR devoid of LBD. COS-1 cells were cotransfected with pARE2-TATA-luc and an expression vector encoding LBD-truncated version of AR (ARΔLBD). After 42 h, cells were cultured for 6 h with indicated concentrations of 15d-PGJ2. Relative luciferase activity of wtAR in the absence of 15d-PGJ2 is set as 100, and other values are expressed in relation to that value. The columns represent the mean ± sd values of a representative experiment with samples in triplicate. D, Effect of 15d-PGJ2 on the activity of AR LBD evoked by GRIP1. COS-1 cells were cotransfected with pSG5-luc, pM-LBD, and pSG5-GRIP1, and after 42 h, T and increasing concentrations of 15d-PGJ2 were added for 6 h as indicated. Relative luciferase activity in the presence of T and the absence of 15d-PGJ2 was set as 100, and other values are expressed in relation to that value. The columns represent the mean ± sd values of a representative experiment with the triplicate samples.
The AR LBD contains hormone-dependent activation function 2 domain that is barely functional in isolation as assessed by the activity of GAL4 DBD-LBD fusion on pG5-Luc, but the activity of activation function 2 can be measured when the LBD is coexpressed together with the coactivator GRIP1 (glucocorticoid receptor-interacting protein 1, steroid receptor coactivator-2) (34). In contrast to the AR NTD and the AR form devoid of the LBD, 15d-PGJ2 was effective in repressing the activity of the AR LBD evoked by the GRIP1, with already 2.5 μm 15d-PGJ2 inhibiting its activity by 71 ± 3% (Fig. 7D). Taken together, these results strongly suggest that the DNA-binding function is not the sole AR activity targeted by the 15d-PGJ2, but the prostaglandin also targets the LBD and thereby the androgen-dependent interaction between the amino and carboxy termini of the receptor.
The inhibitory effect of 15d-PGJ2 is additive with bicalutamide in VCaP cells
The widely used antiandrogen, bicalutamide, is relatively inefficient at inhibiting the activity of agonist-bound AR in VCaP cells (36) (Fig. 8). To test whether the antagonistic effect of bicalutamide could be potentiated by concomitant administration of 15d-PGJ2, VCaP cells were treated with dihydrotestosterone (DHT; 1 nm) and/or bicalutamide (10 μm) in the presence or absence of 5 μm 15d-PGJ2. As shown in Fig. 8, the bicalutamide displayed agonistic activity in VCaP cells, and its inhibitory effect was only modest in repressing the activity of DHT-bound AR on FKBP51 and TMPRSS2 (44 and 25% repression of mRNA levels, respectively). Interestingly, the agonistic activity of bicalutamide was completely abolished by the prostaglandin. Moreover, when the DHT-treated cells were exposed to bicalutamide together with 15d-PGJ2, these compounds had an additive effect, and the remaining activity of DHT-bound AR was reduced to less than 10%. These results suggest that the prostaglandin and bicalutamide do not target the same molecular function in the AR.
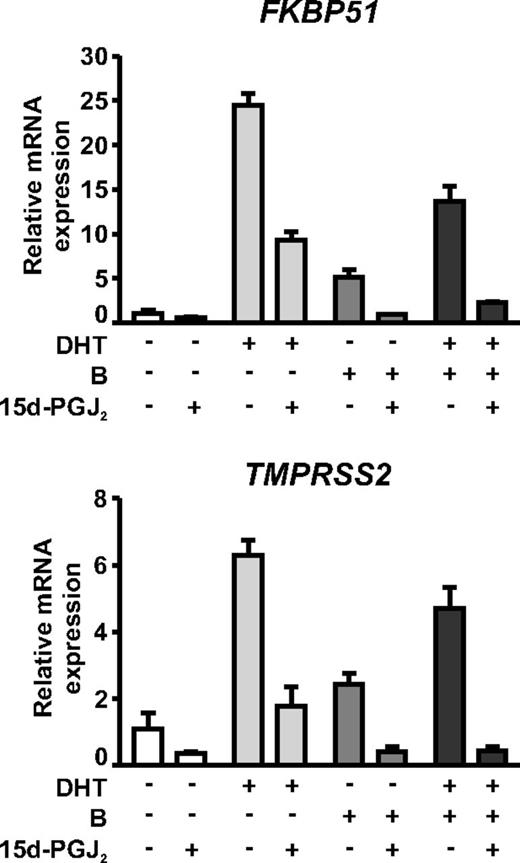
15d-PGJ2 and bicalutamide have an additive effect on AR-dependent gene expression in VCaP cells. VCaP cells were treated with or without 1 nm DHT and/or 10 μm bicalutamide in the presence or absence of 5 μm 15d-PGJ2 for 6 h as indicated. Expressions of FKBP51 and TMPRSS2 were analyzed as described in Fig. 3.
Discussion
The cyclopentenone prostaglandin, 15d-PGJ2, is an endogenous electrophile derived from the nonenzymatic dehydration of prostaglandin D2, a major product of the cyclooxygenase pathway (45, 46). Seminal fluid is the richest source of prostaglandins, and also 15d-PGJ2 has been found in the normal seminal fluid (47). This study demonstrated that exposure of prostate cancer cells to 15d-PGJ2 can rapidly and efficiently inhibit the activity of holo-AR. The inhibition appeared to be due to the ability of the prostaglandin to form adducts with the AR. The inhibitory effect of 15d-PGJ2 (IC50 ∼2 μm) both on the expression of endogenous AR target genes in prostate cancer cells and on an androgen signaling system reconstituted by ectopic expression of reporter genes in heterologous cells was more efficient than that of the antiandrogen, bicalutamide. The prostaglandin was also capable of abolishing the residual agonism of bicalutamide and having an additive effect with the antiandrogen in VCaP prostate cancer cells containing the AR gene amplification. Interestingly, the inhibition on AR target gene expression by 15d-PGJ2 closely paralleled with the inhibition of the AR N/C interaction as well as that with the AR-chromatin interaction in vivo. However, the inhibitory effect of the prostaglandin on the binding of the receptor to naked DNA was weaker than that seen with its binding to chromatin. In keeping with the concept that the compound does not principally target the AR DBD and that the AR LBD plays a role as an important target of 15d-PGJ2, an AR form devoid of the LBD, was markedly less sensitive to the inhibition by 15d-PGJ2 than the full-length receptor. Because the compound inhibited the AR activity in a similar manner independently of the receptor was exposed to 15d-PGJ2 before or concomitantly with the androgen and that bicalutamide still had an inhibitory effect in the presence of 15d-PGJ2, it is unlikely that the prostaglandin inhibits the ligand binding to the ligand-binding pocket. Instead, it may render the conformation of the LBD incapable of interacting with the NTD, which is needed for an efficient interaction of the receptor with the chromatin.
15d-PGJ2 is a well-known physiological ligand for PPARγ, binding covalently to its LBD and activating the expression of PPARγ target genes (21, 22). Similarly to the AR, but in contrast to the situation with PPARγ, the transcriptional activity of ERα was recently shown to be inhibited by 15d-PGJ2. However, in the case of ERα, the prostaglandin was reported to covalently modify and thereby disrupt the second zinc finger of the DBD, which is crucial in the dimerization of the receptor (27). The PPARγ, the AR, and the ERα belong to the same nuclear receptor superfamily, all sharing a similar protein architecture in which the cysteine-rich DBD is the best conserved domain. It is therefore interesting that 15d-PGJ2 displays such dissimilar effects on the PPARγ vs. the ERα and the AR. Agonist binding induced intramolecular NTD/LBD (N/C) interaction and folding, which occurs in the AR and the ERα, but not in the PPARγ, may at least in part explain the differential reactivity of these three nuclear receptors toward 15d-PGJ2 (48). According to our results, the intramolecular folding, the N/C interaction, of the AR, but not the DBD-mediated dimerization, is the molecular function primary disrupted by 15d-PGJ2.
Interestingly, in addition, other important DNA-binding TFs, p50 (nuclear factor-κB subunit), c-Jun (activator protein-1 subunit), and TP53 (tumor protein p53), that do not possess zinc-finger structures but bind to DNA as dimers or multimers have been reported to be sensitive to 15d-PGJ2 inhibition (24–26). Covalent adduct formation at cysteine residues, which causes an inactivation of DNA binding, was identified as the mechanism underlying their inactivation. As in the case of the AR, the electrophilic α,β-unsaturated carbonyl group of 15d-PGJ2 was crucial for the ability of 15d-PGJ2 to inhibit the activity of ERα and TP53 (26, 27). 15d-PGJ2 possesses two electrophilic carbons, both of which can potentially form adducts with reactive thiols. It can therefore also covalently cross-link protein dimers (49), as has been shown to occur with the c-Jun (25).
4-Hydroxynonenal, a lipid oxidation-derived electrophile formed in oxidative stress, is known to increase protein SUMOylation (29). Here we show that in addition to targeting both apo- and holo-AR, 15d-PGJ2 rapidly, but transiently, induced SUMOylation of the holo-AR, with modification by SUMO-2/3 in particular. These novel data add for the first time 15d-PGJ2 to the list of agents that can trigger protein SUMOylation. Interestingly, disruption of AR SUMOylation sites rendered the receptor somewhat less sensitive toward the prostaglandin, suggesting that receptor SUMOylation can at least transiently contribute to the inhibitory effect of 15d-PGJ2 on the AR. Exposure of colon carcinoma cells to 15d-PGJ2 (9 h, 20 μm) has been reported to induce endoplasmic reticulum stress and protein ubiquitylation (50). However, in VCaP prostate cancer cells, the effect of 15d-PGJ2 on the AR activity or its SUMOylation is distinct from any endoplasmic reticulum stress because the well-established endoplasmic reticulum stress inducer, thapsigargin, neither induced SUMOylation of AR nor inhibited the AR activity in these cells (S. Kaikkonen, unpublished observations).
Although 15d-PGJ2 has been shown to trigger cell signaling cascades via direct binding with proteins containing redox active cysteine residues (24–26), it has also been shown to increase the secondary production of ROS (51). The mechanism by which 15d-PGJ2 increases intracellular ROS is likely to occur via alterations in the redox status, because it has been reported to decrease intracellular glutathione (51) and impair the function of proteins involved in antioxidant defense, such as thioredoxin (52) and thioredoxin reductase (53). Intriguingly, cross talk between AR signaling and ROS has been suspected because AR activity can be increased by exposure to ROS (for a review, see Ref. 54). Because 15d-PGJ2 decreased the AR activity rather than the opposite, it seems unlikely that the secondary production of ROS plays a role in the inhibition of the AR signaling observed in this study.
15d-PGJ2 is formed during the late phase of inflammation, and it has protective effects on experimentally induced inflammation models. The effects have been linked to the inhibition of proinflammatory TFs, including nuclear factor-κB, activator protein-1, and signal transducer and activator of transcription-3, and the activation of antiinflammatory TFs, Nrf2 and PPARγ (55). Previously 15d-PGJ2 has been shown to inhibit the proliferation of cancer cells, including both AR-negative and AR-positive prostate cancer cells (50, 56–58). However, these data are somewhat contradictory in terms of whether prolonged exposure to 15d-PGJ2 can induce apoptosis or some other form of nonapoptotic cell death. Moreover, inactivation of AR function has not been previously linked to the action of 15d-PGJ2. It is noteworthy that 15d-PGJ2-targeted repression of AR activity is an early response to the prostaglandin exposure, occurring in VCaP cells far before any signs of the retardation of cell proliferation (S. Kaikkonen, unpublished observations).
There exists some controversy about whether 15d-PGJ2 is endogenously formed by cyclooxygenase-dependent synthesis in concentrations that can elicit biological responses because the concentrations of free 15d-PGJ2 in the cell media have been reported to lie within the nanomolar range (46, 59). However, given the inherent reactivity of the cyclopentenone prostaglandins, measuring free 15d-PGJ2 may represent a gross underestimation of concentrations that are formed in cells. Interestingly, it has been demonstrated that as much as 97–99% of exogenously added 15d-PGJ2 is inactivated by the cell culture medium constituents and do not gain access to the inside of the cells to initiate cell signaling (60). It is therefore conceivable that the intracellular concentrations of 15d-PGJ2 causing inhibition on AR signaling are within the same range as those formed via enzymatic reactions. Additionally, 15d-PGJ2-like isoprostanes that are formed during oxidant stress are likely to exert similar effects on AR signaling, further strengthening the hypothesis that the observed effects are physiologically relevant.
Several lines of epidemiological, pathological, and molecular evidence support the concept that chronic inflammation is causally linked to prostate carcinogenesis (1). Inflammation-triggered formation of 15d-PGJ2 may thus influence the AR activity in prostate cancer cells, contributing to deregulated androgen signaling during prostate carcinogenesis. Our findings showing that the adduct formation by 15d-PGJ2 efficiently inhibits the N/C interaction and chromatin interaction of the AR may also provide novel approaches for the development of inhibitors targeting the AR in prostate cancer.
Acknowledgments
We thank Eija Korhonen and Merja Räsänen for skillful technical assistance.
This study was supported by the Academy of Finland, the Sigrid Jusélius Foundation, The Finnish Cancer Organisations, Kuopio University Foundation, The Emil Aaltonen Foundation, and the strategic funding of the University of Eastern Finland.
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- AR
Androgen receptor
- ARE
androgen response element
- ChIP
chromatin immunoprecipitation
- DBD
DNA-binding domain
- DHT
dihydrotestosterone
- 15d-PGJ2
15-deoxy-Δ12,14-prostaglandin J2
- ER
estrogen receptor
- FBS
fetal bovine serum
- GRIP1
glucocorticoid receptor-interacting protein 1
- LBD
ligand-binding domain
- luc
luciferase
- N/C
N-terminal/C-terminal
- NTD
N-terminal domain
- Nrf2
nuclear factor erythroid 2-related factor 2
- PPAR
peroxisome proliferator-activated receptor
- ROS
reactive oxygen species
- RT-qPCR
quantitative real-time RT-PCR
- SUMO
small ubiquitin-related modifier
- SUMOylation
conjugation of SUMO
- TF
transcription factor
- VCaP
vertebral cancer of the prostate
- wtAR
wild-type AR.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}