
Abstract
Human autoimmune lymphoproliferative syndrome (ALPS) is caused by defective CD95-mediated apoptosis of lymphocytes. In most patients, heterozygous mutations within the CD95 gene are found. Mutated proteins interfere with CD95-signaling in a dominant-negative way. However, the penetrance of clinical disease is variable. We describe 13 patients out of nine families with the clinical presentation of ALPS. Eight different mutations were detected. Sensitivity to CD95-induced cell-death, assembly of the CD95-death-inducing signaling complex (DISC), and activity of initiator caspases-8 and -10 were compared in EBV-transformed B-lymphoblastoid cells of these patients. All CD95-mutations led to a reduced DISC formation and diminished initiator caspase activity upon CD95-stimulation, whereas a marked heterogeneity in sensitivity to CD95-induced killing was found. Residual apoptosis sensitivity to almost normal levels could be achieved upon cross-linking by addition of protein A. Thus, no correlation between residual CD95 sensitivity and clinical phenotype or genotype of ALPS was found. This observation is only partially explained by the variable effects of the CD95-mutations themselves. It also points to a pronounced influence of additional factors, such as modifier pathways or exogenous effects apart from the CD95 pathway in the pathogenesis of ALPS.
Similar content being viewed by others


Main
Apoptosis via CD95 is mediated by a signal transduction cascade that causes cell death. Binding of the CD95-ligand to cell-surface CD95 leads to CD95-oligomer formation (1). This allows binding of CD95-associated protein with death domain (FADD) to the intracellular homologous death domain of CD95. Through death effector domains, the initiator-caspases-8 and -10 and the FLICE-like inhibitory protein (FLIP) are recruited into the death-inducing signaling complex (DISC) (2). Initiator-caspases are activated by autocatalytic cleavage (3). This finally leads to activation of downstream executioner caspases-3, -7, and -9 and subsequently induces cell death via mitochondrial dependent and independent pathways.
Defective CD95-signaling is the cause of the autoimmune lymphoproliferative syndrome (ALPS) (4). ALPS is characterized by nonmalignant lymphoproliferation and by the expansion of the double negative T cell receptor [(TCR)α/β+/CD4−/CD8−] T cells. Patients suffer from autoimmunity, especially from immune thrombocytopenia and hemolytic anemia (5) and have an increased risk to develop lymphomas (6).
Mutations of CD95 account for the majority of ALPS (type Ia) (4,7). Mutations in caspase-10 have been described with a similar clinical picture (type II) (8). Mutations in the CD95-ligand were detected in a patient with lupus erythematodes-like symptoms and in two patients with ALPS-like picture (type Ib) (9). Two patients with homozygous mutation in caspase-8 were described with defective T cell activation and an immunodeficiency different from ALPS (10).
In some patients with clinical ALPS, no genetic defect was identified so far (type III) (11).
With rare exceptions, CD95-mutations are heterozygous germline or somatic mutations and are present in patients as well as in unaffected family members (12–16). Mutations are found predominantly in exon 9 of the CD95 gene encoding the death domain, which in its normal configuration associates with the adaptor molecule FADD (16) and therefore, interfere in a dominant-negative way with CD95-signaling (14,15,17,18). Because this complex then recruits caspase-8 or caspase-10 as initiator caspases to form the DISC and initiate apoptosis execution, an impairment of DISC formation is considered to be the main consequence of CD95-mutations (14–16). Transfection enforced overexpression of mutated receptors has demonstrated a dominant negative effect in cell lines disrupting DISC formation (14,15). Taken together, a dominant negative effect is considered for defective CD95-mediated apoptosis in T cells from ALPS patients. However, some mutations cause haploinsufficiency rather than interfering by a dominant negative effect (14,17).
We describe 13 patients of nine families with ALPS as a result of intracellular or extracellular CD95 mutations. To analyze the effect of different CD95-mutations on the CD95 pathway, we immortalized B cells of our ALPS-patients by transformation with EBV. In these cell lines, we studied DISC formation, activation of the initiator caspases-8 and -10, respectively, and sensitivity to cell death.
METHODS
Patients.
Heparinized and EDTA-blood samples were referred for the analysis of defective T cell apoptosis and mutations in the CD95 gene because of suspected ALPS. Informed consent was obtained for Epstein-Barr-Virus transformed B-lmphoblastoid-cell-line (EBV-BLCL) generation and scientific analysis of clinical data. The study was approved by the IRB University of Ulm 289/2004. The diagnosis of ALPS was based upon nonmalignant lymphoproliferation, increase in double negative (DN) T cells (>2%) and impaired lymphocyte apoptosis in addition to autoimmunity. Clinical data were based upon medical reports.
Sequencing of the CD95 gene.
Genomic DNA was isolated from EDTA-blood using the QiAmp DNA Mini Kit (Qiagen, Hilden). DNA was amplified by PCR with primer pairs flanking the coding region of all nine exons as described in (13). After purification, PCR products were analyzed with the ABI PRISM Dye Terminator Cycle Sequencing KIT (Applied Biosystems, Darmstadt) on an ABI 373A DNA-sequencer using the same primers applied for amplification.
RNA-isolation and sequencing.
RNA was isolated using the RNeasy RNA purification kit (Qiagen, Hilden). RT-PCR and PCR was performed using the Gene Amp RNA PCR Core Kit (Applied Biosystems, Darmstadt). For PCR amplification of a 311 bp fragment of the CD95 cDNA, the primers 5′-TCAAGGAATGCACACACTCACCAGC-3′and5′-GGCTTCCATTGACACCATTCTTTCG-3′ were used.
Immunophenotyping of blood T cells.
Blood lymphocyte populations were identified by flow cytometry (FACS; FACScan Becton Dickinson, Heidelberg) using combinations of fluorochrome-labeled MAb in a standardized whole-blood-lyse and wash assay (FACSLyse, Becton-Dickinson, Heidelberg). AntiCD3-FITC and antiCD4-PE were from Coulter Immunotech (Krefeld), antiCD4-PerCP, antiCD8-PerCP, antiTCR-αβ, and antiCD95-PE were from Becton Dickinson (Heidelberg).
Blood mononuclear cell culture.
Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll density centrifugation (100 × g, 20 min) and cultured in RPMI1640 (GIBCO BRL, Karlsruhe)/10% FCS (Sigma Chemical, Deisenhofen)/2000 U/mL Penicillin/streptomycin (GIBCO, Karlsruhe)/2 mM Glutamine (GIBCO, Karlsruhe) with 1 mg/mL phytohemagglutinine [(PHA), L-9132, Sigma Chemical, Deisenhofen] and 100 U/mL IL-2 (Cetus, Norwalk) for 10–14 d.
EBV cell transformation.
PBMCs (1 × 107) were added to supernatant of exponentially growing EBV-producing B95-8 cells (ATCC CRL 1612) 2 h at 37°C. Cyclosporine A (1 μg/mL) (Novartis, Nürnberg) was added to the medium to inhibit T cell proliferation for at least 2 mo.
Induction of apoptosis.
PHA-blasts or EBV-BLCLs were harvested. Cells of 2 × 105 were stimulated by adding APO-1 IgG3 in the absence or presence of protein A (10 ng/mL, Sigma Chemical, Deisenhofen). Apoptosis was determined by Annexin V staining (Alexis, Günberg) or by propidium-iodide (50 mg/mL; Sigma Chemical, Deisenhofen) staining of nuclei in 0.1% NaCitrate/0.1% TritonX-100, (Sigma Chemical, Deisenhofen) for 18 h at 4°C and analyzed by FACS.
Caspase activity assays.
EBV-BLCLs (5 × 105) were stimulated with 1 μg/mL APO-1 IgG3 in the presence or absence of protein A (10 ng/mL, Sigma Chemical, Deisenhofen) for 2 h at 37°C. The caspase-8 and caspase-10 activity was determined by using a specific fluorometric substrate Kit (IETD-AFC, AcAEVD-AFC, Biocat, Heidelberg).
DISC-analysis.
EBV-BLCLs or PHA blasts were stimulated for 10 min with APO-1 IgG3 (1 μg/mL) or an IgG3 isotype control (1 μg/mL, Becton-Dickinson, Heidelberg) as indicated. After washing with ice cold PBS and centrifugation (100 × g; 4°C), cells were lysed in 1 mL lysis buffer (1% TritonX-100 (Merck, Ulm)/150 mM NaCl/50 mM Tris/pH 8) for 15 min on ice. By centrifugation at 10,000 × g for 5 min, nuclei were spun down and supernatants were harvested. APO-1 and the coimmunoprecipitated DISC was bound by PAN-mouse-IgG-magnetic beads (Dynal, Hamburg) for 10 h at 4°C. After washing in 1% Igepal Ca-630 (Sigma Chemical, Deisenhofen)/500 mM NaCl/Tris 50 mM/pH8, the beads were resuspended in 1 × SDS-sample buffer (Sigma Chemical, Deisenhofen) and boiled for 5 min. After SDS-Page (10–20%), proteins were blotted onto nitrocellulose membranes. After blocking (skimmed milk) bound proteins were detected by specific antibodies [mouse anti-Caspase-8 (Alexis)/mouse anti-FADD (Transduction, Heidelberg), rabbit anti-CD95 (Santa Cruz, California), and mouse anti-CASP 10 (MBL, Woburn)], and visualized by isotype-specific hrp-goat anti-mouse IgG1 or IgG2b (Southern Biotechnology, Birmingham) or hrp-goat anti-rabbit IgG (Santa Cruz, California).
RESULTS
Clinical feature and functional analysis of ALPS.
We identified 13 patients with ALPS of nine different families. TCRα/β+ (DN) T cells were increased in numbers in all patients (Table 1). Lymphadenopathy and/or hepatosplenomegaly were present in all patients. The presence of autoimmunity was indicated by autoimmune thrombocytopenia (n = 12), autoimmune hemolytic anemia (n = 8), autoimmune neutropenia (n = 4), and juvenile chronic arthritis (n = 1; Table 1).
CD95 mutational studies.
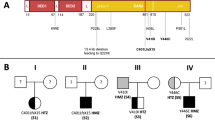
Mutational analysis of all nine exons and all splice-sites of the CD95 gene revealed eight different mutations (Fig. 1). Six mutations were found within exon 9. The heterozygous point mutation at position c748 (C>T) causes a frameshift with a premature stop codon at amino acid position 234 (c748C>T, R234X) and was found in three patients of two families (23.1, 23.2, and 3). This mutation was described before (19). The heterozygous insertion of T (c696_697insT; I217fsX230) in patients 39.1, and 39.2 leads to a frameshift with a premature stop at position 230. In family 14, a novel mutation (c706delA, T219fsX224) was identified. This frameshift leads to a stop codon at position 224. The heterozygous deletion mutation (c809_810delCA; D253fsX263) found in patient 1 causes frameshift with a premature stop at position 263 and was described earlier (4). The heterozygous point mutation in patient 8 (c749G>A; R234Q) with the exchange of Arg to Gln is located in a highly conserved position within the death domain of exon 9, leading to loss of function and was described earlier (14,16).

CD95-mutations of patients with ALPS type Ia. Mutations of 13 patients of 10 families are shown with respect to the signal peptide, cystein rich domains transmembrane region and death domain according to the GenBank AcNo.NM-000043.
Three mutations were found within different splice sites. In family 16, a heterozygous point mutation at the splice-donor site of intron 1 was found (IVS1 + 1G>A; c30 + 1G>A). We failed to detect abnormal mRNA. CD95-surface-expression was reduced by approximately 50% in the B-lmphobastoid-cell-lines (BLCLs) and in the PHA blasts. Intracellular staining of CD95 did not differ from that of EBV-BLCLs of healthy blood donors. The mutation in the splice donor site of intron 3 of patient 22 (IVS3 + 3A>C; c334 + 3A>C) might lead to alternative splicing or skipping of exon 3 as described before (20). The heterozygous mutation at the splice acceptor site of intron 5 in patient 27 (IVS5 + 1G>A, c506-1G>C) was not described yet.
Sensitivity of EBV-transformed B cells to CD95-induced apoptosis.
To test the influence of different mutations on the CD95-pathway the sensitivity to apoptosis of the EBV-BLCLs was assessed after treatment with the stimulatory antibody APO-1 IgG3 in the presence or absence of protein A. As expected, a reduction in sensitivity for cell death was found in all 11 patient cell lines compared with cell lines of healthy donors (Fig. 2). Nevertheless, the cell lines of patients 14.1, 14.2, 22, 3, 1, 16.1, and 16.2 displayed residual sensitivity to CD95-induced cell death. The variable residual sensitivity was enhanced upon cross-linking using protein A (Fig. 2). No residual sensitivity was seen in the cell line of patient 23.1, 39.1, 39.2, and 8.

Sensitivity toward CD95-mediated apoptosis of different EBV-BLCL of ALPS type Ia patients. Immortalized B cells of patients or of six healthy controls (C) were incubated with increasing concentrations of Apo1-IgG3 in the presence (□) or absence (▪) of protein A. DNA fragmentation was assessed by staining of the nuclei with propidium iodide. Mean and SD of triplicates are given of one out of three experiments with comparable results.
DISC-formation in ALPS EBV-BLCLs.
To analyze, whether variable residual CD95 sensitivity was associated with differences in DISC-formation, we immunoprecipitated CD95 complexes after pretreatment with APO-1 antibody and protein A. We found a diminished DISC-formation in all cell lines (Fig. 3). FADD, caspase-10, caspase-8, and FLIP seemed to be incorporated into the DISC to an equally reduced extend. No major differences could be detected between high- and low-residual sensitivity cell lines. DISC-precipitation in the EBV cell line of patient 22 was almost completely abolished.

DISC formation in EBV-BLCLs of ALPS type Ia patients and C. EBV-B cells were stimulated with APO-1 IgG3 or a IgG3-isotype-control. After lysation, antibody-bound proteins were immunoprecipitated. APO-1 IgG3 was added after cell lysation in unstimulated samples. After SDS-Page, the co-immunoprecipitated molecules were detected by specific antibodies. The experiment is representative for more than three experiments with comparable results.
Initiator caspases in ALPS EBV-BLCLs.
To test whether the variable sensitivity for apoptosis is mediated via variations in DISC function, we determined the activity of the initiator-caspases-8 and -10. By using the specific fluorometric substrates IETD-AFC and AcAEVD-AFC, the increase in caspase activity was determined after stimulation with the APO-1 IgG3 antibody in the presence or absence of protein A (Fig. 4). A high-residual apoptosis sensitivity was associated with high-initiator caspase activity after CD95 cross-linking (14.1, 14.2, 22, 3, 1, 16.1, and 16.2). A particular low-caspase activity was found in the EBV cell line of patient 23.1, 39, and 8. Caspase-8 and -10 activity was diminished in equal proportions in each cell line compared with the wild-type EBV cell lines. Initiator caspase activity strongly increased in cell lines 14, 22, 3, and 16 upon CD95 cross-linking using protein A (Fig. 4).

Caspase-8 and caspase-10 activity of EBV-BLCL of ALPS patients. 1 × 106 B cells of ALPS patients or C were stimulated using APO-1 IgG3 (1 μg/mL) in the absence (▪) or presence (□) of protein A. The caspase-8 (A) and caspase-10 (B) activity was determined from cell lysates by using the specific fluorometric substrate IETD-AFC (A) and AcAEVD-AFC (B). Mean and SD of triplicates are given. The experiment is representative for three (caspase 8) and two (caspase 10) experiments, respectively with comparable results. p-values were determined using the unpaired t test. * p < 0.05; ** p < 0.01; §not significant.
Sensitivity to apoptosis and DISC formation in primary T cells of ALPS patients in the absence or presence of protein A.
To evaluate our findings in primary patient cells, we tested CD95 killing and DISC formation in the absence or presence of protein A in PHA-activated T cells of two patients. As shown for EBV cell lines, we found heterogeneous residual sensitivity to APO-1 induced apoptosis. Cross-linking by protein A led to a marked increase in sensitivity to CD95-induced apoptosis (Fig. 5). Although DISC formation in these patients' T cells was reduced, significant recruitment of caspase-8 and FADD upon stimulation could be detected (Fig. 5).

Sensitivity to apoptosis and DISC-formation in T cells of ALPS type Ia patients. In activated T cells of patient 22 (A, C) and patient 8 (B, D) sensitivity to apoptosis was assessed by incubation with APO1 IgG3 in the presence (□) or absence (▪) of protein A. (A, B). DISC formation was determined in patient and control (C) T cells (1 × 107) after stimulation with APO-1 IgG3 in the absence or presence of protein A by immunoprecipitation. (C, D).
DISCUSSION
In this report, we have analyzed CD95 sensitivity, DISC formation, and initiator caspase activity in 11 of 13 patients with clinical presentation of ALPS and mutations in the CD95 gene. We found high-variation in CD95 sensitivity and DISC formation irrespective of clinical phenotype and CD95-mutation.
In nine patients, the mutations were located in exon 9 and caused disruption of the death domain either by insertion of a stop codon in eight patients or amino acid exchanges in one patient. In one family (16.1 and 16.2), splice site mutations between exons one and two were found, which should affect expression of the protein rather than impairment of function. A donor splice site mutation in exon 3 (22) affects the CD95 ligand-binding domain.
The remaining novel mutation of patient 27 involved a splice acceptor site in exon 6 (IVS5-1G>C). Unfortunately, advanced functional analysis of this mutation was impossible. However, its effect on apoptosis might be comparable to the IVS5-3C>G mutant described before (17). This mutation caused skipping of exon-6 leading to excessive production of soluble CD95 (14,21). However, haploinsufficiency of membrane CD95 expression was shown to be responsible for the apoptosis defect in this mutation.
Transfection enforced overexpression of mutated receptors has demonstrated a dominant negative effect in cell lines disrupting DISC formation (14,15). This dominant negative effect is considered to mediate defective CD95-mediated apoptosis in T cells from ALPS patients. Therefore, we expected DISC formation and apoptosis to be severely disabled or absent. However, when we started to screen T cells from patients for apoptosis defects, we found residual to almost normal CD95-mediated apoptosis.
For further analysis, we transformed patients B-cells with EBV. In these cell lines, a wide range of CD95 sensitivity from virtually absent (e.g., 8) to normal apoptosis (e.g., 1) was found. In addition, crosslinking of the CD95 agonistic antibody APO-1 with protein A increased apoptosis in most cases and even restored sensitivity to normal levels (e.g., 14.1, 14.2, 3, 22, and 16).
The availability of EBV-BLCLs from patients enabled us to analyze DISC formation upon CD95 cross-linking in more detail. Recruitment of FADD and caspase-8/-10 to cross-linked CD95 was variably reduced in cells from all patients.
FADD recruitment in cell lines from patients 39.2 and 22 was weak or almost absent. This was associated in patient 39.2 with high-resistance to apoptosis and absence of inducible caspase 8 or 10 activity. In patient 39.2, FADD recruitment is particularly affected by the most upstream frameshift mutation in the death domain.
The IVS3 + 3A>C slice mutation of patient 22 leads to deletion of exon 3, which encodes the cystein-rich domains 1 and 2 necessary for binding of CD95 ligand and anti-APO-1. Mutations in this domain might partially explain the constantly low amount of immunoprecipitated CD95. However, this patient showed residual apoptosis sensitivity and reasonable caspase activity, despite impaired DISC formation. Eventually, the mutated receptors may be excluded because of low antibody binding, allowing DISC formation of the remaining wild-type receptors, sufficient to mediate apoptosis, especially upon cross-linking. Also, haploinsufficiency due to unstable mRNA or rapid degradation of the according protein might be possible. In addition, alternative splicing at a cryptic splice site or weakened normal splicing due to the mutation might allow for residual functional activity in splice site mutations generally.
With the exception of the cell lines from these two patients, recruitment of an intact DISC complex and processing of caspase-8 to its active form could be detected. Accordingly, almost normal (e.g., 14) to reduced (e.g.,8) caspase-8/-10 activity was detectable in all cell lines after CD95 cross-linking. These findings suggest a considerable residual CD95-pathway function in primary untransfected cells from patients with the clinical phenotype of ALPS that exceeds previous findings on disabled CD95 signaling in ALPS using transfection enforced overexpression of CD95 mutants.
Trimerization of wild-type receptors appears to be crucial for unfolding of the death domain. With a heterozygous CD95-mutation, 3 wt receptors would interact with a chance of one in eight with each other thus reducing the number of functional DISC complexes. However, in almost all cases formation of an intact DISC complex with all components and significant functional activity of initiator caspases could be detected.
High residual sensitivity to apoptosis might be explained by overexpression of the wild-type allele. However, in cell lines with residual sensitivity, we did not find enhanced DISC formation compared with resistant cells. Variations in sensitivity would rather be explained by an incomplete recruitment of initiator caspases by different CD95-mutations leading to variation in autoprocessing of caspase-8/-10 or in formation of SPOTS (21). Thus, increased specific caspase activity could be enhanced in some cell lines upon crosslinking of receptors by protein A.
Moreover, mutational changes may lead to unstable mRNA with rapid degradation. For example, we did not detect abnormal mRNA in cells from the patients 16 with a mutation at the splice donor site of intron 1 (signal peptide). CD95 expression was reduced to approximately 50% and no abnormal intracellular accumulation of CD95 found. This mutation might cause haploinsufficiency, which was shown earlier to cause the clinical ALPS phenotype, though associated with low-penetrance (17).
Alternatively, alterations in the amino acid composition of proteins may cause misfolding and accelerated degradation by proteasomes. Deletion and insertion mutants 703delA (14) and 809_810delCA (1) may lead to a short protein survival. This effect could modify the degree of dominant negative interference.
The penetrance and severity of clinical presentation in ALPS was suggested to be influenced by the type of mutation (14,16,22). Mutations in the extracellular domain of CD95 were linked to low-penetrance whereas death domain mutations were linked to high-penetrance. The patients with a homozygous mutation described so far suffered from a more extended disease leading to early death and even hydrops fetalis (23–25). The less severe disease in heterozygotes substantiates that the wild-type allele might be functional and ameliorates disease manifestation. However, in our patient cohort, no correlation between the magnitude of residual CD95 sensitivity, genotype, and disease expression was obvious.
In most ALPS families, there are affected and nonaffected heterozygous carriers of the CD95-mutation. This points to additional factors modifying the clinical phenotype and apoptosis sensitivity apart from the CD95 pathway. These may include genetic or acquired factors such as compensatory death pathways or viral infections (16,22,26–29). Interestingly, even the lpr phenotype in mice is modified by the genetic background of the mouse strain (30). Moreover, CD95 engagement may also mediate caspase independent death involving adaptor molecules different from “classical DISC” (31,32).
As ALPS is considered to be caused by T cell dysregulation, the use of EBV transformed BLCLs from patients is a major limitation of this study. However, these oligoclonal cell lines seemed reasonably close to the patient's primary B-cells, they have an intact CD95 pathway and enabled us to perform biochemical studies with sufficient cell numbers. The function of the CD95 pathway was comparable between BLCLs of different family members harboring the same CD95-mutation. In addition, we compared the results from BLCLs with primary T cells in two patients. Similar results for apoptosis sensitivity and DISC formation were obtained for the primary T cells compared with the EBV-BLCLs showing residual function of the CD95 pathway. An increase in cell death was seen after cross-linking with protein A similar to the observations in the EBV-BLCLs.
In conclusion, we demonstrate a remarkable heterogeneity in both apoptosis sensitivity and DISC formation in a group of patients with heterozygous CD95-mutations and clinical ALPS. However, apoptosis sensitivity and DISC formation exhibited no clear correlation to each other, to the clinical course or disease expression. This highlights a crucial role for additional factors that are possibly not related to the CD95-pathway, which need further clarification to fully understand CD95-mediated cell death and its defects in ALPS.
Abbreviations
- ALPS:
-
autoimmune lymphoproliferative syndrome
- DISC:
-
death-inducing signaling complex
- EBV-BLCL:
-
Epstein-Barr-Virus transformed B-lymphoblastoid-cell-lines
- FADD:
-
Fas-associated protein with death domain
References
Siegel RM, Frederiksen JK, Zacharias DA, Chan FK, Johnson M, Lynch D, Tsien RY, Lenardo MJ 2000 Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science 288: 2354–2357
Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME 1995 Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J 14: 5579–5588
Golks A, Brenner D, Schmitz I, Watzl C, Krueger A, Krammer PH, Lavrik IN 2006 The role of CAP3 in CD95 signaling: new insights into the mechanism of procaspase-8 activation. Cell Death Differ 13: 489–498
Rieux-Laucat F, Le Deist F, Hivroz C, Roberts IA, Debatin KM, Fischer A, de Villartay JP 1995 Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science 268: 1347–1349
Sneller MC, Wang J, Dale JK, Strober W, Middelton LA, Choi Y, Fleisher TA, Lim MS, Jaffe ES, Puck JM, Lenardo MJ, Straus SE 1997 Clincal, immunologic, and genetic features of an autoimmune lymphoproliferative syndrome associated with abnormal lymphocyte apoptosis. Blood 89: 1341–1348
Straus SE, Jaffe ES, Puck JM, Dale JK, Elkon KB, Rosen-Wolff A, Peters AM, Sneller MC, Hallahan CW, Wang J, Fischer RE, Jackson CM, Lin AY, Baumler C, Siegert E, Marx A, Vaishnaw AK, Grodzicky T, Fleisher TA, Lenardo MJ 2001 The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood 98: 194–200
Fisher GH, Rosenberg FJ, Straus SE, Dale JK, Middleton LA, Lin AY, Strober W, Lenardo MJ, Puck JM 1995 Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 81: 935–946
Wang J, Zheng L, Lobito A, Chan FK, Dale J, Sneller M, Yao X, Puck JM, Straus SE, Lenardo MJ 1999 Inherited human Caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type II. Cell 98: 47–58
Bi LL, Pan G, Atkinson TP, Zheng L, Dale JK, Makris C, Reddy V, McDonald JM, Siegel RM, Puck JM, Lenardo MJ, Straus SE 2007 Dominant inhibition of Fas ligand-mediated apoptosis due to a heterozygous mutation associated with autoimmune lymphoproliferative syndrome (ALPS) Type Ib. BMC Med Genet 8: 41
Chun HJ, Zheng L, Ahmad M, Wang J, Speirs CK, Siegel RM, Dale JK, Puck J, Davis J, Hall CG, Skoda-Smith S, Atkinson TP, Straus SE, Lenardo MJ 2002 Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature 419: 395–399
Dianzani U, Bragardo M, DiFranco D, Alliaudi C, Scagni P, Buonfiglio D, Redoglia V, Bonissoni S, Correra A, Dianzani I, Ramenghi U 1997 Deficiency of the Fas apoptosis pathway without Fas gene mutations in pediatric patients with autoimmunity/lymphoproliferation. Blood 89: 2871–2879
Holzelova E, Vonarbourg C, Stolzenberg MC, Arkwright PD, Selz F, Prieur AM, Blanche S, Bartunkova J, Vilmer E, Fischer A, Le Deist F, Rieux-Laucat F 2004 Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N Engl J Med 351: 1409–1418
Beltinger C, Bohler T, Schrappe M, Ludwig WD, Debatin KM 1998 [The role of CD95 (APO-1/Fas) mutations in lymphoproliferative and malignant lymphatic diseases]. Klin Padiatr 210: 153–158
Vaishnaw AK, Orlinick JR, Chu JL, Krammer PH, Chao MV, Elkon KB 1999 The molecular basis for apoptotic defects in patients with CD95 (Fas/Apo-1) mutations. J Clin Invest 103: 355–363
Martin DA, Zheng L, Siegel RM, Huang B, Fisher GH, Wang J, Jackson CE, Puck JM, Dale J, Straus SE, Peter ME, Krammer PH, Fesik S, Lenardo MJ 1999 Defective CD95/APO-1/Fas signal complex formation in the human autoimmune lymphoproliferative syndrome, type Ia. Proc Natl Acad Sci USA 96: 4552–4557
Jackson CE, Fischer RE, Hsu AP, Anderson SM, Choi Y, Wang J, Dale JK, Fleisher TA, Middelton LA, Sneller MC, Lenardo MJ, Straus SE, Puck JM 1999 Autoimmune lymphoproliferative syndrome with defective Fas: genotype influences penetrance. Am J Hum Genet 64: 1002–1014
Roesler J, Izquierdo JM, Ryser M, Rosen-Wolff A, Gahr M, Valcarcel J, Lenardo MJ, Zheng L 2005 Haploinsufficiency, rather than the effect of an excessive production of soluble CD95 (CD95{Delta}TM), is the basis for ALPS Ia in a family with duplicated 3′ splice site AG in CD95 intron 5 on one allele. Blood 106: 1652–1659
Bettinardi A, Brugnoni D, Quiros-Roldan E, Malagoli A, La Grutta S, Correra A, Notarangelo LD 1997 Missense mutations in the Fas gene resulting in autoimmune lymphoproliferative syndrome: a molecular and immunological analysis. Blood 89: 902–909
Drappa J, Vaishnaw AK, Sullivan KE, Chu JL, Elkon KB 1996 Fas gene mutations in the Canale-Smith syndrome, an inherited lymphoproliferative disorder associated with autoimmunity. N Engl J Med 335: 1643–1649
van der Werff ten Bosch J, Schotte P, Ferster A, Azzi N, Boehler T, Laurey G, Arola M, Demanet C, Beyaert R, Thielemans K, Otten J 2002 Reversion of autoimmune lymphoproliferative syndrome with an antimalarial drug: preliminary results of a clinical cohort study and molecular observations. Br J Haematol 117: 176–188
Siegel RM, Muppidi JR, Sarker M, Lobito A, Jen M, Martin D, Straus SE, Lenardo MJ 2004 SPOTS: signaling protein oligomeric transduction structures are early mediators of death receptor-induced apoptosis at the plasma membrane. J Cell Biol 167: 735–744
Rieux-Laucat F, Blachere S, Danielan S, de Villartay JP, Oleastro M, Solary E, Bader-Meunier B, Arkwright P, Pondare C, Bernaudin F, Chapel H, Nielsen S, Berrah M, Fischer A, Le Deist F 1999 Lymphoproliferative syndrome with autoimmunity: a possible genetic basis for dominant expression of the clinical manifestations. Blood 94: 2575–2582
van der Burg M, de Groot R, Comans-Bitter WM, den Hollander JC, Hooijkaas H, Neijens HJ, Berger RM, Oranje AP, Langerak AW, van Dongen JJ 2000 Autoimmune lymphoproliferative syndrome (ALPS) in a child from consanguineous parents: a dominant or recessive disease?. Pediatr Res 47: 336–343
Kasahara Y, Wada T, Niida Y, Yachie A, Seki H, Ishida Y, Sakai T, Koizumi F, Koizumi S, Miyawaki T, Taniguchi N 1998 Novel Fas (CD95/APO-1) mutations in infants with a lymphoproliferative disorder. Int Immunol 10: 195–202
Le Deist F, Emile JF, Rieux-Laucat F, Benkerrou M, Roberts I, Brousse N, Fischer A 1996 Clinical, immunological, and pathological consequences of Fas-deficient conditions. Lancet 348: 719–723
Shi X, Xie C, Kreska D, Richardson JA, Mohan C 2002 Genetic dissection of SLE: SLE1 and FAS impact alternate pathways leading to lymphoproliferative autoimmunity. J Exp Med 196: 281–292
Do Y, Rafi-Janajreh AQ, McKallip RJ, Nagarkatti PS, Nagarkatti M 2003 Combined deficiency in CD44 and Fas leads to exacerbation of lymphoproliferative and autoimmune disease. Int Immunol 15: 1327–1340
Vacek MM, Schaffer AA, Davis J, Fischer RE, Dale JK, Adams S, Straus SE, Puck JM 2006 HLA B44 is associated with decreased severity of autoimmune lymphoproliferative syndrome in patients with CD95 defects (ALPS type Ia). Clin Immunol 118: 59–65
Clementi R, Chiocchetti A, Cappellano G, Cerutti E, Ferretti M, Orilieri E, Dianzani I, Ferrarini M, Bregni M, Danesino C, Bozzi V, Putti MC, Cerutti F, Cometa A, Locatelli F, Maccario R, Ramenghi U, Dianzani U 2006 Variations of the perforin gene in patients with autoimmunity/lymphoproliferation and defective Fas function. Blood 108: 3079–3084
Ogata Y, Kimura M, Shimada K, Wakabayashi T, Onoda H, Katagiri T, Matsuzawa A 1993 Distinctive expression of lprcg in the heterozygous state on different genetic backgrounds. Cell Immunol 148: 91–102
Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J 2000 Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1: 489–495
Peter ME, Krammer PH 2003 The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ 10: 26–35
Acknowledgements
We are grateful to F. Rieux-Laucat for EBV-BLCL of patient 1.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported by the Deutsche Forschungsgemeinschaft (K.-M.D.) and an inhouse grant of the University of Ulm (H.F. and C.P.).
Rights and permissions
About this article
Cite this article
Fuchs, H., Posovszky, C., Lahr, G. et al. Residual CD95-Pathway Function in Children With Autoimmune Lymphoproliferative Syndrome Is Independent From Clinical State and Genotype of CD95 Mutation. Pediatr Res 65, 163–168 (2009). https://doi.org/10.1203/PDR.0b013e318191f7e4
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e318191f7e4
