
Abstract
Placenta requires energy to support its rapid growth, maturation, and transport function. Fatty acids are used as energy substrates in placenta, but little is known about the role played by carnitine in this process. We have investigated the role of carnitine in the expression of the enzymes involved in fatty acid β-oxidation in placenta of OCTN2−/− mice with defective carnitine transporter (OCTN2). Heterozygous (OCTN2+/−) female mice were mated with heterozygous (OCTN2+/−) male mice. Pregnant mice were killed and fetuses and placentas were collected. Carnitine was measured using HPLC and tandem mass spectrometry. Immunohistochemistry was used to detect enzyme expression. Enzyme activities were measured spectrophotometrically. The fetal and placental weights were similar among the three genotypes (OCTN2+/+, OCTN2+/−, and OCTN2−/−). The levels of carnitine were markedly reduced (<20%) in homozygous OCTN2−/− null fetuses and placentas compared with wild-type OCTN2+/+ controls. However, carnitine concentration in placenta was 2- to 7-fold higher than in the fetus in all three genotypes. Immunohistochemistry revealed that β-oxidation enzymes are expressed in trophoblast cells. Catalytic activities of these enzymes were present at comparable levels in wild-type (OCTN2+/+) and homozygous (OCTN2−/−) mouse placentas, with the exception of SCHAD, for which activity was significantly higher in OCTN2−/− placentas than in OCTN2+/+ placentas. These data show that placental OCTN2 is obligatory for accumulation of carnitine in placenta and fetus, that fatty acid β-oxidation enzymes are expressed in placenta, and that reduced carnitine levels up-regulate the expression of SCHAD in placenta.
Similar content being viewed by others


Main
Carnitine is an obligatory cofactor for the transport of long-chain fatty acids into mitochondria for subsequent β-oxidation (1). Carnitine deficiency is defined as a metabolic state in which the carnitine concentration in plasma and tissues falls below 10–20% of normal values. Biologic consequences of carnitine deficiency mimic primary β-oxidation defects (2). OCTN2 (also known as SLC22A5) is a plasma membrane transporter that mediates the Na+-coupled entry of carnitine into mammalian cells (3). Absence of OCTN2 function leads to excessive loss of carnitine in the urine due to defective reabsorption of filtered carnitine and consequently causes carnitine deficiency. Genetic defects in OCTN2 are the cause of primary carnitine deficiency in humans, a condition associated with hypoglycemia, hyperammonemia, skeletal myopathy, and cardiomyopathy (4). The homozygous OCTN2 null (OCTN2−/−) mouse, which lacks a functional carnitine transporter OCTN2 due to a missense mutation (leucine at position 352 mutated to arginine), is an established model of systemic carnitine deficiency (5). The OCTN2 null mouse is also known as the juvenile visceral steatosis (JVS) mouse because of accumulation of fat in viscera due to defects in fatty acid oxidation caused by carnitine deficiency. This mouse is an ideal model to study the biologic consequences of not only carnitine deficiency but also fatty acid oxidation defects. This mouse model is also useful to study how carnitine deficiency modulates various metabolic pathways to compensate for lack of long-chain fatty acid metabolism. Homozygous OCTN2 null mice develop fatty infiltration of liver and other viscera, hypoglycemia, and, without therapeutic intervention with carnitine, die within 3–4 wk after birth with dilated cardiomyopathy. Heterozygous (OCTN2+/−) mice appear healthy at birth but with age they show a similar cardiomyopathic phenotype (5, 6). Most of the inherited defects of long-chain fatty acid oxidation that develop secondary carnitine deficiency have a similar phenotype. The role of carnitine in placental metabolism and consequently in fetal development has not been investigated, although there is scant data suggesting carnitine deficiency leads to intrauterine growth retardation (7, 8).
The placenta is a unique organ in the sense that, although genetically of fetal origin, it has to interact with maternal circulation to provide the fetus with all the nutrients needed for growth and to serve as an excretory organ to eliminate waste products of fetal metabolism. Accordingly, placenta requires a reliable and plentiful source of energy to maintain its functions and sustain optimal fetal growth. It was believed that glucose transported across the placenta from the maternal circulation provided all placental and fetal energy needs via glycolysis and the citric acid cycle. Because this glucose supply is constant, consistent, and reliable, it has been suggested that the placenta and fetus do not need to use alternative energy-producing metabolic pathways (9, 10). But, recent studies have shown that fatty acid oxidation (FAO) disorders in the fetus are associated with maternal complications including preeclampsia, acute fatty liver of pregnancy, and hemolysis, elevated liver enzymes and low platelets (HELLP) syndrome in humans (11–13). Because placenta serves as the sole link between the mother and the developing fetus, it is likely that the placenta plays a key role in the pathogenesis of HELLP syndrome. But, if placental energy metabolism consists entirely of glucose utilization, FAO defects in the fetus are not expected to have any significant effect on placental function despite the fact that the placenta is of fetal origin. We therefore questioned the old dogma that the placental energy metabolism consists primarily of glucose utilization and hypothesized that FAO may serve as a significant source of metabolic energy in this organ. The rationale for this hypothesis was that if FAO is important for placental energy metabolism, FAO defects in the fetus would be associated with FAO defects in the placenta, thus compromising placental function and initiating a cascade of events leading to HELLP syndrome in the mother. Recently, we provided evidence in support of this hypothesis by showing that FAO is active in the human placenta and that several key enzymes of the FAO process are expressed and active in the placenta throughout gestation (14). These findings have been replicated independently by other investigators (15).
The recent findings that placenta is capable of FAO bring up the issue of carnitine status in this tissue. Placenta expresses robust carnitine transport activity (16) and the Na+-dependent carnitine transporter OCTN2 is responsible for this transport process. It has been believed in the past that the primary physiologic role of the placental carnitine transport system is to transfer carnitine from the mother to the developing fetus. The possibility that carnitine may have an essential metabolic role in the placenta itself was never entertained because of the belief that FAO does not occur in this tissue. With the recent data showing that FAO does occur in placenta, it seems that the placental carnitine transport system may have dual functions: to transfer carnitine from the mother to the fetus and to provide carnitine to the placenta for its own metabolic needs. In the present study, we investigated the function of OCTN2 in placenta by comparing the placental and fetal content of carnitine and expression of FAO enzymes in placenta between wild-type (OCTN2+/+) and OCTN2 null (OCTN2−/−) mice.
METHODS
Animals and preparation of RNA and protein samples.
Heterozygous OCTN2+/− mice are viable and fertile. Several pairs of OCTN2+/− heterozygous males and females were mated and the day on which the vaginal plug was noted was identified as 0 dpc (days post coitus). The pregnant mothers were observed closely during gestation and killed near term on 20 dpc.
One pair of pregnant OCTN2+/− mice were allowed to deliver and the pups were observed closely for a period of 3 wk. Homozygous OCTN2−/− mice in the litters could be identified by 2–3 wk of age when they were noted to be much smaller in size and very weak. Wild-type (OCTN2−/−) and homozygous (OCTN2−/−) pups were killed at 3 wk of age to identify the various pathologies evident by this time. A cesarean section was performed on other pregnant females and both the uterine horns opened to identify individual embryo-placenta pairs. Litter size was recorded and individual placentas and corresponding embryos were carefully dissected, weighed, and grossly examined for birth defects. They were later put in chilled PBS to remove maternal blood and snap frozen at −80°C for further analysis or fixed in 10% neutral buffered formalin solution at 4°C for 24 h before paraffin embedding and immunohistochemical studies.
Wild-type (OCTN2+/+) mice were bred and viscera from pregnant OCTN2+/+ mice were collected for measurement of total, free, and acyl carnitine levels for comparison with placental tissue. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Medical College of Georgia.
Genotyping of mice.
The embryo-placenta pairs were genotyped using embryo limb genomic DNA. Direct analysis of mRNA from OCTN2−/− mice has identified a single nucleotide mutation leading to the substitution of leucine (CTG) at codon 352 with arginine (CCG). This mutation in the carnitine transporter causes loss of function (5, 6). This mutation can be detected by RFLP analysis of the genomic DNA using the restriction enzyme Cfr 13 I. First, a gene-specific PCR product (675 bp) is obtained using the primers 5′-TCT ACT AAG GGT CCT TCT CC-3′ (upstream primer) and 5′-ACC AGC TGC ATG AAG AGA AG-3′ (downstream primer) (5). The cutting site of C fr 13 I restriction enzyme is G/GNCC on one allele and CCNG/G on the other allele and RFLP analysis of this PCR product with the restriction enzyme Cfr 13 I yields two fragments (346 and 329 bp) for wild-type DNA, four fragments (346, 329, 201, and 145 bp) for heterozygous OCTN2+/− DNA, and three fragments (329, 201, and 145 bp) for homozygous OCTN2−/− DNA. This is because the mutation creates a new site for the restriction enzyme Cfr 13 I within the 346 bp PCR product. Thus, the 675 bp PCR product contains a single site for Cfr 13 I if derived from the wild-type genomic DNA and two sites for the same enzyme if derived from the mutant genomic DNA.
Immunohistochemistry.
Mouse placentas were fixed in 10% formalin and 2- to 5-μm thick sections of paraffin-embedded tissue were cut, applied to glass slides, deparaffinized in xylene, and rehydrated in an ethanol gradient. Endogenous per-oxidase activity was quenched by incubating the specimens in 3% H2O2 in methanol for 30 min. After equilibrating for 5 min in distilled water, the samples were subjected to heat antigen retrieval using citrate buffer (pH 6.0). The samples were heated at maximum power in a microwave for 5 min, cooled for 5 min, reheated for 5 min, and allowed to stand at room temperature for 20 min.
The slides were then washed and blocked using an avidin/biotin blocking kit (Vector Laboratories, Burlingame, CA, U.S.A.) for 30 min followed by a blocking buffer (PerkinElmer Life Science Products, Boston, MA, U.S.A.) for 30 min. The blocking buffer was removed, and the sections were exposed to primary rabbit polyclonal antisera raised against one of the following enzymes at the indicated dilution: MCAD (1:200), LCAD (1:400), VLCAD (1:200), SCHAD (1:200), LCHAD (1:400), or LKAT (1:400). The primary antibody was applied with 0.3% Triton X-100 in PBS overnight at 4°C. After two washes with PBS the next day, secondary goat anti-rabbit biotinylated antibody (PerkinElmer Life Science Products) was applied at a dilution of 1:800 for 1 h at room temperature. The tertiary reagent was streptavidin horseradish peroxidase (DAKO, Carpinteria, CA, U.S.A.) at a dilution of 1:1000 for 1 h at room temperature followed by application of 3,3-diaminobenzidine substrate for 1–5 min. The slides were rinsed, counterstained with Mayer's hematoxylin, dehydrated in ethanol, cleared with xylene, and mounted with glass coverslips using Histomount (Zymed Laboratories, South San Francisco, CA, U.S.A>). We stained two to five sets of placental tissue from near-term placentas for all six FAO enzymes.
Western blot analyses.
About 50 mg of placental tissue freed of maternal blood was homogenized in a buffer containing 0.1 M sodium phosphate, 0.5 mM EDTA, and 0.5% Triton X-100 with protease inhibitors using a tissue homogenizer. The placental homogenate was sonicated three times for 10 s each on ice. The homogenates were subjected to centrifugation at 3000 × g for 5 min, and the protein concentration of the supernatant was measured by the Bradford method. Fifteen micrograms of protein was analyzed by immunoblotting with rabbit polyclonal antisera raised against one of the six different FAO enzymes at the following dilutions: MCAD (1:1000), LCAD (1:5000), VLCAD (1:500), SCHAD (1:5000), LCHAD (1:3000), and LKAT (1:2000). Incubation with secondary antibody (goat anti-rabbit IgG, 1:1000 dilution) and visualization with diaminobenzidine reagent were done until the protein bands were visible. A total of two to five blots was prepared for each enzyme and the blots were analyzed with an AlphaImager 3400 (Alpha Innotech Corp., San Leandro, CA, U.S.A.) using its AlphaEase image analysis software for densitometry. Densitometry data were subjected to statistical analysis.
Measurement of enzyme activities.
The activities of SCHAD, LCHAD, and LKAT in placental homogenates were measured as described (17, 18). For LCHAD and SCHAD, assays are based on decrease in absorbance of NADH at 340 nm when tissue homogenate is incubated with 3-ketopalmitoyl-CoA and acetoacetyl-CoA, respectively, in the presence of NADH. Analysis of LKAT activity is based on decrease in absorbance at 303 nm due to loss of the 3-keto group when 3-keto-palmitoyl-CoA is incubated with CoA.
HPLC and mass spectrometry.
Total carnitine content was measured using standard HPLC methods; total, free, and acyl carnitine fractions were determined by tandem mass spectrometry as described (19, 20).
Statistical analyses.
Three enzyme activity measurements were performed for each sample and a total of 26 samples were compared with each other after categorizing them by genotype. Data analysis was performed by ANOVA and p values were calculated. Data were later subjected to Student-Newman-Keul's post hoc analysis for multiple comparisons using statistical software SPSS for PC version 11.01. Data are presented as the mean ± SE and, unless stated otherwise, statistical significance was set at p ≤ 0.05.
RESULTS
The heterozygous OCTN2+/− mice are viable and fertile but the homozygous OCTN2−/− mice survive for only about 3–4 wk without carnitine supplementation. At 3 wk of age, the body weight of OCTN2−/− mice was about 50% compared with that of age-matched wild-type mouse. The wild-type mice showed normal viscera sharply contrasting the enlarged fatty liver with steatosis of other organs and a dilated cardiomyopathy in the OCTN2−/− mice (data not shown).
There are no data in the literature on the levels of carnitine in the placenta and on how these levels compare with those in other tissues, especially the tissues that are dependent on FAO for metabolic energy. Therefore, we measured carnitine levels in various tissues, including the placenta, in wild-type mice. Table 1 shows the comparative amounts of total carnitine content of various organs of wild-type mice. The carnitine levels in placenta were 7- to 10-fold higher than in the heart (p < 0.0001).
The litter size of seven heterozygous matings (8.4 ± 1.5) was similar to that of wild-type matings (8.6 ± 1.1). Table 2 shows the data for the weights of embryos and placentas from the litters arising from three heterozygous matings. There was no statistically significant difference between the fetal and placental weights among the three genotypes. However, two of the placentas from homozygous fetuses were hydropic and their weights were 217 g and 140 g, respectively, compared with an average placental weight of 102 g.
Table 3 describes the placental and fetal carnitine content in the three genotypes. The carnitine content of homozygous OCTN2−/− placentas and embryos was <20% of the corresponding wild-type tissue with intermediate amount of carnitine in the heterozygous mice (p < 0.0001). The placental carnitine content was 2- to 7-fold higher than fetal carnitine content in all three genotypes. There was a uniform decrease in the relative amount of total, free, and acyl carnitine content among the heterozygous (OCTN2+/−) and homozygous (OCTN2−/−) placentas and fetuses.
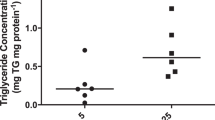
We also determined the activities of three enzymes involved in FAO in placental homogenates obtained from mice with different genotypes (Table 4). There were no differences among the three genotype groups in the enzyme activities of LCHAD and LKAT, which are involved in the metabolism of long-chain fatty acids. In contrast, the enzyme activity of SCHAD, which is required for the metabolism of short-chain fatty acids, was higher in homozygous OCTN2−/− placentas than in wild-type OCTN2+/+ placentas (p = 0.05).
Figure 1 is a representative Western blot analyzing the expression of six different FAO enzymes in near-term placental extracts. Densitometric analysis of Western blots indicated that expression of SCHAD was 1.5- to 2.0-fold higher in the homozygous OCTN2−/− placentas compared with the wild-type OCTN2+/+ placentas. There were no measurable differences among the three genotype groups for MCAD, LCAD, VLCAD, LCHAD, and LKAT expression.

Western blots showing the expression of FAO enzymes in three genotypes of OCTN2 mice near-term placenta. Expression of all six FAO enzymes is evident in placentas of all three genotypes. Expression of SCHAD is significantly higher in the OCTN2−/− homozygous placentas than in OCTN2+/+ wild-type placentas.
Figures 2 and 3 are photomicrographs of expression of six β-oxidation pathway enzymes by immunohistochemistry in near-term wild-type mouse placentas. Minimal nonspecific background staining was observed in control sections processed without primary antibody (panel A in each figure). Figure 2, B–D, shows immunoreactivity for MCAD, LCAD, and SCHAD, respectively. There was expression of all three enzymes in spongiotrophoblast cells facing the maternal side but very faint staining was observed in the trabecular cords, which are mainly composed of precursors to differentiated trophoblast cells. Figure 3, B–D, represents staining for VLCAD, LCHAD, and LKAT, respectively, showing a similar pattern of expression for the enzymes of the β-oxidation pathway.

Immunohistochemical analysis of the expression of FAO enzymes (MCAD, LCAD, and SCHAD) in OCTN2+/+ wild-type near-term placentas (magnification ×20 for A and ×40 for B-D, bar is 20 μm). (A) Negative control with no primary antibody. (B-D) Immunoreactivity for MCAD, LCAD, and SCHAD. All three enzymes are extensively expressed with deep staining of spongiotrophoblast cells and faint staining of trabecular cords.

Immunohistochemical analysis of the expression of FAO enzymes (VLCAD, LCHAD, and LKAT) in OCTN2+/+ wild-type near-term placentas (magnification ×20 for A and ×40 for B–D, bar is 20 μm). (A) Negative control with no primary antibody. (B–D) Immunoreactivity for VLCAD, LCHAD, and LKAT. All three enzymes are extensively expressed in spongiotrophoblast cells and lighter staining of trabecular cords.
DISCUSSION
Our results show that murine placenta has the capability to metabolize fatty acids. In this study, we have shown that carnitine is concentrated 7- to 10-fold higher in the mouse placenta compared with heart and that placental carnitine content modulates the expression and activity of SCHAD, one of the mitochondrial FAO enzymes. This is the first report of direct evidence of the expression of FAO enzymes in placenta in another species besides humans, indicating FAO is important for placental growth and metabolism across species. These findings are in contrast to the established view that glucose, transported to and across the placenta from the maternal circulation, is the major source of energy for the placenta-fetus unit (10). Earlier studies in human placenta provided strong evidence that fatty acid oxidation is an active metabolic pathway in this tissue (14, 15).
The up-regulation of SCHAD in placenta associated with carnitine depletion is of significance. Oxidation of short-chain fatty acids does not require carnitine, whereas oxidation of long-chain fatty acids does. Carnitine deficiency in the placenta is expected to interfere with the utilization of long-chain fatty acids in β-oxidation. Therefore, any alterations in the expression of enzymes involved in the oxidation of long-chain fatty acids represent futile efforts to maintain energy homeostasis under conditions in which carnitine is depleted. On the other hand, facilitation of the use of short-chain fatty acids by up-regulating SCHAD is expected to be an effective compensatory mechanism to maintain the energy status in placenta under conditions of decreased carnitine levels.
Our present study is also the first to examine the carnitine content of placenta in comparison to other body tissues. It was initially surprising to find a 7- to 10-fold higher carnitine content in the placenta than in tissue such as the heart, which preferentially uses fatty acids for energy production. Thus, one wonders about the biologic role of this enormous amount of carnitine in the placenta. It has been proposed by others that carnitine may be involved in fetal growth (21) and differential gene regulation of various metabolic pathways (22–24). Carnitine may also play a protective role for the growing fetus and prevent the free radical damage that causes lipid peroxidation, cell death and apoptosis (25–27). Homozygous OCTN2−/− placental carnitine content is about 20% of the wild-type OCTN2+/+ placenta. There is still a significant amount of carnitine in placentas from OCTN2−/− mice. This could be attributed to residual action of mutated OCTN2 transporter or activity of Na+- and Cl−-coupled amino acid transporter ATB0,+ which has been shown to be highly expressed in rat and mouse placenta and is known to transport carnitine (28, 29). Alternatively there is a possibility that carnitine is biosynthesized in the mouse placenta from trimethyllysine (30) and that this biosynthetic capacity is not compromised when OCTN2 is inactive.
Fatty acids play a critical role in fetal-placental growth and metabolism during gestation; this has recently been unraveled by studies of specific mouse models. Ablation of genes encoding enzymes involved in FAO, such as LCAD, VLCAD, and TFP is associated with increased in utero fetal demise, subfertility, and fetal growth restriction (31–33). Gene knockout mouse models of transcription factors such as the peroxisome proliferator-activated receptors (PPAR) β and γ and their associated co-activator (PGC-1), which are considered the master regulators of fatty acid and glucose metabolism, exhibit embryonic lethality. These defects have specific placental phenotypes where syncytiotrophoblast cells fail to develop and sustain pregnancy (34–36). These observations provide further evidence in support of a critical role of FAO in placental development and function.
Placenta also needs to synthesize fatty acids, and ablation of the gene for fatty acid synthase (FAS) leads to fetal demise and a high rate of in utero deaths in heterozygous mice (37). Other FAO enzymes, such as enoyl-CoA isomerase, which do not appear to have a distinct placental phenotype or affect fetal growth, possibly have alternative compensatory pathways (38, 39). Knowledge about the role of FAO defects in fetal-placental metabolism has expanded over the past decade and these serious metabolic defects can be diagnosed prenatally and in the neonatal period by mass screening using tandem mass spectrometry (40, 41). Thus, it is now clear that FAO has an important metabolic function in placenta that is essential for normal fetal development and there is need to further study the biologic role played by carnitine in placenta.
Abbreviations
- MCAD:
-
medium-chain acyl CoA dehydrogenase
- LCAD:
-
long-chain acyl CoA dehydrogenase
- VLCAD:
-
very-long-chain acyl CoA dehydrogenase
- SCHAD:
-
short-chain l-3-hydroxyacyl CoA dehydrogenase
- LCHAD:
-
long-chain l-3-hydroxyacyl CoA dehydrogenase
- TFP:
-
trifunctional protein
- LKAT:
-
long-chain 3-ketoacyl-CoA thiolase
References
Rinaldo P, Matern D, Bennett MJ 2002 Fatty acid oxidation disorders. Annu Rev Physiol 64: 477–502.
Bennett MJ, Rinaldo P, Strauss AW 2000 Inborn errors of mitochondrial fatty acid oxidation. Crit Rev Clin Lab Sci 37: 1–44.
Tamai I, Ohashi R, Nezu J, Yabuuchi H, Oku A, Shimane M, Sai Y, Tsuji A 1998 Molecular and functional identification of sodium ion-dependent, high affinity human carnitine transporter OCTN2. J Biol Chem 273: 20378–20382.
Tein I 2003 Carnitine transport: pathophysiology and metabolism of known molecular defects. J Inherit Metab Dis 26: 147–169.
Nezu J, Tamai I, Oku A, Ohashi R, Yabuuchi H, Hashimoto N, Nikaido H, Sai Y, Koizumi A, Shoji Y, Takada G, Matsuishi T, Yoshino M, Kato H, Ohura T, Tsujimoto G, Hayakawa J, Shimane M, Tsuji A 1999 Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat Genet 21: 91–94.
Nikaido H, Horiuchi M, Hashimoto N, Saheki T, Hayakawa J 1995 Mapping of jvs (juvenile visceral steatosis) gene, which causes systemic carnitine deficiency in mice, on chromosome 11. Mamm Genome 6: 369–370.
Bernardini I, Evans MI, Nicolaides KH, Economides DL, Gahl WA 1991 The fetal concentrating index as a gestational age-independent measure of placental dysfunction in intrauterine growth retardation. Am J Obstet Gynecol 164: 1481–1487; discussion 1487–1490.
Bayes R, Campoy C, Goicoechea A, Peinado JM, Pedrosa T, Baena RM, Lopez C, Rivero M, Molina-Font JA 2001 Role of intrapartum hypoxia in carnitine nutritional status during the early neonatal period. Early Hum Dev 65:S103–S110.
Kaufmann P, Scheffen I 1999 Placental development. In: Polin RA, Fox F (eds) Fetal and Neonatal Physiology. Saunders, New York, 47–56.
Illsley NP 2000 Glucose transporters in the human placenta. Placenta 21: 14–22.
Ibdah JA, Bennett MJ, Rinaldo P, Zhao Y, Gibson B, Sims HF, Strauss AW 1999 A fetal fatty-acid oxidation disorder as a cause of liver disease in pregnant women. N Engl J Med 340: 1723–1731.
Ibdah JA, Yang Z, Bennett MJ 2000 Liver disease in pregnancy and fetal fatty acid oxidation defects. Mol Genet Metab 71: 182–189.
Rinaldo P, Studinski AL, Matern D 2001 Prenatal diagnosis of disorders of fatty acid transport and mitochondrial oxidation. Prenat Diagn 21: 52–54.
Shekhawat P, Bennett MJ, Sadovsky Y, Nelson DM, Rakheja D, Strauss AW 2003 Human placenta metabolizes fatty acids: implications for fetal fatty acid oxidation disorders and maternal liver diseases. Am J Physiol Endocrinol Metab 284:E1098–E1105.
Oey NA, den Boer ME, Ruiter JP, Wanders RJ, Duran M, Waterham HR, Boer K, van der Post JA, Wijburg FA 2003 High activity of fatty acid oxidation enzymes in human placenta: implications for fetal-maternal disease. J Inherit Metab Dis 26: 385–392.
Wu X, Huang W, Prasad PD, Seth P, Rajan DP, Leibach FH, Chen J, Conway SJ, Ganapathy V 1999 Functional characteristics and tissue distribution pattern of organic cation transporter 2 (OCTN2), an organic cation/carnitine transporter. J Pharmacol Exp Ther 290: 1482–1492.
Bennett MJ, Weinberger MJ, Kobori JA, Rinaldo P, Burlina AB 1996 Mitochondrial short-chain l-3-hydroxyacyl-coenzyme A dehydrogenase deficiency: a new defect of fatty acid oxidation. Pediatr Res 39: 185–188.
Venizelos N, Ijlst L, Wanders RJ, Hagenfeldt L 1994 beta-Oxidation enzymes in fibroblasts from patients with 3-hydroxydicarboxylic aciduria. Pediatr Res 36: 111–114.
Matern D, Magera MJ 2001 Mass spectrometry methods for metabolic and health assessment. J Nutr 131: 1615S–1620S.
Stevens RD, Hillman SL, Worthy S, Sanders D, Millington DS 2000 Assay for free and total carnitine in human plasma using tandem mass spectrometry. Clin Chem 46: 727–729.
Genger H, Enzelsberger H, Salzer H 1988 Carnitine in therapy of placental insufficiency-initial experiences. Z Geburtshilfe Perinatol 192: 155–157.
Tomomura M, Tomomura A, Musa DA, Horiuchi M, Takiguchi M, Mori M, Saheki T 1997 Suppressed expression of the urea cycle enzyme genes in the liver of carnitine-deficient juvenile visceral steatosis (JVS) mice in infancy and during starvation in adulthood. J Biochem (Tokyo) 121: 172–177.
Masuda M, Kobayashi K, Horiuchi M, Terazono H, Yoshimura N, Saheki T 1997 A novel gene suppressed in the ventricle of carnitine-deficient juvenile visceral steatosis mice. FEBS Lett 408: 221–224.
Athanassakis I, Mouratidou M, Sakka P, Evangeliou A, Spilioti M, Vassiliadis S 2001 l-carnitine modifies the humoral immune response in mice after in vitro or in vivo treatment. Int Immunopharmacol 1: 1813–1822.
Vanella A, Russo A, Acquaviva R, Campisi A, Di Giacomo C, Sorrenti V, Barcellona ML 2000 l-propionyl-carnitine as superoxide scavenger, antioxidant, and DNA cleavage protector. Cell Biol Toxicol 16: 99–104.
Hagen TM, Liu J, Lykkesfeldt J, Wehr CM, Ingersoll RT, Vinarsky V, Bartholomew JC, Ames BN 2002 Feeding acetyl-l-carnitine and lipoic acid to old rats significantly improves metabolic function while decreasing oxidative stress. Proc Natl Acad Sci U S A 99: 1870–1875.
Wainwright MS, Mannix MK, Brown J, Stumpf DA 2003 l-carnitine reduces brain injury after hypoxia-ischemia in newborn rats. Pediatr Res 54: 688–695.
Nakanishi T, Hatanaka T, Huang W, Prasad PD, Leibach FH, Ganapathy ME, Ganapathy V 2001 Na+- and Cl−-coupled active transport of carnitine by the amino acid transporter ATB(0,+) from mouse colon expressed in HRPE cells and Xenopus oocytes. J Physiol 532: 297–304.
Cramer S, Beveridge M, Kilberg M, Novak D 2002 Physiological importance of system A-mediated amino acid transport to rat fetal development. Am J Physiol Cell Physiol 282:C153–C160.
Carter AL, Abney TO, Lapp DF 1995 Biosynthesis and metabolism of carnitine. J Child Neurol 10:S3–S7.
Kurtz DM, Rinaldo P, Rhead WJ, Tian L, Millington DS, Vockley J, Hamm DA, Brix AE, Lindsey JR, Pinkert CA, O'Brien WE, Wood PA 1998 Targeted disruption of mouse long-chain acyl-CoA dehydrogenase gene reveals crucial roles for fatty acid oxidation. Proc Natl Acad Sci U S A 95: 15592–15597.
Cox KB, Hamm DA, Millington DS, Matern D, Vockley J, Rinaldo P, Pinkert CA, Rhead WJ, Lindsey JR, Wood PA 2001 Gestational, pathologic and biochemical differences between very long-chain acyl-CoA dehydrogenase deficiency and long-chain acyl-CoA dehydrogenase deficiency in the mouse. Hum Mol Genet 10: 2069–2077.
Ibdah JA, Paul H, Zhao Y, Binford S, Salleng K, Cline M, Matern D, Bennett MJ, Rinaldo P, Strauss AW 2001 Lack of mitochondrial trifunctional protein in mice causes neonatal hypoglycemia and sudden death. J Clin Invest 107: 1403–1409.
Barak Y, Liao D, He W, Ong ES, Nelson MC, Olefsky JM, Boland R, Evans RM 2002 Effects of peroxisome proliferator-activated receptor delta on placentation, adiposity, and colorectal cancer. Proc Natl Acad Sci U S A 99: 303–308.
Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, Koder A, Evans RM 1999 PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell 4: 585–595.
Lim H, Gupta RA, Ma WG, Paria BC, Moller DE, Morrow JD, DuBois RN, Trzaskos JM, Dey SK 1999 Cyclo-oxygenase-2-derived prostacyclin mediates embryo implantation in the mouse via PPARdelta. Genes Dev 13: 1561–1574.
Chirala SS, Chang H, Matzuk M, Abu-Elheiga L, Mao J, Mahon K, Finegold M, Wakil SJ 2003 Fatty acid synthesis is essential in embryonic development: fatty acid synthase null mutants and most of the heterozygotes die in utero. Proc Natl Acad Sci U S A 100: 6358–6363.
Janssen U, Stoffel W 2002 Disruption of mitochondrial beta-oxidation of unsaturated fatty acids in the 3,2-trans-enoyl-CoA isomerase-deficient mouse. J Biol Chem 277: 19579–19584.
Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SF, Silverstein RL 1999 A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem 274: 19055–19062.
Yang Z, Yamada J, Zhao Y, Strauss AW, Ibdah JA 2002 Prospective screening for pediatric mitochondrial trifunctional protein defects in pregnancies complicated by liver disease. JAMA 288: 2163–2166.
Ibdah JA, Zhao Y, Viola J, Gibson B, Bennett MJ, Strauss AW 2001 Molecular prenatal diagnosis in families with fetal mitochondrial trifunctional protein mutations. J Pediatr 138: 396–399.
Acknowledgements
The authors thank Arnold Strauss, M.D., of Vanderbilt University, Nashville, TN, for supplying us with antibodies raised against the FAO enzymes.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported in part by a combined intramural grant program grant of University System of Georgia (P.S.S.) and National Institutes of Health grant HL 64196 (V.G.).
Rights and permissions
About this article
Cite this article
Shekhawat, P., Yang, HS., Bennett, M. et al. Carnitine Content and Expression of Mitochondrial β-Oxidation Enzymes in Placentas of Wild-Type (OCTN2+/+) and OCTN2 Null (OCTN2−/−) Mice. Pediatr Res 56, 323–328 (2004). https://doi.org/10.1203/01.PDR.0000134252.02876.55
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000134252.02876.55
This article is cited by
-
The Placental Barrier: the Gate and the Fate in Drug Distribution
Pharmaceutical Research (2018)
