
Abstract
This study was undertaken to clarify whether seizures in the newborn cause damage to the healthy brain and, more specifically, to determine the extent to which seizures may contribute to the brain-damaging effects of hypoxia-ischemia (HI). Seizures were induced in 10-d-old rat pups with kainic acid (KA). Seizure duration was determined electrographically. HI was induced by common carotid artery ligation followed by exposure to 8% oxygen for either 15 or 30 min. Six groups of animals were assessed:1) controls [neither KA nor HI (group I)];2) group II, KA alone;3) group III, 15 min HI alone;4) group IV,15 min HI plus KA;5) group V, 30 min HI alone; and 6) group VI, 30 min HI plus KA. Animals were assessed neuropathologically at 3 (early) and 20 (late) d of recovery. KA injection without hypoxia resulted in continuous clinical and electrographic seizures lasting a mean of 282 min. No neuropathologic injury was seen in groups I (no HI or KA), II (KA alone), III (15 min HI alone), or IV (15 min HI and KA). Animals in group V (30 min HI alone) displayed brain damage with a mean score of 2.3 and 0.60 at 3 and 20 d of recovery, respectively. Animals in group VI (30 min HI and KA) had a mean score of 12.1 and 3.65 at 3 and 20 d of recovery, respectively. Compared with group V, the increased damage as a result of the seizure activity in group VI occurred exclusively in the hippocampus. Status epilepticus in the otherwise “healthy” neonatal brain does not cause neuropathologic injury. However, seizures superimposed on HI significantly exacerbate brain injury in a topographically specific manner.
Similar content being viewed by others

Main
Seizures are the most frequent of neurologic signs in the newborn infant and usually reflect a serious underlying derangement of the brain. Because of the difficulty in recognizing and diagnosing seizures in this age group, their exact incidence is uncertain, but was estimated to be approximately 0.5% in one large population-based study from the National Collaborative Perinatal Project (1). In premature and low birth weight infants the incidence rises dramatically to upward of 25% (2, 3).
Controversy exists as to whether seizures per se damage the developing brain (4, 5). Certainly, the immature brain appears to be much less vulnerable than its mature counterpart in its susceptibility to seizure-induced brain injury, and experimental data from seizures induced in mature animals are clearly not applicable to the developing brain (4). Despite this uncertainty, neonatal seizures are perceived as medical emergencies and treated aggressively with antiepileptic agents, which may in themselves be detrimental to the newborn infant and ongoing brain development.
In studies published to date, seizures have been induced in immature animals that have otherwise healthy brains, and with no coexistent insult. However, in clinical practice, most newborns who experience seizures do so as a result of significant underlying brain pathology. Although the healthy immature brain appears quite resistant to injury from prolonged seizures, the situation may be quite different if the brain has already been compromised by another insult.
Perinatal HI is the most common cause for neonatal seizures, accounting for ≥40% of all cases. When combined with other forms of perinatal cerebral ischemic events such as bland and hemorrhagic infarction, cerebrovascular disease accounts for 50 to 60% of all seizures in the term and preterm infant (6, 7). Seizures are seen in the majority of babies with significant hypoxic-ischemic encephalopathy, and usually occur within the first 24 h of life (8). They are often prolonged and frequent, and status epilepticus is not rare. The neuropathology of perinatal HI varies depending on the gestational age of the infant and the nature of the insult, but has been well documented by Volpe (9). An animal model of perinatal hypoxic-ischemic brain damage in the immature rat has been established by Rice et al. (10), and neuropathologic changes of selective neuronal necrosis or infarction of the cerebral hemisphere ipsilateral to the arterial ligation have been described (10–12).
In this study, we determined first whether prolonged seizures were associated with neuronal cell death in the immature rat, and second, using an established model of perinatal hypoxic-ischemic brain damage, whether seizures superimposed on a mild to moderate hypoxic-ischemic insult would accentuate the neuronal injury seen with HI alone.
METHODS
Female Wistar rats (Charles River, Montreal, Canada) were bred in our laboratory. The rat pups were reared with their dams in a temperature-controlled environment at 21 ± 0.5°C, with a relative humidity of 30%, and a 12-h on/off lighting schedule. The date of experimentation was postnatal d 10 (date of birth, d 1). This study received ethical approval from the Animal Care Committee at the University of Saskatchewan.
Induction of Cerebral HI
Cerebral HI was induced in 10-d-old rat pups using the modified Levine preparation described by Rice et al. (10) and previously used by our laboratory (13, 14). Rat pups were anesthetized with halothane (4% induction, 1% maintenance) and placed in the supine position, and their right common carotid artery was permanently ligated through a midline neck incision measuring no more than 1 cm. The incision was then sutured. After a 2-h recovery period with their dam, HI was induced by placing each animal in individual 500-mL glass jars through which a gas mixture of 8% oxygen/balance nitrogen was delivered via inlet and outlet portals. The pups were exposed to hypoxia for either 15 or 30 min, after which they were allowed to recover for an additional 30 min before the induction of seizures. Thermoregulation was maintained by placing each animal, within the glass jars, in a single-walled neonatal incubator (Air Shields; Healthdyne Co., Hatford, PA, U.S.A.), thermocontrolled to 34 ± 0.2°C (nesting temperature).
Placement of Electrodes
After the surgical procedure required for ligation of the right common carotid artery, selected rat pups were rotated to the prone position and placed in a Kopf stereotactic frame adapted for neonatal rats. While still under anesthesia (1% maintenance), a bifrontooccipital scalp flap was made and temporarily secured with sutures. The membrane was removed from the skull with microdissecting forceps, and the skull landmark bregma was identified (the point that the coronal suture crosses the sagittal suture). Using stereotaxic coordinates from Snead et al. (15), burr holes were made with a 25-gauge needle, and electrodes were placed in the right hemispheric sensorimotor cortex (−2.5 mm from bregma; −2.0 mm from midsagittal suture; −2.0 mm from skull) and the left hemispheric dorsal hippocampus (−2.9 mm from bregma; +2.5 mm from midsagittal suture; and −3.5 mm from skull) and held in place with glass polymer. Referential electrodes were placed along the nasal ridge. ECG was monitored via an s.c. electrode placed in the left abdominal wall. Temperatures were simultaneously and continuously monitored using a flexible YSI 511 probe (YSI Inc., Yellow Springs, OH, U.S.A.) inserted rectally to 0.25 cm and secured to the tail of the animal. All temperature probes were connected to a multichannel temperature monitor that conditioned the signals to the computer (A/D conversion, Dycor) and software analysis (Lab Notebook 4.3), which converted voltages into degrees centigrade. Temperatures were displayed on a VGA monitor in real time. The average time for complete surgery was 25 min.
Preliminary Studies
Seizures.
In the immature human brain, clinical seizure activity infrequently correlates with electrographic events (16). Because the goals of this study were to determine the neuropathologic effects of prolonged seizures in the healthy and ischemic brain, preliminary studies were conducted to ensure accurate correlation of electrographic and clinical seizures. Moreover, we defined seizures by continuous paroxysmal discharges on EEG, as we considered this to be a more accurate reflection of abnormal neuronal activity, and, therefore, more likely to be associated with neuropathologic injury.
KA (Sigma Chemical Co., St. Louis, MO, U.S.A.), a powerful glutamate analog, was used to induce seizures. To determine the quantity of KA required to induce prolonged electrographic status epilepticus, groups of rat pups underwent stereotactic implantation of cortical electrodes as described above. The KA was dissolved in normal saline and pH adjusted to 7.40 using HCO3−. The proconvulsant was then injected s.c. at concentrations of between 1 and 8 mg/kg as a single dose, at a fixed volume of 0.1 mL. The animals were then placed within the thermocontrolled neonatal incubator and continuously monitored for heart rate and temperature. Electrographic seizures, and their correlation to clinical manifestations, were also documented throughout.
These studies indicated that single injections of KA at doses of ≤8 mg/kg resulted in prolonged, although intermittent, seizures, and at high doses, unacceptable mortality. Electrographic status epilepticus began 27 min after single injections of KA. After 1 h the previously continuous EEG seizure activity became intermittent, and after 2 h, prolonged periods of inactivity lasting 5–10 s were frequent. In all cases, electrographic seizures no longer occurred 3 h after injection. Mortality rates increased with increasing concentrations of KA such that at doses of 1–3 mg/kg there was no mortality, whereas doses of 4, 6, and 8 mg/kg yielded mortality rates of 25, 50, and 100%, respectively.
Given that with the above protocol we were unable to produce continuous prolonged electrographic seizures without high mortality rates, the following experimental paradigm was developed whereby continuous electrographic seizures could be maintained with consistent clinically evident seizures. Before the injection of the KA, a small-bore polyethylene catheter (PE10) was inserted s.c. between the scapula of each rat pup, and held in place with acrocyanate adhesive. Rat pups each received a single s.c. injection of 3 mg/kg followed by a continuous s.c. infusion of 2 mg·kg−1·h−1 for 3 h. Control animals received normal saline in the same fashion, in a volume equal to that of the KA (150 μL followed by an infusion of 100 μL/h for 3 h).
Animals in whom electrographic recordings were undertaken were anesthetized after the insult, their implants were removed, and they were allowed to recover with their dams to 30 d postnatal age. Subsequently the rat pups were killed as described below, and their brains were assessed for electrographic probe placement. Animals who were part of the neuropathologic assessment were monitored clinically as we found consistent correlation between clinical and electrographic seizures with above-described experimental paradigm and probe placement was not required.
Hypoxia-ischemia.
To determine whether seizures contribute to the injury incurred by a hypoxic-ischemic injury in the immature brain, studies were undertaken to assess that level of HI required to ascertain the “minimal lesion.” A minimal lesion injury was used for two reasons. First, in the immature rat, prolonged HI inevitably results in moderate to severe injury in 92% of animals (10, 11). Enhancement of lesions with this degree of underlying brain damage would not be possible to detect, and hence may inappropriately lead to a false-negative study. Second, from a clinical perspective, seizures occur most frequently in those infants who experience a stage II or stage III encephalopathy (moderate to severe degree of asphyxia) (8), many of whom, particularly after stage II, however, experience only minor late neurologic sequelae (9). Hence groups of 10-d-old rat pups underwent unilateral common carotid artery ligation and exposure to hypoxia, as described above, for periods of 15, 30, 45, and 60 min. The pups were then allowed to recover with their dams to 30 d postnatal age, after which they were assessed for neuropathologic damage.
Recordings.
Electrodes were connected to a Grass polygraph (electroencephalograph model 8-18D, Grass Instrument Co., Quincy, MA, U.S.A.), and recordings were made for 2 h during the stabilization period before the onset of seizures (baseline), continuing throughout the KA infusion, and until the electrographic seizure activity became intermittent or abated. Paper speed was 30 mm/s and sensitivity was 10 μV/mm. ECGs were simultaneously recorded by placing an electrode s.c. within the abdominal wall of selected animals.
Study Groups
Six groups of animals were assessed: Group I consisted of control animals who did not receive HI and who were injected with normal saline rather than KA (n = 8). Group II consisted of animals injected with KA but not exposed to HI (n = 22). Group III consisted of animals exposed to 15 min of HI and injected with normal saline rather than KA (n = 18). Group IV consisted of animals injected with KA 30 min after exposure to 15 min of HI (n = 21). Group V consisted of animals exposed to 30 min of HI and injected with normal saline rather than KA (n = 18). Group VI consisted of animals injected with KA 30 min after exposure to 30 min of HI (n = 24). As indicated below, early and late pathologic assessment was obtained for each of the groups, and as such, have been designated as early (a) or late (b). The number of animals in either the early or late neuropathologic categories is indicated in the relevant section in results.
Neuropathologic Analysis
Surviving animals in all groups were examined neuropathologically at 3 (early pathology) or 20 d (late pathology) of recovery. All animals were recovered with their dams. On the day of sacrifice, each animal was deeply anesthetized with 5% halothane in a 20% O2/80% N2 environment. The animals were then decapitated, and the brains quickly removed and placed in formaldehyde-glacial acetic acid-methanol 1:1:8 (FAM) for a minimum of 24 h before being embedded in paraffin.
Brains were then sectioned coronally from anterior to posterior. Serial sections of 6 μm in thickness were taken every 0.5 mm and stained with hematoxylin and eosin (3- and 20-d pathology), and GFAP (20-d pathology only). Evaluation of brain injury was performed on each section by two investigators (E.W. and J.Y.) who were blinded to group assignment.
The degree of brain damage was graded on four sections ranging from normal to >75% damage, including infarction (Table 1), using a modification of the method described by Cataltepe et al. (17). Anteriorly, sections were taken starting with a cut through the midportion of the body of the anterior commissure, and included the anterior portions of cerebral cortex and caudate nucleus. Subsequently, caudal sections were taken every 1 mm such that each included the cerebral cortex, dorsal hippocampus, thalamus, and amygdaloid nucleus. Evidence of irreversible neuronal injury included eosinophilia or clearing of the perikaryon, pyknosis and karyorrhexis of the nucleus at 3 d after insult, and positive GFAP staining at 20 d after insult. The histologic evaluation was made under a 40× objective, and each subdivision of the sections was scored on a scale of 1–5 (Table 1). The most anterior section was divided into the cerebral cortex (subdivided into anterior, middle, and posterior artery circulatory distributions) and caudate. The posterior sections were divided into cerebral cortex (subdivided as described above), thalamus, striatum, and the hippocampus (further subdivided into dentate gyrus, CA1, medial and lateral CA2, medial and lateral CA3, and CA4 (Fig. 3A). The total sum of the scores given each of the segments were added together, and then divided by the number of sections that was included for that brain, to derive a score that represented the mean histologic damage score for the entire brain (maximum whole brain damage score, 70).

Representative examples of hippocampal injury using hematoxylin and eosin staining at 3 d of recovery (early pathology). Regions of the hippocampus used for neuropathologic assessment are demarcated in A. A, B, represent low- and high-power photomicrograph sections after 30 min of HI alone (group Va). Bright pink-staining cells indicate eosinophilic dying cells. Note paucity of dying cells. C, D, represent low- and high-power photomicrographs of animals exposed to 30 min of HI followed by KA injection and seizures (group VIa). Note marked increase in eosinophilic-staining cells. Thick arrow shows bright-staining dying pyramidal cell, thin arrow points to normal cell (D) (magnification × 100).
Statistical Analysis
Data regarding neuropathologic outcome were analyzed using the Mann-Whitney U test. Heart rate and temperature were compared with two-tailed ANOVA with repeated measures followed by Tukey comparison. Differences in mortality rates among the age groups were analyzed using Fisher's exact test. Significance was considered at p < 0.05.
RESULTS
Preliminary Studies
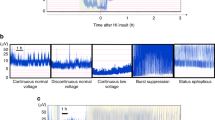
Injection of 3 mg/kg of KA, followed immediately by a 2 mg·kg−1·h−1 s.c. infusion, resulted in the onset of clinical seizures within 5 min. These were characterized by scratching, loss of balance, hyperactivity, upper limb tremors, and salivation, and persisted for the duration of monitored electrographic seizure discharges. Electrographic seizures were manifest by continuous high-voltage spikes, which contrasted markedly with the low-voltage activity seen during the baseline recording (Fig. 1). In those animals receiving KA, continuous electrographic seizure activity persisted for a mean duration of 4 h 42 min (range, 4 h 0 min to 5 h 27 min).

A, electrocortical activity arising from a 10-d-old control rat pup. B, electrocortical activity after injection of KA s.c. Note continuous high-voltage sharp wave activity. Clinical seizures with scratching, loss of balance, hyperactivity, upper limb tremors, and salivation appeared. HP-R, right hippocampus to reference;NC-R, left neocortex to reference;HP-NC, hippocampus to neocortex.
Neuropathologic outcome for rat pups in our preliminary study to determine the minimal lesion was assessed at 20 d of recovery (30 postnatal d of age), at which time the animals were killed and their brains were fixed in FAM for gross neuropathologic assessment as previously described (13, 18). Brains were rated into one of five groups ranging from normal, through mild, moderate, and severe atrophy, to infarction. Animals exposed to HI for periods of 15 min revealed mild atrophy in only one of six animals examined. Of the seven animals exposed to HI for 30 min, two were normal and five displayed mild atrophy. Damage was more significant in those animals experiencing HI for 45 and 60 min, with >50% of both groups showing moderate atrophy to infarction. Inasmuch as the purpose of the present study was to examine the effects of seizures on a background of a minimal lesion after HI, the experimental paradigm included only those groups exposed to HI for either 15 or 30 min.
Experimental Paradigm
Physiologic variables including heart rate, as a reflection of cardiac vulnerability, and temperature were ascertained in those animals in which EEGs were recorded in each of the six groups. Recordings were made during each of the four phases (pretreatment, HI, before KA injection, and after KA injection) of the experiment to determine the individual contribution of HI or KA seizures to cardiac status. As shown in Table 2, heart rate increased with the onset of HI in all groups. Injection of KA resulted in a significant increase in heart rate above homologous controls. No difference in heart rate, however, was noted among any of the KA groups.
Core temperatures were monitored continuously throughout the experimental paradigm (Fig. 2). Those animals receiving KA injections had significantly higher temperatures throughout the duration of their seizures, compared with their homologous controls (p < 0.01). The overall mean core temperature during status epilepticus for those rats injected with KA was 37.76 ± 0.12°C compared with 36.29 ± 0.13°C for those animals given normal saline (p < 0.0001).

Core (rectal) temperatures obtained in 10-d-old rat pups during status epilepticus. Rat pups were swaddled to reduce movement during seizures and decrease the risk of dislodging the rectal probe. Temperatures were taken every 60 s. Rat pups receiving s.c. injections of KA had significantly higher core temperatures (*p < 0.0001) than those who did not.
The mean electrographic seizure duration for those animals receiving either 30 min of HI followed by KA (group VI) or 15 min of HI followed by KA (group IV) was 4 h 29 min and 4 h 42 min, respectively. There was no significant difference in the mean seizure duration among any of the groups receiving KA.
In only those groups of animals receiving KA did we see any death. Mortality rates were 0 for groups I, III, and V, and 13, 34.9, and 29.73%, respectively, for groups II, IV, and VI, being significantly higher in those animals previously exposed to HI in combination with KA (groups IV and VI) compared with those animals receiving HI alone (groups III and V;p < 0.05).
Neuropathology
Early pathology (3 d).
No pathologic damage was seen in the brains of any of the animals in group Ia (control, n = 4), group IIa (KA alone, n = 11), group IIIa (15 min HI alone, n = 7), or group IVa (15 min HI plus KA, n = 10).
Neuropathologic outcome at 3 d of recovery, as a function of the mean damage scores, is displayed in Figs. 3 and 4. Seven of 10 animals exposed to 30 min of HI alone (group Va) displayed damage. Of these seven, five of the animals showed both cortical and hippocampal damage whereas an additional two animals had hippocampal damage alone. None of the animals displayed thalamic damage, whereas the caudate was minimally damaged in three, and the striatum in one. The mean damage scores in the cortex and the hippocampus were 0.68 ± 0.29 and 1.33 ± 0.66, respectively. The overall damage score for the brains of the animals in group Va was 2.34 ± 0.89. Comparison of damage between the cortex and hippocampus within this group revealed no significant differences.

Mean damage scores for rat pups receiving 30 min HI alone (group V) or 30 min HI and KA (group VI). A, values represent scores ±SEM at 3 d (early) recovery from insult. B, values represent scores ±SEM at 20 d (late) recovery from insult. *p < 0.001 from homologous region of the brain.
The early pathologic (3 d) outcome of those animals exposed to 30 min of HI followed by KA-induced seizures (group VIa) showed a significant increase in damage compared with those animals not experiencing seizures. Of the 13 animals in this group, all showed damage. Twelve of 13 showed damage in both the cortex and hippocampus, and only one animal had hippocampal damage alone. Seven of the rat pups displayed minimal damage within the striatum, and one had damage in the thalamus. Mean damage scores in group VIa were 1.04 for the cortex, 10.08 for the hippocampus, and 12.14 for the whole brain. Interestingly, the increase in damage between homologous regions of groups V and VI were accounted for entirely by significant increases in damage for the hippocampus (p < 0.001), whereas damage to the cortex by those animals experiencing seizures was no greater than in those animals who sustained an HI alone (Fig. 4A).
Late pathology (20 d).
All groups were also assessed neuropathologically at 20 d of recovery to determine whether a progression of damage occurred as a result of delayed neuronal death (Figs. 4B and 5). Again, no animal in groups Ib (n = 4), IIb (n = 12), IIIb (n = 11), or IVb (n = 11) displayed damage in any of the regions examined.

Representative examples of hippocampal injury at 20 d of recovery (late pathology) using GFAP immunohistochemistry. A, representation of hippocampal injury from an animal in group Vb (30 min HI alone). B, representation of hippocampal injury from an animal in group VIb (30 min HI alone followed by KA seizures).
Mean brain damage scores for animals in groups Vb and VIb after 20 d of recovery are shown in Fig. 4B. Of the eight brains examined in group Vb, only four showed damage, two of which were isolated to the hippocampus. None of the eight displayed any neuronal injury in the thalamus or striatum. The mean damage scores for the cortex and hippocampus was 0.29 ± 0.25 and 0.31 ± 0.21, respectively, and for the whole brain, 0.60 ± 0.34. Comparisons of damage scores between the cortex and hippocampus of animals within group Vb again revealed no significant differences.
Of the 11 rats who experienced HI for 30 min and subsequently had KA-induced seizures (group VIb), 10 displayed damage. Of these, four had neuronal injury isolated to the hippocampus, and three displayed damage in the caudate nucleus. The mean damage scores for those animals in group VIb was 0.32 ± 0.17, 3.06 ± 0.93, and 3.65 ± 0.94 for the cortex, hippocampus, and whole brain, respectively. Comparisons of brain damage between homologous regions of the brain again revealed no differences in cortical injury between groups Vb and VIb, whereas there was a significant increase in hippocampal damage in group VIb compared with that seen in group Vb (p < 0.05).
Because the increase in damage at both 3 d (early) and 20 d (late) was shown to be exclusive to the hippocampus, comparisons were made to delineate intraregional differences within the CA1 to CA4 subfields including the dentate gyrus. With the exception of the dentate gyrus, regions CA1 through CA4 showed significantly greater damage for those animals in group VIa compared with group Va at 3 d of recovery. At 20 d, CA2 through CA4 displayed a greater degree of neuronal injury in group VIb compared with Vb. Within the subfields of group VI, CA2, CA3, and CA4 were all significantly more damaged than CA1 (p < 0.01) when assessed for both early and late neuropathologic outcome.
As indicated above, damage scores were assessed for all groups at both 3 d (early) and 20 d (late) of recovery. Comparing the early and late outcomes for both groups V and VI showed that the extent of neuropathologic injury actually diminished significantly with time, but only in those groups exposed to HI and KA injection, compared with those exposed to HI alone. Hence, the mean damage scores for group VI revealed significantly worse damage early in the evolution of the neuropathologic assessment in all three areas: the cortex (p < 0.05), hippocampus (p < 0.01), and whole brain (p < 0.01), compared with that seen at the later (20 d) stages of assessment (Fig. 4).
DISCUSSION
The above described series of experiments 1) confirms previous studies (19–22) that indicate that seizures per se do not cause neuronal injury to the immature brain, 2) indicates a reduction in the extent of damage over time with HI in association with prolonged seizures, and 3) most importantly, shows the profound exacerbation of damage by neonatal seizures caused to the newborn brain previously compromised by the effects of HI.
Several investigators have reported on the benign nature of seizures in the immature brain. Albala et al. (19) induced seizures with KA in rats aged 15–18 d (pups), 33–37 d (pubescent), and >90 d (adults). No histologic damage was seen in the rat pups. Nitecka et al. (20) similarly found no damage in rat pups <18 d of age, even after 2 h of tonic-clonic convulsions. A progressive increase in damage was seen, however, from 18 to 35 d of age. Stafstrom et al. (21, 22) had earlier reported an absence of neuropathologic findings, long-term cognitive and behavioral deficits, and a lack of spontaneous recurrent seizures in postnatal d 5–10 rats receiving KA (21), although damage was seen in those rats at 30 and 60 d of age. Nor was hippocampal injury seen on light microscopy in seizures induced by electrical stimulation (23) or pilocarpine (24–26) in immature rat pups.
Recently, however, Holmes et al. (27) induced seizures in immature rats, beginning on postnatal d 0, for 5 d by exposing them to flurothyl. In this study, there was no difference in CA3 cell counts between controls and the flurothyl-treated rats. However, there was an increase in axonal sprouting, as well as a reduction in seizure threshold in later adult life. Behaviorally, rats subjected to recurrent flurothyl seizures were slower in locating the platform in a water maze test, although this difference was only present on the first day of testing and subsequently disappeared. Several other authors have shown seizures to cause brain damage in immature animals of different species (5, 28).
Although our own study supports the above findings, it adds to the previous literature in several important ways. First, the current experimental paradigm resulted in continuous documented electrographic seizures, without any interictal phases, which lasted well over 4 h. To our knowledge, persistent electrographic seizures of this duration have not previously been shown. In this regard, during the time of seizure activity there would have been no ability for the animal to recover, even briefly, either from a cerebral metabolic or systemic compromise. The latter remark is substantiated by our monitoring of heart rate throughout the ictal phase. As indicated above, no differences were noted among the experimental and control groups, suggesting that in the presence of well-preserved cerebral blood flow, and presumably substrate delivery, seizures of this duration do not appear to cause direct neuronal damage. Finally, of the available models, the cerebral development of the postnatal d 10 rat pup appears to most closely parallel the human term infant (5, 29, 30). Although many studies have used rat pups ≥15 d of age, cerebral development at this age more likely parallels that of a more mature human infant rather than a neonate.
Several possible explanations for the resistance to damage in immature brains have been proposed. Among these is the finding that the immature brain has low concentrations of glutamate (31) and glutamate-related enzymes (32). In addition, the postsynaptic action of glutamate differs in that there is less Ca2+ entry through N-methyl-d-aspartate receptors and greater Ca2+ buffering capabilities (33–35). Finally, the immature brain is less metabolically active than its more mature counterpart, leading presumably to a greater time duration before ATP depletion (5, 36, 37).
Most intriguing about the current study is the finding that prolonged neonatal seizures do exacerbate the damage incurred by an already compromised brain. This is particularly relevant as neonatal seizures are invariably the result of an underlying brain abnormality, cerebrovascular disease accounting for 50 to 60% of all seizures in the term and preterm infant (16). Only one previous study has examined the effect of status epilepticus on hypoxic-ischemic brain damage in the immature rat. Cataltepe et al. (17) induced repetitive bouts of status epilepticus in postnatal d 7 rat pups using injections of bicuculline at 2, 6, and 12 h after induction of an hypoxic-ischemic insult. Mortality was 53%. Among the survivors, there was no statistically significant difference in the degree of brain damage, assessed histopathologically, between those animals exposed to bicuculline and HI versus those exposed to HI alone. Although these results appear contradictory to ours, several methodological differences are important. Most relevant is the fact that Cataltepe's group used a model (2 h HI) in which damage is severe (infarction) in >50% of animals (11, 12, 17), particularly in the area of the hippocampus. Determining an increase in damage, particularly on a background of severe infarction, under these circumstances might prove difficult and lead to falsely negative results. We induced a much milder hypoxic-ischemic insult that caused little damage to the hippocampus or neocortex. Hence, any increase in damage would be relatively more easy to detect. Second, our animals recovered for only 30 min after HI, compared with 2 h in the experimental paradigm of Cataltepe et al. (17). It has been previously shown that recovery of energy reserves in the immature brain requires at least 60 min (38). Therefore, the shorter period of recovery time in the current study may have left the brain more vulnerable to the superimposed metabolic perturbations caused by the seizures. Finally, our study used 10-versus 7-d-old rats. Given that seizure-induced brain damage is clearly age dependent, it is certainly possible that the 10-d-old rat pup is more susceptible to damage after HI and seizures compared with the younger 7-d-old animal.
Our results clearly point to a prerequisite requirement of injury to the immature brain for seizures to cause further damage. The underlying perturbation required, however, to initiate the cascade of events known to culminate in brain injury (39) under this paradigm of HI complicated by seizures is not known. Because ATP is the critical regulator of cell function and is responsible for cell membrane integrity through the active transport of Na+, K+, and Ca2+ ions, it has been suggested by many investigators that it is the depletion of high-energy phosphates that must occur as a necessary prerequisite to those mechanisms underlying cellular dysfunction and death (40–43). Hence, whereas prolonged seizures alone do not deplete the energy reserves of the immature brain (36, 37, 44), HI certainly does (38, 45) and may act to trigger further metabolic disruption and cell death via the excitatory amino acid pathway, as is reported to occur in the adult brain (46). Whether this theory holds true of the newborn is uncertain. Severe hypoxia to 4% has been shown to increase the epileptogenicity of the immature 10-d-old rat brain (47) and to enhance the susceptibility of the brain to seizures later in life (48, 49). In addition, Young et al. (50) reported that, in juvenile rabbits, the combination of hypoxia and bicuculline-induced seizures acted in a complementary fashion to cause a marked and progressive rise in extracellular fluid concentrations of the excitatory amino acids, whereas either of the insults alone had no effect.
Another possibility lay in our finding that those groups of rats that had experienced seizures also showed a significant increase in their core temperature. Hyperthermia has been shown to exacerbate brain injury in both the human and experimental animal when induced either during or after a hypoxic-ischemic insult (51–55). It would not be surprising, therefore, to find that the increase in temperature among our animals injected with KA was contributory to the exacerbation of damage. Had the increase in temperature been the sole reason for the differences in brain damage seen among our treatment groups, however, we would have expected all groups injected with KA to display brain damage or an increase in brain damage. Because this only occurred in those animals in whom a mild degree of brain damage was already expected (KA plus 30 min HI), temperature alone could not have been the sole underlying cause for the detrimental effect, although it certainly may have contributed. Further studies controlling for the effect of temperature under these circumstances are required.
Prolonged seizures after HI preferentially damaged the hippocampus in our animals, with no significant increase in cell injury seen in the neocortex, despite the fact that the KA was administered systemically. These finding are consistent with the metabolic ontogeny of the hippocampus. Elegant work by Ben-Ari and colleagues (20, 56, 57) outlined the developmental changes that occur with KA-induced seizures. With specific reference to the observed metabolic perturbations, using the technique of glucose utilization, they found a rise in metabolism during KA-induced seizures, which was restricted to the hippocampus from d 3 to the third week of age, after which a more diffuse enhancement of glucose utilization was noted (57). These findings correspond to the ontogenic development of KA binding receptors in rats, which have been shown to have active high-affinity receptors in the hippocampal region as early as 10 d, whereas they were undetected in other regions of the brain until the end of the third week (56). Similarly, postsynaptic receptor ontogeny for N-methyl-d-aspartate receptors peaks at 150 to 200% of adult values in the hippocampus between 6 and 14 d (58, 59). The concept of enhanced damage to the hippocampus is also in keeping with the increased propensity of this region for seizures over other areas of the neocortex. Hence, investigators have clearly shown the immature hippocampus to develop early calcium conductance and have a reduced number of inhibitory synapses, which allow it to generate prolonged and organized afterdischarges, compared with the neocortex (6, 60).
Finally, it was intriguing to note that assessment of the brain damage in our groups revealed a reduction in damage to the hippocampus at 20 d of recovery compared with 3 d of recovery. It is likely that these findings are an indication of the degree to which the additive seizure-induced injury may be recoverable in the immature brain. Certainly, others have witnessed a similar resolution of injury. Owens et al. (61) reported on the morphologic and electrophysiologic consequences of hypoxia-induced seizures. In their groups of immature animals (postnatal d 8–10), 3% oxygen, causing bradycardia (47), near complete energy depletion (62), and seizures, resulted in neuronal injury detected acutely in the dentate and hilar regions of the hippocampus, but not when the animals were examined at 60–80 d of recovery. Similarly, Toth et al. (63) found that in their model of febrile seizures in the immature rat, hyperthermic seizures resulted in the appearance of cell damage in the limbic system within 24 h, but by 4 wk of recovery no significant neuronal dropout was evident. With respect to HI alone, data from the work by Towfighi et al. (12) also revealed an overall decrease in damage with time. In their study, 7-d-old rat pups were exposed to 2 h of HI and assessed histopathologically at recovery periods of from 0 to 3 wk. Damage was seen almost immediately and peaked in the hippocampal region at 24 h of recovery. Subsequently, the overall extent of neuronal injury diminished. Hence, in the newborn, the evolution of neuronal necrosis occurs much more rapidly than in the adult, and delayed neuronal necrosis, if indeed it occurs in the immature brain, is accelerated compared with its adult counterpart (10, 11).
An additional question not to be ignored is whether the described experimental paradigm may be responsible for the hippocampal sclerosis presumed to evolve after an episode of status epilepticus. VanLandingham et al. (64) found evidence of hippocampal sclerosis on magnetic resonance imaging in 40% of children with focal or lateralized complex febrile seizures. Harvey et al. (65) also found that in a series of 53 children with temporal lobe epilepsy, 57% displayed hippocampal sclerosis on magnetic resonance imaging. Because hippocampal sclerosis is often associated with infants and children who have experienced status epilepticus, it is possible that early neonatal seizures, complicating HI, may predispose the individual to the onset of sclerosis.
In summary, our findings substantiate those of others who have found that prolonged seizures alone do not cause neuronal cell death in the immature brain. However, seizures superimposed on mild to moderate hypoxic-ischemic injury markedly accentuate neuropathologic injury, particularly in the hippocampus. From a clinical perspective, the prognosis of a hypoxic-ischemic insult to the term human infant depends on the degree of encephalopathy (66). The majority of those infants who are severely encephalopathic will die or be severely disabled (9). Those with moderate encephalopathy have an intermediate outcome, with sequelae being seen in approximately 20%, and those with only mild encephalopathy do well. The study by Cataltepe et al. (17) suggests that prevention of seizures may not be helpful in those infants with severe hypoxic-ischemic insults. However, if superimposed seizures do accentuate brain injury in those with mild to moderate hypoxic-ischemic encephalopathy, prevention of seizures would be extremely important in the large proportion of infants with less severe insults.
Abbreviations
- KA:
-
kainic acid
- HI:
-
hypoxia-ischemia
- GFAP:
-
glial fibrillary acidic protein
References
Holden KR, Mellits ED, Freeman JM 1982 Neonatal seizures I. Correlation of prenatal and perinatal events with outcomes. Pediatrics 70: 165–176
Scher MS, Kosaburo A, Beggarly ME, Hamid MY, Steppe DA, Painter MJ 1993 Electrographic seizures in preterm and fullterm neonates: clinical correlates, associated brain lesions, and risk of neurological sequelae. Pediatrics 91: 128–134
Yager JY, Vannucci RC 1997 Neonatal seizures. In: Fanaroff AA, Martin RI (eds) Neonatal-Perinatal Medicine, 6th Ed. Mosby-Year Book, Inc., Philadelphia, pp 1891–1911
Camfield PR 1997 Recurrent seizures in the developing brain are not harmful. Epilepsia 38: 735–737
Wasterlain CG 1997 Recurrent seizures in the developing brain are harmful. Epilepsia 38: 728–734
Holmes GL 1997 Epilepsy in the developing brain: lessons for the laboratory and clinic. Epilepsia 38: 12–30
Mizrahi EM, Kellaway P 1998 Diagnosis and Management of Neonatal Seizures. Lippincott-Raven, Piladelphia, pp 47–60
Sarnat HB, Sarnat MS 1976 Neonatal encephalopathy following fetal distress—A clinical and electroencephalographic study. Arch Neurol 33: 695–706
Volpe JJ 1995 Neurology of the Newborn, 3rd Ed. WB Saunders, Philadelphia, pp 172–207
Rice JE, Vannucci RC, Brierly JB 1981 The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 9: 131–141
Towfighi J, Yager JY, Housman C, Vannucci RC 1991 Neuropathology of remote hypoxic-ischemic damage in the immature rat. Acta Neuropathol 81: 578–587
Towfighi J, Zec N, Yager J, Housman C, Vannucci RC 1995 Temporal evolution of neuropathologic changes in an immature rat model of cerebral hypoxia: a light microscopic study. Acta Neuropathol 90: 375–386
Yager JY, Towfighi J, Vannucci RC 1993 Influence of mild hypothermia on hypoxic-ischemic brain damage in the immature rat. Pediatr Res 34: 525–529
Yager JY, Asselin J 1996 The effect of mild hypothermia on cerebral energy metabolism during evolution of hypoxic-ischemic brain damage in the immature rat. Stroke 27: 919–926
Snead OC, Stephens HI 1983 Ontogeny of cortical and subcortical electroencephalographic events in unrestrained neonatal and infant rats. Exp Neurol 82: 249–269
Mizrahi EM, Kellaway P 1987 Characterization and classification of neonatal seizures. Neurology 37: 1837–1844
Cataltepe O, Vannucci RC, Hietgan DF, Towfighi J 1995 Effect of status epilepticus on hypoxic-ischemic brain damage in the immature rat. Pediatr Res 38: 251–257
Yager JY, Heitjan DF, Towfighi J, Vannucci RC 1992 Effect of insulin induced and fasting hypoglycemia on perinatal hypoxic-ischemic brain damage. Pediatr Res 31: 138–142
Albala BJM, Moshe SL, Okada R 1984 Kainic acid induced seizures: a development study. Dev Brain Res 13: 139–148
Nitecka L, Tremblay E, Charton G, Bouillot JP, Berger ML, Ben-Ari Y 1984 Maturation of kainic acid seizure-brain damage syndrome in the rat. II. Histopathological sequelae. Neuroscience 13: 1073–1094
Stafstrom CE, Thompson JL, Holmes GL 1992 Kainic acid seizures in the developing brain: status epilepticus and spontaneous recurrent seizures. Dev Brain Res 65: 227–236
Stafstrom CD, Holmes GL, Chronopoulos A, Thurber S, Thompson JL 1993 Age-dependent cognitive and behavior deficits following kainic acid-induced seizures. Epilepsia 33: 420–432
Thurber S, Chronopoulos A, Stafstrom CE, Holmes GL 1992 Behavioral effects of continuous hippocampal stimulation in the developing rat. Dev Brain Res 68: 35–40
Cavalheiro EA, Silva DF, Turski WA, Calderazzo-Filho LS, Bortolotto ZA, Turski L 1987 The susceptibility of rats to pilocarpine-induced seizures is age-dependent. Brain Res 465: 43–58
Hirsch E, Baram TZ, Snead OC 1992 Ontogenic study of lithium-pilocarpine-induced status epilepticus in rats. Brain Res 583: 120–126
Liu Z, Gatt A, Werner SJ, Mikati M, Holmes GL 1995 Long-term behavioral deficits following pilocarpine-induced seizures in immature rats. Epilepsy Res 19: 191–204
Holmes GL, Gairsa J-L, Chevassus-Au-Louis N, Ben-Ari Y 1998 Consequence of neonatal seizures in the rat: morphological and behavioral effects. Ann Neurol 44: 845–857
Franck JE, Schwartzkroin PA 1984 Immature rabbit hippocampus is damaged by systemic but not intraventricular kainic acid. Dev Brain Res 13: 219–227
Dobbing J, Sands J 1979 Comparative aspects of the brain growth spurt. Early Human Dev 3: 79–83
Hagberg H, Bona E, Gilland E, Puka-Sundvall M 1997 Hypoxia-ischaemia model in the 7-day old rat: possibilities and shortcomings. Acta Paediatr Suppl 422: 85–88
Campochiaro P, Coyle JT 1978 Ontogenic development of kainate neurotoxicity: correlates with glutamatergic innervation. Proc Natl Acad Sci USA 75: 2025–2029
Rothe F, Schmidt W, Wolf G 1983 Postnatal changes in the activity of glutamate dehydrogenase and aspartate aminotransferase in the rat nervous system with special reference to the glutamate transmitter metabolism. Dev Brain Res 11: 67–74
Bickler PE, Gallego SM, Hansen BM 1993 Developmental changes in intracellular calcium regulation in rat cerebral cortex during hypoxia. J Cereb Blood Flow Metab 13: 811–819
Carmant L, Liu Z, Werner SJ, Mikati MA, Holmes GL 1995 Effect of kainic acid-induced status epilepticus on inositol-triphosphate and seizure-induced brain damage in mature and immature animals. Dev Brain Res 89: 67–72
Sperber EF, Weireter KK, Kubova H, Romero MT 1995 Age-related changes in parvalbumin immunoreactivity following kainic acid seizure. Soc Neurosci Abstr 21: 1473
Young RSK, Osbakken MD, Briggs RW, Yagel SK, Rice DW, Goldberg S 1985 31P NMR study of cerebral metabolism during prolonged seizures in the neonatal dog. Ann Neurol 18: 14–20
Young RSK, Petroff OAC, Chen B, Gore JC, Aquila WJ 1991 Brain energy state and lactate metabolism during status epilepticus in the neonatal dog. In vivo 31P and 1H nuclear magnetic resonance study. Pediatr Res 29: 191–195
Yager JY, Brucklacher RM, Vannucci RC 1992 Cerebral energy metabolism during hypoxia-ischemia and early recovery in immature rats. Am J Physiol 262: H672–H677
Vannucci RC 1990 Experimental biology of cerebral hypoxia-ischemia: relation to perinatal brain damage. Pediatr Res 27: 317–326
Yager JY, Kala G, Hertz L, Juurlink BHJ 1994 Correlation between ATP content and hypoxic-ischemic damage in immature and mature astrocytes. Dev Brain Res 82: 62–68
Jurkowitz-Alexander MS, Altschuld RA, Hohl CM, Johnson JD, McDonald JS, Simmons TD, Horrocks 1992 Cell swelling, blebbing, and death are dependent on ATP depletion and independent of calcium during chemical hypoxia in a glial cell line (ROC-1). J Neurochem 59: 344–352
Lipton P, Whittingham TS 1982 Reduced ATP concentration as a basis for synaptic transmission failure during hypoxia in the in-vitro guinea-pig hippocampus. J Physiol Lond 325: 51–65
Novelli A, Reilly JA, Lysko PG, Henneberry RC 1988 Glutamate becomes neurotoxic via the N-methyl-d-aspartate receptor when intracellular energy levels are reduced. Brain Res 451: 205–212
Chapman AG, Meldrum BS, Siesjo BK 1977 Cerebral metabolic changes during prolonged epileptic seizures in rats. J Neurosci 28: 1025–1035
Palmer C, Brucklacher RM, Christensen MA, Vannucci RC 1990 Carbohydrate and energy metabolism during the evolution of hypoxic-ischemic brain damage in the immature rat. J Cereb Blood Flow Metab 10: 227–235
Siesjo BK, Ingvar M, Wieloch T 1986 Cellular and molecular events underlying epileptic brain damage. Ann NY Acad Sci 462: 208–223
Jensen FE, Applegate CK, Holtzman D, Belin TR, Burchfiel JL 1991 Epileptogenic effect of hypoxia in the immature rodent brain. Ann Neurol 29: 629–637
Jensen FE, Wang C, Stafstrom CE, Liu Z, Geary C, Stevens MC 1998 Acute and chronic increases in excitability in rat hippocampal slices after perinatal hypoxia in vivo. J Neurophysiol 79: 73–81
Sato K, Morimoto K, Ujike H, Yamada T, Yamada N, Kuroda S, Hayabara T 1994 The effects of perinatal anoxia or hypoxia on hippocampal kindling development in rats. Brain Res Bull 35: 167–170
Young RSK, During MJ, Aquila WJ, Tendler D, Ley E 1992 Hypoxia increases extracellular concentrations of excitatory and inhibitory neurotransmitters in subsequently induced seizures: in vivo microdialysis study in the rabbit. Exp Neurol 117: 204–209
Busto R, Dietrich D, Globus MY-T, Ginsberg MD 1989 The importance of brain temperature in cerebral ischemic injury. Stroke 20: 1113–1114
Chen H, Chopp M, Welch KMA 1991 Effect of mild hyperthermia on the ischemic infarct volume after middle cerebral artery occlusion in the rat. Neurology 41: 1133–1135
Castillo J, Davalos A, Marrugat J, Noya M 1998 Timing for fever-related brain damage in acute ischemic stroke. Stroke 29: 2455–2460
Schwab S, Spranger M, Aschoff A, Steiner T, Hacke W 1997 Brain temperature monitoring and modulation in patients with severe MCA infarction. Neurology 48: 762–767
Jorgenson HS, Reith J, Pedersen PM, Hakayama H, Olsen TS 1996 Body temperature and outcome in stroke patients. Lancet 348: 193
Berger ML, Tremblay E, Nitecka L, Ben-Ari Y 1984 Maturation of kainic acid seizure-brain damage syndrome in the rat. III. Postnatal development of kainic acid binding sites in the limbic system. Neuroscience 13: 1095–1104
Tremblay E, Nitecka L, Berger ML, Ben-Ari Y 1984 Maturation of kainic acid seizure-brain damage syndrome in the rat. I. Clinical, electrographic and metabolic observations. Neuroscience 13: 1051–1072
Hattori H, Wasterlain CG 1990 Excitatory amino acids in the developing brain: ontogeny, plasticity, and excitotoxicity. Pediatr Neurol 6: 219–228
Tremblay E, Roisin MP, Represa A, Charriaut-Marlangue C, Ben-Ari Y 1988 Transient increased density of NMDA binding sites in the developing rat hippocampus. Brain Res 461: 393–396
Moshe SL 1993 Seizures in the developing brain. Neurology 43( suppl 5): S3–S7
Owens J, Robbins CA, Wenzel HJ, Schwartzkroin PA 1997 Acute and chronic effects of hypoxia on the developing hippocampus. Ann Neurol 41: 187–199
Jensen F, Tsuji M, Offutt M, Firkusny I, Holtsman D 1993 Profound, reversible energy loss in the hypoxic immature rat brain. Dev Brain Res 73: 99–105
Toth Z, Yan X-X, Haftoglou S, Ribak C, Baram TZ 1988 Seizure-induced neuronal injury: vulnerability to febrile seizures in an immature rat model. J Neurosci 18: 4285–4292
VanLandingham KE, Heinz ER, Cavazos JE, Lewis DV 1998 Magnetic resonance imaging evidence of hippocampal injury after prolonged focal febrile convulsions. Ann Neurol 43: 413–426
Harvey AS, Grattan-Smith JD, Desmond PM, Chow CW, Berkovic SF 1995 Febrile seizures and hippocampal sclerosis: frequent and related findings in intractable temporal lobe epilepsy of childhood. Pediatr Neurol 12: 201–206
Robertson C, Finer N 1985 Term infants with hypoxic-ischemic encephalopathy: outcome at 3.5 years. Dev Med Child Neurol 27: 473–484
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by grants from the Health Services Utilization and Research Commission of Saskatchewan, and the Toronto Hospital for Sick Children Foundation.
Rights and permissions
About this article
Cite this article
Wirrell, E., Armstrong, E., Osman, L. et al. Prolonged Seizures Exacerbate Perinatal Hypoxic-Ischemic Brain Damage. Pediatr Res 50, 445–454 (2001). https://doi.org/10.1203/00006450-200110000-00005
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200110000-00005
This article is cited by
-
Neuroprotective therapies in the NICU in term infants: present and future
Pediatric Research (2023)
-
Difluoroboron β-diketonate polylactic acid oxygen nanosensors for intracellular neuronal imaging
Scientific Reports (2021)
-
Akut symptomatische Anfälle bei Neonaten und Einsatz des amplitudenintegrierten EEGs (aEEG)
Zeitschrift für Epileptologie (2021)
-
Pilot study of a single-channel EEG seizure detection algorithm using machine learning
Child's Nervous System (2021)
-
Investigating and managing neonatal seizures in the UK: an explanatory sequential mixed methods approach
BMC Pediatrics (2020)
