
Abstract
Insulin-like growth factor binding protein (IGFBP)-3 binds to IGF and modulates their actions and also possesses intrinsic activities. We investigated its effects on insulin action and found that when IGFBP-3 was added to fully differentiated 3T3-L1 adipocytes in culture, insulin-stimulated glucose transport was significantly inhibited to 60% of control in a time- and dose-dependent manner. Tumor necrosis factor (TNF)-α treatment also inhibited glucose transport to the same degree as IGFBP-3 and, in addition, increased IGFBP-3 levels 3-fold. Co-treatment with TNF-α and IGFBP-3 antisense partially prevented the inhibitory effect of TNF-α on glucose transport, indicating a role for IGFBP-3 in cytokine-induced insulin resistance. Insulin-stimulated phosphorylation of the insulin receptor was markedly decreased by IGFBP-3 treatment. IGFBP-3 treatment suppressed adiponectin expression in 3T3-L1 adipocytes. Infusion of IGFBP-3 to Sprague-Dawley rats for 3 h decreased peripheral glucose uptake by 15% compared with controls as well as inhibiting glycogen synthesis. Systemic administration of IGFBP-3 to rats for 7 d resulted in a dramatic 40% decrease in peripheral glucose utilization and glycogen synthesis. These in vitro and in vivo findings demonstrate that IGFBP-3 has potent insulin-antagonizing capability and suggest a role for IGFBP-3 in cytokine-induced insulin resistance and other mechanisms involved in the development of type-2 diabetes.
Similar content being viewed by others


Main
IGF-I and -II are involved in the regulation of cell growth and differentiation in a variety of cell types (1). However, the IGF also mimic some of the metabolic actions of insulin and act as insulin sensitizers (2). IGF-I has approximately 1/12th the glucose-lowering capacity of insulin (3), in vivo and an equipotent effect on ex vivo muscle strips (4), as well as being an insulin sensitizer, and has been considered as a putative treatment agent for both type-1 and type-2 diabetes (5,6). The IGFBP are a family of six binding proteins that bind to IGF with high affinity and specificity. A variety IGFBP profiles are observed in different tissues, presumably regulating specific cellular activities. IGFBP-3 is the most abundant circulating IGF binding protein and is expressed in most tissues. IGFBP-3 not only regulates IGF bioavailability and action (so-called IGF-dependent actions), but also mediates IGF independent actions on cell survival and apoptosis (7–9). By binding IGF in the circulation, the IGFBP reduce the levels of free IGF and antagonize their insulin-like activity; in addition, they may be involved in carbohydrate metabolism in ways that remain poorly characterized (10). IGFBP-3 levels are regulated by multiple factors, including cytokines that have been implicated in insulin resistance, such as TNF-α. Recently, it has been shown that IGFBP-3 reduces insulin-stimulated glucose uptake in both rodent and human adipocytes (11). We carried out a series of experiments to elucidate the effects of IGFBP-3 on insulin sensitivity in vitro and in vivo and the mechanisms involved in its actions. In addition, we show here that the insulin-antagonistic effects of tumor necrosis factor (TNF)-α are mediated in part by IGFBP-3.
MATERIALS AND METHODS
Materials.
Recombinant hIGFBP-3 was a generous gift from Celtrix (Mountain View, CA). Human recombinant insulin was obtained from Sigma Chemical Co. (Saint Louis, MO). 2-[3H (G)] deoxy-d-glucose was purchased from New England Nuclear, Inc. (Boston, MA). Anti-human IGFBP-3 antibodies, which were affinity purified on an IGFBP-3 column, were purchased from Diagnostic Systems Laboratories (Webster, TX). 125-I-labeled IGF-I and IGF-II were purchased from Amersham (Piscataway, NJ). Anti-phospho-insulin receptor beta subunit antibody was purchased from BioSource International (Camarillo, CA). Anti-adiponectin antibody was purchased from Chemicon International (Temecula, CA) The Bradford protein assay kit and all electrophoresis chemicals were obtained from Bio-Rad (Richmond, CA). All other chemicals were purchased form Sigma Chemical Co. The antisense oligonucleotide designed to flank the initiation codon of murine IGFBP-3 (12) was GCGCGCGGGATGCATGGCGCCGGGTGGACG, with the corresponding sense oligonucleotide being 5′- CGTCCAC-CCGGCGCCATGCATCCCGCGCGC. Thio-ester bonds linked the first three and final three nucleotides of each oligo (Sigma-Genosys, Ltd., The Woodlands, TX).
Cell culture.
All cell lines and tissue culture reagents were purchased from ATCC (Rockville, MD). 3T3-L1 adipocytes were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and an antibiotic mixture containing penicillin and streptomycin. For the experiments, cells were cultured in 12-well dishes and differentiated into adipocytes using 3-isobutyl 1-methylxanthine, dexamethasone and insulin, according to methods previously described (13). Briefly, confluent cultures were incubated with the differentiation medium containing dexamethasone (25 μM), isobutyl methylxanthine (0.5 mM), and insulin (100 nmol/L) in DMEM with 10% FBS for 48 h. The cells were then maintained in a medium containing 10% FBS and insulin (10nmol/L). Experiments were performed when greater than 90% of the cells were differentiated into adipocytes.
Glucose transport assay.
Before glucose transport assays, the cells were incubated in serum-free media, with or without treatment (IGFBP-3 or TNF-α). Treatment with IGFBP-3 was performed at various concentrations and durations (see “Results”). TNF-α treatment was performed at a concentration of 10 ng/mL for 24 h. All experiments were done in triplicate unless otherwise indicated. The procedure for glucose transport measurement was modified from methods previously described (14). After the treatment period, the cells were washed twice with PBS and incubated in the same buffer for 30 min with insulin (10 nmol/L). The transport reaction was started by addition of 10 μL substrate (3H-2-deoxyglucose 0.1 μCi to produce a final concentration of 0.1 mM) and halted after 5 min by aspirating the reaction mixture and rapidly rinsing each well five times with 4-degree PBS. Cells were solubilized by addition of 0.5 mL 0.1 N NaOH and incubated with shaking. An aliquot (100 μL) of the suspension was removed for protein analysis using Bio-Rad reagent (Richmond, CA) (15). After solubilization, 400 μL of the suspension was placed in a scintillation vial and neutralized with 1.0 N HCL and scintillation fluid was added. Radioactivity in this lysate was determined by scintillation counting.
Western immunoblots.
Phosphorylated insulin receptor beta subunit levels were detected using cell lysate from differentiated 3T3-L1 adipocytes that were treated with or without IGFBP-3 (1 μg/mL) for 24 h. Each of these experiments was performed in the presence or absence of insulin (10 nmol/L) for the last 30 min of the treatment period. Adiponectin was detected using cell lysates from 3T3-L1 adipocytes that were treated with or without IGFBP-3 (1 μg/mL) and rosiglitazone (10 μM/L) for 24 h. All experiments were repeated three times. Samples of 50 μL were separated by nonreducing 8% SDS-PAGE overnight at constant voltage and electroblotted onto nitrocellulose. The membranes were then sequentially washed with NP40, 1%BSA, and Tween 20, blocked with 5% nonfat dry milk in Tris-buffered saline, probed with the specified antibody and detected using a peroxidase-linked enhanced chemiluminescence detection system (Amersham, Arlington Heights, IL).
Immunofluorescence confocal microscopy.
Fully differentiated 3T3-L1 adipocytes, 1 × 104, were plated on cover-glass in serum containing media for 2 d. The cells were then incubated in serum-free medium for 24 h. For the last 6 h, half of the wells were treated with IGFBP-3 in a dose of 1 μg/mL for 6 h. The cells were then exposed to insulin (10 nmol/L) for 30 min. After three washes in PBS, fixation and permeabilization of the cells were performed with 1% paraformaldehyde in PBS for 15 min at room temperature and 0.2% Triton X-100 in PBS for 15 min on ice, and cells were washed twice with PBS. Specimens were incubated with primary antibodies in PBS for 1 h at room temperature, with secondary antibodies in PBS for 40 min at room temperature, and then incubated with Hoechst from Electron Microscopy Sciences (Ft. Washington, PA) for 2 min. Samples were analyzed using the Inverted Confocal Microscope (Leica, Wetzlar, Germany), equipped by digital camera (Hamamatsu, Hamamatsu City, Japan), and operated by QED-image software. DAPI (blue) identifies the nuclei.
To examine IGFBP-3 induction by TNF-α, 1 × 104 fully differentiated 3T3-L1 adipocytes were plated on cover-glass in serum containing media for 2 d. The cells were then incubated in serum-free media with or without TNF-α at a concentration of 10 ng/mL for 48 h, before staining for immunofluorescence as described above. IGFBP-3 protein localization was detected using the DSL hIGFBP-3 goat polyclonal antibody (which was previously purified on an IGFBP-3 column), diluted 1:200, followed by fluorescein anti-goat antibody (Vector Laboratories, Burlingame, CA). Samples were then analyzed using the Inverted Confocal Microscope at 60× magnification (Leica), equipped by digital camera (Hiramitsu), and operated by QED-image software.
Western ligand blotting.
IGFBP-3 protein levels were assessed using cell lysate from 3T3-L1 adipocytes that were treated with or without TNF-α (10 ng/mL) for 48 h. Samples of 50 μL were separated by nonreducing 10% SDS-PAGE overnight at constant voltage and electroblotted onto nitrocellulose. The membranes were then sequentially washed with NP40, 1% BSA, and Tween 20, incubated with 106 cpm each of 125I-labeled IGF-I and IGF-II for 12 h, dried, and exposed to film for 5 d. All experiments were done in triplicate.
IGFBP-3 antisense treatment.
3T3-L1 adipocytes were grown and differentiated in 12-well plates (in quadruplicates) as described above. The cells were then preincubated with sense or antisense IGFBP-3 oligos (detailed above), at concentrations of 500 ng/plate for 30 min in the presence of LipofectAMINE (Invitrogen). Following the preincubation, the cells were incubated in serum-free medium for 24 h with and without TNF-α (10 ng/mL). At the end of the 24 h, the cells were treated with insulin (10 nmol/L) and a glucose transport assay was performed as described above.
In vivo hyperinsulinemic euglycemic clamps.
The principles of laboratory animal care set out by the National Institutes of Health were followed strictly. The study protocol was reviewed and approved by the Animal Care and Use Committee of the Albert Einstein College of Medicine. Male Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA) were housed in individual cages and subjected to a standard light (0600–1800 h) and dark (1800–0600 h) cycle. They were fed ad libitum using regular rat chow that consisted of 64% carbohydrate, 30% protein, and 6% fat with a physiologic fuel value of 3.3 kcal/g chow.
To study the acute effects of an infusion of IGFBP-3, two groups of awake, unstressed, chronically catheterized Sprague-Dawley rats (0.3 kg) were studied for 300 min. All rats received a primer continuous infusion (15–49 μCi/min bolus, 0.4 μCi/min) of [3-3H] glucose throughout the study. After establishing rates of basal glucose turnover, a primed infusion of somatostatin (1.5 μg/kg/min), insulin (3 mU/kg/min), and a variable infusion of 25% glucose to clamp the plasma concentration of euglycemic levels of 140 mg/dL was administered for 2 h. At 120 min, the rats received a primed continuous infusion of IGFBP-3 (60 μg/kg/h) or saline (control) for an additional 3 h.
For the chronic IGFBP-3 infusion experiments, Sprague-Dawley rats (0.3 kg) received either IGFBP-3 (40 μg/kg/h) or saline as control by osmotic minipumps for 7 d and clamp studies were performed on d 7. All rats received a primed continuous infusion (15–49 μCi/min bolus, 0.4 μCi/min) of [3-3H] glucose throughout the study. After establishing rates of basal glucose turnover, a primed infusion of somatostatin (1.5 μg/kg/min), insulin (3 mU/kg/min), and a variable infusion of 25% glucose to clamp the plasma glucose concentration at euglycemic levels of 140 mg/dL were administered for 2 h. Recombinant human IGFBP-3 levels in rat sera were measured by ELISA (DSL, Webster, TX). There was no cross-reactivity between human and rat IGFBP-3 in this assay.
Statistical analysis.
Statistical significance was evaluated using t tests and ANOVA and two-tailed p values were calculated. Significance was accepted at the p < 0.05 level.
RESULTS
IGFBP-3 inhibits glucose uptake in 3t3-L1 adipocytes.
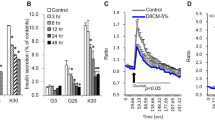
Addition of 1 μg/mL IGFBP-3 to 3T3 adipocytes for 24 h resulted in a >40% decrease in insulin-stimulated glucose transport compared with serum-free controls. When adipocytes were exposed to IGFBP-3 for 24 h at a concentration of 1 μg/mL, glucose transport decreased by >40% (Fig. 1A). This is similar to the decrease in insulin-stimulated glucose transport observed when adipocytes are exposed to 10 ng/mL TNF-α over the same time period.

Effect of IGFBP-3 on insulin-stimulated glucose uptake in 3T3-L1 adipocytes. (A) 3T3-L1 adipocytes treated (n = 3 per condition) with 1 μg/mL IGFBP-3 or with 10 ng/mL TNF-α for 24 h and pulsed with insulin (10 nmol/L) ×30 min before measurement of glucose transport as described. Glucose transport expressed as percentage of serum-free conditions. Dose response (B) and time course (C) are also shown. The effects of 1 μg/mL IGFBP-3 on basal glucose transport (n = 4 per group) are shown in (D). *p < 0.05.
Treatment with IGFBP-3 for 24 h suppressed glucose uptake in adipocytes in a dose-dependent manner. The effect was maximal at an IGFBP-3 concentration of 1 μg/mL where glucose transport decreased by 40% compared with serum-free controls. Treatment with IGFBP-3 at a concentration of 1.5 μg/mL did not increase the response (Fig. 1B). In addition, a time course treatment with IGFBP-3 at a concentration of 1 μg/mL demonstrated suppression of glucose uptake in a time-dependent manner. The suppression was greatest after 24 h of treatment; however, an effect was detectable as early as 30 min of treatment (Fig. 1C). These results indicate that IGFBP-3 induces insulin resistance in vitro, in a time- and dosage-dependent manner. IGFBP-3 inhibited basal glucose transport by 20% (Fig. 1D).
TNF-α induces the production of IGFBP-3 in 3T3-L1 adipocytes.
To test whether TNF-α induces IGFBP-3 production in 3T3-L1 adipocytes, adipocytes in serum-free media were treated with TNF-α for 48 h at a concentration of 10 ng/mL. Production of IGFBP-3 was then detected by immunofluorescence confocal microscopy using a rodent IGFBP-3-specific antibody. Following TNF-α treatment, the overall levels of IGFBP-3, (stained in green) in the cells rose dramatically compared with serum-free controls (Fig. 2), implying increased endogenous production. Interestingly, IGFBP-3 located within the nuclei at low levels in basal conditions appears to increase to a greater extent in response to treatment with TNF-α.

Exposure to TNF-α induces IGFBP-3 production in 3T3-L1 adipocytes that is involved in glucose transport regulation. (A) Confocal microscopy of 3T3-L1 adipocytes in culture treated with TNF-α (10 ng/mL) × 48 h, then stained with IGFBP-3 antibody and counterstained with fluorescein-labeled anti-rabbit antibody before confocal microscopy. (B) Cells similarly treated with TNF-α (10 ng/mL) × 72 h were lysed and IGFBP-3 content was assayed using Western-ligand blotting and quantified densitometrically. (C) 3T3-L1 adipocytes were pretreated with IGFBP-3 sense and antisense oligos (n = 4 per group) for 30 min as described. They were then treated with TNF-α 10 ng/mL for 24 h and pulsed with insulin 10nmol/L and glucose transport was assayed as described. Results are expressed as percentage of serum-free. *p < 0.05.
The increase in IGFBP-3 protein levels in response to treatment with TNF-α was also demonstrated by densitometric analysis of Western ligand blots using 125I-IGF-I and -II. We quantified the protein levels of IGFBP-3 in total cell lysates of adipocytes that were exposed to treatment with TNF-α at a concentration of 10 ng/mL for 72 h. When the blot was assessed by densitometric analysis, it was found that TNF-α induced a 3-fold increase in production of IGFBP-3 compared with serum-free conditions (p < 0.05, Fig. 2B).
Insulin-antagonistic effect of TNF-α is partially blocked by pretreatment with IGFBP-3 antisense.
To test whether the insulin-antagonistic action of TNF-α may be mediated via induction of IGFBP-3, 3T3-L1 adipocytes were exposed to TNF-α after transfection of IGFBP-3 antisense or sense oligonucleotides. Cells pretreated with antisense IGFBP-3 oligos and then treated with TNF-α 10 ng/mL exhibited a significantly smaller decrease in insulin-mediated glucose transport (p < 0.05, Fig. 2C). Cell extracts transfected with antisense IGFBP-3 demonstrate >50% reduction in IGFBP-3 content by immunoblot with no change noted in the sense-transfected cells.
IGFBP-3 inhibits insulin-receptor-phosphorylation in 3T3-L1 adipocytes.
To examine whether IGFBP-3 induces insulin resistance by decreasing the tyrosine phosphorylation of insulin receptors on the cell membrane, phosphorylated insulin receptor levels were assayed. Cells were treated with and without IGFBP-3 (1 μg/mL) for 24 h in serum-free media. Each of these conditions was performed in the presence or absence of insulin (10 nmol/L) for the final 30 min of the treatment period. Western blotting of phosphorylated insulin receptor beta subunit proteins from these insulin-treated cells, which were also treated with IGFBP-3, revealed a 70% decrease in insulin-stimulated phosphorylation of the insulin receptor compared with cells treated with insulin alone (Fig. 3).

Effect of IGFBP-3 on insulin-stimulated phosphorylation of the insulin receptor. 3T3-L1 adipocytes (n = 3 per group) treated with 1 μg/mL IGFBP-3 for 24 h and pulsed with insulin (10 nmol/L) × 30 min. Cells were lysed and lysates were electrophoresed and blotted with phospho-specific insulin receptor antibody. Western blot densitometry was expressed as percent of SF. *p < 0.05.
IGFBP-3 inhibit adiponectin expression in mature adipocyte.
To determine whether IGFBP-3 regulates additional adipocyte functions, we examined the effects of IGFBP-3 on adiponectin expression. Adiponectin immunoblots were performed on cell lysates from differentiated 3T3-L1 adipocytes that were treated with and without IGFBP-3 and the PPARγ agonist rosiglitazone for 24 h in serum-free media. Western blotting of adiponectin from IGFBP-3 treated cells revealed a 55% decrease in adiponectin expression compared with control. PPARγ-stimulated adiponectin was also inhibited by IGFBP-3 (Fig. 4).

Effect of IGFBP-3 on adiponectin expression in 3T3-L1 adipocytes. 3T3-L1 adipocytes (n = 3 per group) treated with 1 μg/mL IGFBP-3 for 24 h with or without rosiglitazone. Cells were lysed and lysates were electrophoresed and blotted with adiponectin antibody. Western blot densitometry was expressed as percentage of control. *p < 0.05.
Infusion of IGFBP-3 impairs glucose metabolism in Sprague-Dawley rats.
To examine the effect of elevated IGFBP-3 levels upon insulin sensitivity in vivo, we studied Sprague-Dawley rats utilizing the insulin clamp technique. Acute effects were studied by infusing IGFBP-3 (60 μg/kg/h) for 3 h. Because maximal effect in vitro is seen after several days, we also performed IGFBP-3 infusion for 7 d and the effects of prolonged exposure were studied by continuously infusing 40 μg/kg/h over 7 d.
Infusion of IGFBP-3 for 3 h decreased peripheral glucose uptake by 15% compared with controls (19.0 ± 0.7 versus 22.8 ± 0.3 mg/kg/min, treatment versus control; p < 0.05). Glycogen synthesis was decreased by 25% (7.3 ± 0.3 mg/kg/min vs 10.8 ± 1.4, treatment versus control; p < 0.05) (Fig. 5, A and B).

Effects of a 3 h infusion of IGFBP-3 (A and B) and of 7 d of IGFBP-3 infusion (C and D) on peripheral glucose uptake and glycogen synthesis in Sprague-Dawley rats. Rats (n = 6 per group) were infused with IGFBP-3 (60 μg/kg/h) for 3 h or with IGFBP-3 40 μg/kg/h for 7 d. Glucose uptake and glycogen synthesis were assayed as described. *p < 0.05.
Seven days of IGFBP-3 infusion decreased the peripheral glucose uptake (Rd) by 40% in rats treated with IGFBP-3 compared with control (14.0 ± 0.2 versus 23.2 ± 0.5 mg/kg/min, treatment versus control; p < 0.01). This decrease in Rd was primarily accounted for by a 50% decrease in glycogen synthesis (4.5 ± 1.0 versus 10.8 ± 1.4 mg/kg/min, treatment versus control; p < 0.005) (Fig. 5, C and D). Human IGFBP-3 levels were undetectable in preinfusion sera and achieved levels of 1200 ± 225 ng/mL at the end of 7 d, well within the physiologic range.
DISCUSSION
In this study, we have shown that IGFBP-3 rapidly induces insulin resistance in vivo and in vitro and that this effect occurs at physiologic concentrations of IGFBP-3. We have also demonstrated a link between IGFBP-3 induction and the insulin-antagonistic effect of inflammatory cytokines.
It has previously been shown that transgenic mice that over-express human IGFBP-3 cDNA exhibit fasting hyperglycemia, impaired glucose toleranc, and insulin resistance, without an apparent change in total IGF-I levels, and this was not clearly explained by disturbances in growth hormone secretion or adiposity (16).
A relationship of elevated circulating IGFBP-3 levels to hyperglycemia is also suggested in certain clinical states characterized by impaired insulin action including puberty (17), acromegaly (18), and treatment with recombinant hGH (19). These conditions are associated with the development of insulin resistance and glucose intolerance despite a concomitant elevation of circulating IGF-I levels in each case. We show in this paper that IGFBP-3 is a potent insulin antagonist in 3T3-L1 adipocytes and in Sprague-Dawley rats. Our results also show that the magnitude of this effect is similar to the effect of TNF-α, a multifunctional cytokine that may be an important mediator of insulin resistance linked to obesity (20).
It was not directly tested in our studies whether IGFBP-3 insulin-antagonizing effects are dependent or independent of IGF-I. But Chan et al. (13) recently showed that IGFBP-3 mutants with reduced binding to IGF-I and -II were still able to reduce insulin-stimulated glucose uptake and both that group's as well as our data demonstrated that other IGFBP do not mediate this effect. This suggests the possibility that the inhibitory effect of IGFBP-3 may be independent of its binding to IGF, although an IGF-inhibitory mechanism is also possible. Distinguishing between these possibilities in vivo will require additional studies involving the systemic administration of non-IGF-binding mutants of IGFBP-3. Hyperglycemia has been convincingly demonstrated after the injection of IGFBP-1 (21) in rats and in transgenic mice, which over-express IGFBP-1 (22), however, an IGF-dependent mechanism for this phenomena has been postulated.
IGFBP-3 in serum inhibits the effects of IGF on IGF-activated glucose consumption in mouse fibroblasts (23). In addition, glucose intolerance is observed in liver-specific IGF-I knockout mice (24) where circulating IGF-I levels are reduced to a larger degree than those of IGFBP-3. In these situations, the hyperglycemia could be attributable to a reduction in free IGF-I levels. It is possible that excess IGFBP-3 binds free (unbound) IGF-I, thereby decreasing its bioavailability and its hypoglycemic effect.
However, in addition to modulating the availability of IGF-I, IGFBP-3 has been shown to have independent effects in a variety of cell lines. For example, IGFBP-3 has antiproliferative effects on breast cells that are unresponsive to IGF (8) and on mouse fibroblasts that lack IGF-1 receptors (9). IGFBP-3 has been shown to inhibit type-1 IGF receptor activation independently of its IGF binding affinity in breast cancer cells (25). In addition, proteolytic fragments of IGFBP-3 that have markedly reduced affinity for IGF-I retain antiproliferative effects in vitro. These IGFBP-3 effects may be mediated via cell surface binding proteins (26), nuclear binding sites (27), or other pathways (28). The IGFBP-3 NLS mutant, which is known to not translocate to the nucleus, also inhibited insulin-stimulated glucose uptake (13) Thus, it is possible that some of the insulin-antagonistic effects of IGFBP-3 are mediated via IGF-independent pathways that does not require nuclear localization. For example, a previously published study reports that IGFBP-3 is capable of activating a phosphotyrosine phosphatase independent of its IGF binding affinity (29). Activation of a phosphotyrosine phosphatase and subsequent de-phosphorylation could be a mechanism responsible for the decreased phosphorylated insulin receptor levels observed in our study. Of note is that Chan et al. (13) did not observe reduced IR phosphorylation but did see less Thr (308) phosphorylation of Akt after IGFBP-3 treatment. These investigators also did not observe an effect of IGFBP-3 on baseline (insulin-free) glucose transport, which we have. The possible differences between these studies may include either subtle differences in the experimental conditions or a different behavior of the substrain of the 3T3-L1 cells, which may be susceptible to biologic drift over time in repeat culturing as described for many other cell lines (30).
TNF-α is thought to play an important role in the pathogenesis of insulin resistance associated with obesity. In adipose tissue, levels of TNF-α and its mRNA correlate positively with the degree of obesity or hyperinsulinemia (31). TNF-α's main mechanism of action is unknown, although it has been shown to decrease insulin receptor and the insulin-receptor substrate IRS-1 phosphorylation (32) by attenuating tyrosine kinase activity and/or activating phospho-protein phosphatase-1. Here, we report that TNF-α induces the production of IGFBP-3 in 3T3-L1 adipocytes and that the insulin-antagonistic effect of TNF-α on cultured adipocytes is partially blocked by the presence of IGFBP-3 antisense. This raises the possibility that some of the insulin antagonistic activity of TNF-α may be mediated via induction of IGFBP-3. Such a role is not unexpected as IGFBP-3 has been shown to mediate other effects of TNF-α in various cell types (33) and has also been implicated in mediating the effects of other growth-inhibitory and apoptosis-inducing agents such as tumor suppressor gene p53 (34), retinoic acid, (35), and transforming growth factor-beta (36).
We have previously reported that IGFBP-3 is a binding partner for the ligand-dependent nuclear receptor, retinoid X receptor-α (RXR-α) and modulates its transcriptional activity (37). RXR-α is the obligate heterodimeric partner for the nuclear receptor PPAR-γ (38), which controls the transcription of genes important in the regulation of carbohydrate and lipid metabolism. TNF-α has previously been shown to antagonize PPAR-γ (39). Our observation that TNF-α appears to increase the nuclear localization of IGFBP-3 hints at the possibility that TNF-α may exert some of its insulin-antagonizing effects by modulating the transcriptional activity of PPAR-γ via induction and nuclear translocation of IGFBP-3.
Regardless of the possible numerous interactions of IGFBP-3 with the insulin-signaling pathway, the strength of this report is in demonstrating that IGFPB-3 rapidly induces peripheral insulin resistance in rodents, which is evident within hours. Furthermore, after several days, the degree of insulin resistance induced by IGFBP-3 is similar in magnitude to that seen in many diabetic states. This suggests that IGFBP-3 effects are important in vivo and may explain the alterations in insulin action during pubertal development, and in pathophysiological conditions such as in acromegaly, when IGFBP-3 levels are high.
Adiponectin, an important adipocytokine that is induced by thiazolidinediones (TZD), is closely related to insulin sensitivity (40). We also show here that IGFBP-3 inhibits adiponectin expression both at the basal state and after PPAR-γ agonist stimulation. These findings may further explain how IGFBP-3 induces insulin resistance in vivo.
In conclusion, our results show that IGFBP-3 is a potent inhibitor of insulin action in cultured adipocytes as well as in vivo. This effect is of the same magnitude as the insulin antagonistic effect of TNF-α. In addition, we show that TNF-α induces IGFBP-3 production in cultured adipocytes and that IGFBP-3 may mediate some of the insulin-antagonistic activity of TNF-α. We also show that IGFBP-3 suppresses adiponectin expression. The mechanisms of IGFBP-3-induced insulin resistance action are as yet fully uncharacterized and are likely multiple. Further studies of the role of IGFBP-3 in insulin resistance will shed light on the molecular mechanisms of insulin resistance in general and the physiologic significance of IGFBP-3 in particular.
Abbreviations
- IGFBP:
-
insulin-like growth factor binding protein
- PPAR:
-
peroxisome proliferator activated receptor
References
Baker J, Liu JP, Robertson EJ, Efstratiadis A 1993 Role of insulin-like growth factors in embryonic and postnatal growth. Cell 75: 73–82
Moses AC, Young SC, Morrow LA, O'Brein M, Clemmons DR 1996 Recombinant human insulin-like growth factor I increases insulin sensitivity and improves glycemic control in type II diabetes. Diabetes 45: 91–100
Guler HP, Zapf J, Froesch ER 1987 Short-term metabolic effects of recombinant human insulin-like growth factor I in healthy adults. N Engl J Med 317: 137–140
Dohm GL, Elton CW, Raju MS, Mooney ND, DiMarchi R, Pories WJ, Flickinger EG, Atkinson SM Jr, Caro JF 1990 IGF-I–stimulated glucose transport in human skeletal muscle and IGF-I resistance in obesity and NIDDM. Diabetes 39: 1028–1032
Simpson HL, Umpleby AM, Russell-Jones DL 1998 Insulin-like growth factor-I and diabetes. Growth Horm IGF Res 8: 83–95
Cusi K, DeFronzo R 2000 Recombinant human insulin-like growth factor I treatment for 1 week improves metabolic control in type 2 diabetes by ameliorating hepatic and muscle insulin resistance. J Clin Endocrinol Metab 85: 3077–3084
Firth SM, Baxter RC 2002 Cellular actions of the insulin-like growth factor binding proteins. Endocr Rev 23: 824–854
Oh Y, Gucev Z, Ng L, Muller HL, Rosenfeld RG 1995 Antiproliferative actions of insulin-like growth factors binding protein (IGFBP)-3 in human breast cancer cells. Prog Growth Factor Res 6: 505–512
Valentinis B, Bhala A, DeAngeleis T, Baserga R, Cohen P 1995 The human insulin-like growth factor (IGF) binding protein-3 inhibits the growth of fibroblasts with a targeted disruption of the IGF-I receptor gene. Mol Endocrinol 9: 361–367
Murphy LJ 2003 The role of the insulin-like growth factors and their binding proteins in glucose homeostasis. Exp Diabesity Res 4: 213–224
Chan SS, Twigg SM, Firth SM, Baxter RC 2005 Insulin-like growth factor binding protein-3 (IGFBP-3) leads to insulin resistance in adipocytes. J Clin Endocrinol Metab 90: 6588–6595
Schuller AG, Groffen C, Van Neck JW, Zwarthoff EC, Drop SL 1994 cDNA cloning and mRNA expression of the six mouse insulin-like growth factor binding proteins. Mol Cell Endocrinol 104: 57–66
Clancy BM, Czech MP 1990 Hexose transport stimulation and membrane redistribution of glucose transporter isoforms in response to cholera toxin, dibutyryl cyclic AMP and insulin in 3T3- adipocytes. J Biol Chem 265: 12434–12443
Ranganathan S, Davidson MB 1996 Effect of tumor necrosis factor-a on basal and insulin-stimulated glucose transport in cultured muscle and fat cells. Metabolism 45: 1089–1094
Bradford MM 1976 A rapid and sensitive method for the quantitation of microgram quantities of protein using the principle of protein dye binding. Anal Biochem 72: 248–254
Silha JV, Gui Y, Murphy LJ 2002 Impaired glucose homeostasis in insulin-like growth factor-binding protein-3-transgenic mice. Am J Physiol Endocrinol Metab 283: E937–E945
Caprio S, Amiel SA, Merkel P, Tamborlane WV 1993 Insulin-resistant syndromes in children. Horm Res 39: 112–114
Grinspoon S, Clemmons D, Swearingen B, Klibanski A 1995 Serum insulin-like growth factor-binding protein-3 levels in the diagnosis of acromegaly. J Clin Endocrinol Metab 80: 927–932
Alford FP, Hew FL, Christopher MC, Rantzau C 1999 Insulin sensitivity in growth hormone (GH)-deficient adults and effect of GH replacement therapy. J Endocrinol Invest 22: 28–32
Ruan H, Lodish HF 2003 Insulin resistance in adipose tissue: direct and indirect effects of tumor necrosis factor-α. Cytokine Growth Factor Rev 14: 447–455
Lewitt MS, Denyer GS, Cooney GJ, Baxter RC 1991 Insulin-like growth factor binding protein-1 modulates blood glucose levels. Endocrinology 129: 2254–2256
Rajkumar K, Krsek M, Dheen ST, Murphy LJ 1996 Impaired glucose homeostasis in insulin-like growth factor binding protein-1 transgenic mice. J Clin Invest 98: 1818–1825
Okajima T, Iwasshita M, Takeda Y, Sakamoto S, Tanabe T, Yasuda T, Rosenfeld RG 1993 Inhibitory effects of insulin-like growth factor (IGF)-binding proteins-1 and-3 on IGF-activated glucose consumption in mouse BALB/ c 3T3 fibroblasts. J Endocrinol 136: 457–470
Yakar S, Liu JL, Fernandex AM, Wu Y, Schally AV, Frystyk J, Chernausek SD, Mejia W, LeRoith D 2001 Liver-specific IGF-I gene deletion leads to muscle insulin insensitively. Diabetes 50: 1110–1118
Ricort JM, Binoux M 2001 Insulin-like growth factor (IGF) Binding protein-3 inhibits type 1 IGF receptor activation independently of its IGF binding affinity. Endocrinology 142: 108–113
Oh Y, Muller HL, Phan H, Rosenfeld RG 1993 Demonstration of receptors for insulin-like growth factor binding protein-3 on Hs578T human breast cancer cells. J Biol Chem 268: 26045–26048
Schedlich LJ, LePage SL, Firth SM, Briggs LJ, Jans DA, Baxter RC 2000 Nuclear import of insulin-like growth factor-binding protein-3 and-5 is mediated by the importin beta subunit. J Biol Chem 275: 23462–23470
Butt AJ, Fraley KA, Firth SM, Baxter RC 2002 IGF-binding protein-3 induced growth inhibition and apoptosis do not requires cell surface binding and nuclear translocation in human breast cancer cells. Endocrinology 143: 2693–2699
Ricort JM, Binoux M 2002 Insulin-like growth factor-binding protein-3 activates a phosphotyrosine phosphatase. Effects on the insulin-like growth factor signaling pathway. J Biol Chem 277: 19448–19454
Welch DR, Milas L, Tomasovic SP, Nicolson GL 1983 Heterogeneous response and clonal drift of sensitivities of metastatic 13762NF mammary adenocarcinoma clones to gamma-radiation in vitro. Cancer Res 43: 6–10
Saghizadeh M, Ohg JM, Garvey WT, Henry RR, Kern PA 1996 The expression of TNF by human muscle: relationship to insulin resistance. J Clin Invest 97: 1111–1116
Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM 1994 Tumor necrosis factor-a inhibits signaling from the insulin receptor. Proc Natl Acad Sci U S A 91: 4854–4858
Rajah R, Lee KW, Cohen P 2002 Insulin-like growth factor binding protein-3 mediates tumor necrosis factor-alpha-induced apoptosis: role of Bcl-2 phosphorylation. Cell Growth Differ 13: 163–171
Buckbinder L, Talbott R, Velasco-Miguel S, Takenaka I, Faha B, Seizinger BR, Kley N 1995 Induction of the growth inhibitor IGF binding protein 3 by p53. Nature 377: 646–649
Gucev ZS, Oh Y, Kelley KM, Rosenfeld RG 1996 Insulin-like growth factor biding protein 3 mediates retinoic acid- and transforming growth factor B2-induced growth inhibition in human breast cancer cells. Cancer Res 56: 1545–1550
Rajah R, Valentinis B, Cohen P 1997 Insulin-like growth factor binding protein-3 induces apoptosis and mediates the effects of transforming growth factor B1 on programmed cell death through a p53-and IGF- independent mechanism. J Biol Chem 272: 12181–12188
Liu B, Lee HY, Weinzimer SA, Powell DR, Clifford JL, Kurie JM, Cohen P 2000 Direct functional interactions between the insulin-like growth factor-binding protein-3 and retinoid X receptor-a regulate transcriptional signaling and apoptosis. J Biol Chem 275: 33607–33613
Mukherjee R, Davies PJ, Crombie DL, Bischoff ED, Cessario RM, Jow L, Hamann LG, Boehm MF, Mondon CE, Nadzan AM, Paterniti JR, Heyman RA 1997 Sensitization of diabetic and obese mice to insulin by retinoid X receptor agonists. Nature 386: 407–410
Tanaka T, Itoh H, Doi K, Fukunaga Y, Hosoda K, Shintani M, Yamashita J, Chun TH, Inoue M, Masatwugu K, Sawada N, Saito T, Inoue G, Nishimura H, Yoshimasa U, Nakao K 1999 Down regulation of peroxisome proliferator-activated receptor expression by inflammatory cytokines and its reversal by thiazolidinediones. Diabetologia 42: 702–710
Kern PA, Di Gregorio GB, Lu T, Rassouli N, Ranganathan G 2003 Adiponectin expression from human adipose tissue: relation with obesity, insulin resistance, and tumor necrosis factor-α expression. Diabetes 52: 1779–1785
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported, in part, by National Institutes of Health grants R01AG20954, R01HD047013, P50CA92131, P30DK063491 (to PC), and P01-AG021654 (to NB).
Rights and permissions
About this article
Cite this article
Kim, H., Ali, O., Shim, M. et al. Insulin-Like Growth Factor Binding Protein-3 Induces Insulin Resistance in Adipocytes In Vitro and in Rats In Vivo. Pediatr Res 61, 159–164 (2007). https://doi.org/10.1203/pdr.0b013e31802d8a30
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/pdr.0b013e31802d8a30
This article is cited by
-
Maternal serum placental growth hormone, insulin-like growth factors and their binding proteins at 20 weeks’ gestation in pregnancies complicated by gestational diabetes mellitus
Hormones (2017)
-
Maternal serum insulin-like growth factor-binding protein-3 (IGFBP-3) at 11–13 weeks in preeclampsia
Journal of Human Hypertension (2012)
-
Chromosome 7p linkage and association study for diabetes related traits and type 2 diabetes in an African-American population enriched for nephropathy
BMC Medical Genetics (2010)
-
Insulin-like growth factor binding protein: a possible marker for the metabolic syndrome?
Acta Diabetologica (2010)
