
Abstract
There is some evidence that fetuses of diabetic rats (FDR) are hypomineralized. To explore the pathogenic role of decreased maternal duodenal Ca absorption, fetal hypotrophy, and decreased placental calbindin-D9K, respectively, spontaneously diabetic rats fed a 1.0% Ca diet were compared with diabetic rats treated with 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] (15 ng/100 g) during week 3 of pregnancy, which restored duodenal calbindin-D9K concentrations to normal; with nondiabetic rats semistarved during week 3, which resulted in similar fetal hypertrophy; and with nondiabetic rats fed high cation diets (1.5% Ca-1.5% Sr and 1.5% Ca-3.5% Sr) during week 3, the latter of which repressed duodenal and placental calbindin-D9K toward concentrations measured in diabetic rats. In addition, fetal tibiae were studied histologically. Ca content was lower in 21.5-d-old FDR than in control fetuses. FDR had lower plasma osteocalcin (OC) levels and, on histomorphometry, increased hypertrophic cartilage width, indicating retarded bone maturation. Maternal 1,25(OH)2D3 treatment did not change Ca content and hypertrophic cartilage width in FDR. Fetuses of semistarved rats had plasma OC levels and hypertrophic cartilage width comparable to those of control fetuses. Fetuses of rats fed the 1.5% Ca-3.5% Sr diet were more severely hypomineralized than FDR but had higher plasma OC than both FDR and control fetuses, compatible with fetal Ca deficiency. Whereas diabetic placentas showed weak but homogeneous staining of calbindin-D9K in the labyrinth on immunohistology, degenerative zones were present in placentas of rats fed the 1.5% Ca-3.5% Sr diet. Thus, there is no mineralization defect in FDR caused by disturbed maternal duodenal Ca absorption or transplacental Ca transport, but a delay in bone maturation that is unexplained by their lower body weight.
Similar content being viewed by others

Main
IDM have a higher risk of developing hypocalcemia in the first days after birth (1), the incidence and severity of which depend on the level of glycemic control during pregnancy (2). Early neonatal hypocalcemia in IDM is not the consequence of fetal hypercalcemia with suppression of PTH secretion; rather, both Ca and PTH levels were found to be lower in umbilical cord serum of IDM than in controls (3). The pathogenesis of fetal/neonatal hypocalcemia in IDM remains uncertain. IDM have been reported to have a decreased bone mineral mass: the number and mean diameter of the ossification centers at the knee (4) and the BMC measured at a distal site of the radius (5) were lower in IDM than in age-matched controls despite higher birth weight. In a recent study, however, whole body BMC was not found to be decreased in IDM (6). We found that cord serum concentrations of OC, a 6-kD glycoprotein produced by maturing osteoblasts, are below 1 SD of the normal range in most IDM (7). Biochemical parameters of bone resorption at birth were found to be either normal (8) or increased (9).
FDR show decreased mineralization as measured by the number of ossification centers and total ash weight (10), Ca and phosphate (P) content/fetus, and Ca and P content/fetal body weight (11–13). The impairment in mineralization is most pronounced in large litters (11). Maternal-fetal Ca transport is decreased in perfused placentas from diabetic rats (13).
In the present study, we investigated the pathogenesis of decreased mineralization in FDR. We identified a number of pathogenic variables that may explain hypomineralization. First, because 92% of fetal Ca has been calculated to originate from the maternal diet (14), the lack of increase in 1,25(OH)2D3 levels during late pregnancy observed in both diabetic women (15) and rats (11) may hamper the pregnancy-induced stimulation of intestinal Ca absorption that coincides with maximal fetal mineralization. Indeed, calbindin-D9K concentrations in duodenal mucosal scrapings tended to be lower in pregnant diabetic rats (16). Calbindin-D9K is a cytosolic 9-kD protein that binds and ferries Ca through the absorptive cells of the duodenum (17). Second, FDR have a lower body weight that at least in part is caused by impaired uteroplacental blood flow (18). In guinea pigs, placental blood flow correlates with fetal Ca content (19). Third, calbindin-D9K concentrations (16) and gene expression (13) are lower in diabetic rat placentas. Placental calbindin-D9K concentrations increase rapidly in the last week of pregnancy in rats (20) and correlate with transplacental Ca transfer (21). In fact, cytosolic calbindin-D9K, but not the Ca2+-ATPase activity at the basolateral membrane, is the rate-limiting step for Ca transport across the syncytiotrophoblast in the rat (21). Fourth, chondrocyte proliferation is decreased in FDR in vitro and in vivo (22), and plasma OC concentrations are lower than in control fetuses (23), confirming data obtained in IDM (7). These data are compatible with a delay in bone growth and bone mineralization.
Thus, we designed experimental models and methods to clarify these possible mechanisms: 1) Diabetic pregnant rats were treated with 1,25(OH)2D3 (15 ng/100 g/d). We previously found that this dose increased calbindin-D9K concentrations and active Ca absorption in duodenum of male diabetic BB rats (24). 2) Nondiabetic rats were semistarved during the last week of pregnancy to reproduce decreased uteroplacental blood flow and fetal hypotrophy (25). 3) Nondiabetic rats were fed high cation diets containing extra Ca and Sr. High Ca-Sr diets have been shown to repress intestinal and placental calbindin-D9K concentrations in pregnant mice (26), and Sr inhibits maternal-fetal Ca transport in dually perfused rat placentas (27). 4) Histology of fetal tibiae was performed to study endochondral mineralization and mineralized bone volume in the diaphysis.
METHODS
Animals. The experiments described below were approved by the ethical committee for animal research. Female diabetic rats were obtained from the Leuven BB/pfd Wistar rat colony, inbred since 1983. Approximately one third of these rats develop spontaneous type 1-like diabetes at a mean age of 13 wk (10,28). Diabetes was diagnosed by checking 3 times weekly for glycosuria, which invariably corresponds to a glycemia of >300 mg/dL. After onset of diabetes, the rats were mated overnight with a nondiabetic male from the same litter. When a vaginal plug was detected in the morning (defined as d 0.5), the rat was kept in a separate cage. Diabetic rats were untreated unless they showed ketonuria (checked 5 times weekly), in which case they received 1 U of Ultralente MC (Novo Nordisk, Bagsvaerd, Denmark); this regime leaves the rats untreated from a biologic viewpoint while preventing deaths due to ketosis (24). No insulin was given in the last 2 d of pregnancy. The rats were fed a standard rat chow (Trouw, Ghent, Belgium) containing 1.034% Ca, 0.758% P, and 2000 IU/kg vitamin D3.
A group of nine diabetic rats was compared with five other groups: 1) a group of seven diabetic pregnant rats infused s.c. with 1,25(OH)2D3 (15 ng/100 g/d) starting on d 14.5 by use of a model 2002 Alzet miniosmotic pump (Charles River, Palo Alto, CA) inserted in the neck region under light ether anesthesia; 2) a group of seven nondiabetic control pregnant rats fed ad libitum; 3) a group of nine pregnant rats that received 9 g/d of chow from d 14.5 [we previously found that nondiabetic rats consumed 21.6 ± 0.6 g (n = 12) and diabetic rats 27.4 ± 1.6 g of chow (n = 9) on d 20-21 (29)]; 4) a group of seven pregnant rats that received a high cation diet from d 14.5; the diet was supplemented with CaCO3 5 g/kg and SrCl2 15 g/kg. 5) In the course of the experiments, a group of eight pregnant rats fed an even higher Sr diet was added, because the previous group did not show decreased duodenal and placental calbindin-D9K concentrations. The diet was supplemented with CaCO3 5 g/kg and SrCl2 35 g/kg from d 14.5.
On d 20.5-21.5, the rats were placed in metabolic cages and a 24-h urine sample was collected. Cesarean section was performed on d 21.5 under pentobarbital anesthesia (60 mg/kg, intraperitoneally). The uterus was exposed from the left horn. Each fetus was bled from the axilla, and blood sampled from one litter was pooled. All fetuses and placentas were weighed separately. The tibiae of the first fetus were fixed in paraformaldehyde (4%, wt/vol) and glutaraldehyde (1%, vol/vol) in cacodylate buffer (0.1 M, pH 7.2). The other fetuses were pooled and eviscerated, dried for 24 h at 100°C, and ashed for 24 h at 600°C; the ash was dissolved in 12 M HCl. The first two placentas were fixed in 4% paraformaldehyde in cacodylate buffer. The other placentas were snap-frozen in liquid nitrogen and stored at -80°C. Maternal blood was sampled from the abdominal aorta, and plasma was stored at -20°C. A maternal duodenal mucosal scraping and the lower lobe of the left lung were snap-frozen in liquid nitrogen and stored at -80°C. The left maternal femur was kept at -20°C.
Assays. Glucose was measured by the glucose-oxidase technique (Beckman Glucose Analyzer 2, Fullerton, CA). Insulin was determined by RIA by use of a rat insulin standard (10). Ca in plasma, urine, and fetal ash dilutions was measured by atomic absorption spectroscopy (11,28). Total protein was measured as described previously (28). PTH was determined by an immunoradiometric assay kit that measures PTH1-34 (Immunotopics, San Clemente, CA). Sensitivity of the assay is 1 pg/mL (0.25 pmol/L), and the intra- and interassay coefficients of variation are ≤4.7%; this assay has been validated for in vivo measurements in rats (30). 1,25(OH)2D3 was measured by RIA after HPLC purification and the DBP by single radial immunodiffusion. The "free 1,25(OH)2D3 index" was calculated as the 1,25(OH)2D3/DBP molar ratio × 105 (10,31). OC was measured by RIA by use of a rat standard (23). Calbindin-D9K in homogenates of duodenum, lung, and placenta were determined by RIA by use of a standard purified from rat duodenum and a rabbit antiserum; the results were expressed as µg/mg protein (16,24,32).
Bone studies in dams. The area, BMC, and BMD (BMC/area) at the femur were measured by DXA with a Hologic QDR-4500 absorptiometer (S/N 45262); high-resolution mode was used (line spacing 0.0254 cm; resolution 0.0127 cm) with a collimator of 0.9 cm. We measured the BMC and BMD of the total femur at a middiaphyseal area consisting of cortical bone and at a metaphyseal area at 20% of the distal end consisting mainly of trabecular bone (33).
Histologic studies. Calbindin-D9K immunohistologic staining of the placenta was performed in five placentas of untreated diabetic rats, control rats, and rats fed the 1.5% Ca-3.5% Sr diet. Placentas were fixed for 24 h, rinsed in cacodylate buffer (0.1 M, pH 7.2) for 1-2 d, and then kept in 70% ethanol. Three micrometer-thick paraffin-embedded sections were dewaxed and endogenous peroxidase was blocked for 30 min with hydrogen peroxide (0.5%, vol/vol) and sodium azide (0.1%, wt/vol) in absolute methanol; sections were predigested with pepsin (0.005%, wt/vol) in HCl 0.01 M at 37°C for 10 min and nonspecific binding was blocked with BSA (2%, wt/vol). Subsequently, sections were incubated overnight at 4°C with anti-rat calbindin-D9K rabbit antiserum or normal rabbit serum (1:60 000). After a second blocking with BSA, goat Ig were detected with peroxidase-conjugated goat anti-rabbit Ig (1:100; Jackson Immunoresearch Laboratories, Westgrove, PA) in the presence of 4% complement-inactivated normal rat serum. The peroxidase activity was shown with diaminobenzidine in the presence of 0.1 M imidazole according to standard procedure; sections were lightly counterstained with Mayer's hematoxylin, dehydrated, and mounted. The sections were assessed qualitatively by a blinded examiner (R.P.).
Fetal tibiae were rinsed in cacodylate buffer after 2-h fixation and kept in 70% ethanol. They were embedded in a mixture of methylmetacrylate without removal of the inhibitor and JB-4 embedding kit (Polysciences, Warrington, PA) (25/75, vol/vol). Three micrometer-thick sections were cut with a Sorvall Porter-Blum MT2-B Ultramicrotome (Norwalk, CT); three series of sections per tibia were obtained with 60 µm in between. Consecutive sections were stained (34) according to von Kossa (5% silver nitrate) and by a modified Masson-Goldner trichrome: the red staining was 2 parts of solution A (Ponceau 0.1% in 0.2% acetic acid) and 8 parts of solution B (acid fuchsin 0.1% in 0.2% acetic acid) with Orange G at 0.2%. Histomorphometry of von Kossa-stained sections was performed at magnification ×20 with use of a VIDAS 21 Image Analyzing Computer (Kontron Bildanalyse, Munich, Germany); specific software was developed with the help of the manufacturer. The width of the hypertrophic cartilage at the proximal growth plate of the tibia was calculated by tracing the proximal, distal, and lateral ends of cartilage occupied by cells with the microscopic appearance of hypertrophic chondrocytes. The percentage of trabecular mineralized tissue [bone volume/total volume (BV/TV)] was measured in three consecutive fields in the vertical axis of the diaphysis excluding cortical bone, starting at the distal end of the hypertrophic cartilage of the proximal growth plate; the mean of these fields was calculated. For each parameter, the mean of the three sections was then calculated. Goldner-stained sections were used for qualitative histology.
Statistical analysis. A software program (NCSS, Kaysville, UT) was used. For each parameter, the overall difference was assessed by one-way ANOVA. If p < 0.05, the conservative Scheffé's post hoc test was used to detect significant differences between groups. Unpaired t tests were then used to quantify the level of significance between the diabetic group, the control group, and the other experimental groups. Data are expressed as mean (SEM).
RESULTS
General maternal and fetal parameters. Diabetic and semi-starved pregnant rats weighed less than control rats (Table 1); however, the high cation diets did not affect body weight. Diabetic rats were severely hyperglycemic and hypoinsulinemic; semistarved rats tended to have lower insulin levels than control rats (p = 0.06). Plasma Ca concentrations were higher in diabetic rats treated with 1,25(OH)2D3 than in untreated diabetic rats; semistarved rats had lower Ca levels than control rats. Diffusible Ca levels, a measure of protein-unbound Ca calculated as 6.25 × Ca - 0.375 × total protein (TP)/6.25 + TP (11,28), were not significantly different among groups (p = 0.15). Diabetic rats were polyuric [63 (8) mL/d versus 8.6 (0.8) in control rats; p < 0.001] and hypercalciuric; 1,25(OH)2D3 treatment further increased urinary Ca excretion but not urine volume [62 (6) mL/d]. Rats fed the high cation diets also had markedly increased urinary Ca excretion: daily Ca excretion was 10-fold higher in rats fed the 1.5% Ca-3.5% Sr diet than in control rats, whereas their urine volume was only 2-fold higher [18 (3) mL/d; p = 0.01].
Diabetic rats had fewer implantation sites than control rats, whereas there was no significant difference (p = 0.06) in the number of live fetuses among the six groups. There were more dead/resorbed fetuses in the 1.5% Ca-3.5% Sr group (Table 2). FDR weighed less than control fetuses; a similar mean fetal weight was obtained in semistarved rats. Fetuses of rats fed the 1.5% Ca-3.5% Sr diet weighed 78% of control fetuses. Mean placental weight was higher in the diabetic than in the nondiabetic groups; by contrast, semistarvation and the 1.5% Ca-3.5% Sr diet reduced placental weight compared with control rats.
Maternal and fetal calciotropic hormones. PTH concentrations were lower in diabetic than in control pregnant rats (Table 3). Rats on both high cation diets also had reduced PTH levels, whereas semistarved rats had increased PTH levels. 1,25(OH)2D3 concentrations were lower in diabetic than in control rats and were restored to normal by 1,25(OH)2D3 treatment in diabetic rats. Semistarved rats had slightly higher 1,25(OH)2D3 levels (p = 0.09) compared with rats fed ad libitum. Maternal DBP values were lower in diabetic [5.62 (0.35) µmol/L] and semistarved [5.77 (0.14) µmol/L] rats than in controls [7.33 (0.28) µmol/L]; p < 0.01 and p < 0.001, respectively. The calculated free 1,25(OH)2D3 index was higher in diabetic rats treated with 1,25(OH)2D3 than in untreated diabetic rats and was higher in semistarved than in ad libitum fed rats.
Fetal 1,25(OH)2D3 concentrations were always lower than the respective maternal values. Because the DBP values were even lower relative to maternal values [mean (SEM) of the groups between 1.13 (0.04) and 1.82 (0.08) µmol/L, i.e. between 17 and 25% of the respective maternal values], the free 1,25(OH)2D3 index was higher than the respective maternal value in all groups. There was no significant difference in the fetal 1,25(OH)2D3 concentrations or the free 1,25(OH)2D3 index among the groups (p = 0.22 and 0.12, respectively). However, the maternal and fetal 1,25(OH)2D3 concentrations, but not the free 1,25(OH)2D3 index, were correlated (Fig. 1).

Correlation between maternal and fetal plasma 1,25(OH)2D3 concentrations. o, diabetic rats; δ, diabetic rats treated with 1,25(OH)2D3; •, control rats; ♦, semistarved rats; ▴, rats fed 1.5% Ca-1.5% Sr diet; ▾, rats fed 1.5%-3.5% Sr diet. Regression curve: [OCf] = 37.7 + 0.15 [OCm]; r = 0.62; n = 47; p < 0.001. This correlation confirms our previous data (16).
Calbindin-D9K in duodenum, lung, and placenta. As previously reported (16,24), calbindin-D9K concentrations were lower in the duodenum (49% of controls) and placenta (58% of controls) but not in the lung of diabetic rats (Table 4); 1,25(OH)2D3 treatment increased duodenal but not placental calbindin-D9K concentrations. In rats fed the 1.5% Ca-3.5% Sr diet, duodenal and placental calbindin-D9K concentrations were 45 and 66%, respectively, of control values. The mean placental calbindin-D9K concentration per litter was not different between diabetic rats and rats fed the 1.5% Ca-3.5% Sr diet according to Scheffé's multiple comparison test; however, when the results of all placentas were analyzed separately, the mean concentration in the 1.5% Ca-3.5% Sr group [0.95 (0.02) µg/mg protein; n = 80] was higher (p < 0.01) than in the diabetic group [0.84 (0.03) µg/mg; n = 68]. Lung calbindin-D9K concentrations, which have been shown to be vitamin D independent (35), were higher in the 1.5% Ca-3.5% Sr group than in both diabetic and control groups.
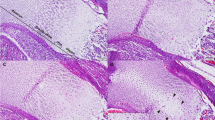
On immunohistology of control placentas, staining for calbindin-D9K concentrations was intense in the intraplacental yolk sac and less intense but homogeneous in the labyrinth zone; there was no discernible staining of the spongiosa. This confirms previous RIA, immunohistologic, and in situ hybridization data (20,36). In all five diabetic placentas, the intensity of staining in the labyrinth was weak, yet the homogeneity of the staining was preserved. By contrast, in rats fed the 1.5% Ca-3.5% Sr diet, the calbindin-D9K staining in the labyrinth was inhomogeneous, with degenerative zones where staining was absent (Fig. 2).

Microphotographs showing the immunohistologic pattern of calbindin-D9K in a typical placenta from a diabetic rat (a), a control rat (b), and a rat fed the 1.5% Ca-3.5% Sr diet (c). In control rats, calbindin-D9K staining was strong in the intraplacental yolk sac (long arrow) and less strong but homogeneous in the labyrinth (L) zone. Control sections from the three groups incubated with normal rabbit serum were all negative (no background staining). In diabetic rats, calbindin-D9K staining was equally strong in intraplacental yolk sac, whereas the staining was weak but still homogeneous in the labyrinth. In rats fed the 1.5% Ca-3.5% Sr diet, degenerative zones (short arrow) were present in the labyrinth with absent calbindin-D9K staining. Bar is 375 µm (control placenta).
Maternal bone mineralization. The total area and the area at the middiaphysis and distal metaphysis of the femur (Table 5) were not different among groups (p = 0.21, 0.71, and 0.18, respectively). Total femoral BMC was not different (p = 0.63). Femoral BMD (p = 0.03) tended to be higher in rats fed the 1.5% Ca-3.5% Sr diet than in diabetic (p = 0.06) rats. There were no differences in the middiaphyseal BMC (p = 0.71) or BMD (p = 0.50). The distal metaphyseal BMD (p = 0.003) was lower in semistarved than in diabetic rats, whereas the distal metaphyseal BMC (p = 0.005) and BMD of rats fed the 1.5% Ca-3.5% Sr diet were higher than in control rats (p = 0.03 for BMC; p = 0.02 for BMD).
OC concentrations in maternal and fetal plasma. OC concentrations were 73% lower in diabetic than in control dams; 1,25(OH)2D3 treatment did not increase plasma OC (Fig. 3). Semistarved dams had higher OC levels than controls fed ad libitum (p = 0.015), whereas rats fed the 1,25(OH)2 diet had decreased OC levels.

Plasma OC concentrations in maternal (M) and fetal (F) rats in the diabetic (D) group, diabetic group treated with 1,25(OH)2D3 (D + 1,25), control group (C), control semistarved group (C-SS), control group fed a 1.5% Ca-1.5% Sr diet (C-1.5% Sr), and the control group fed a 1.5% Ca-3.5% Sr diet (C-3.5% Sr). Statistical analysis was performed by one-way ANOVA followed by Scheffé's post hoc test, and the level of significance was quantified by unpaired t tests: different from diabetic group, maternal or fetal rats (a, p < 0.001; b, p < 0.01); different from control group, maternal or fetal rats (c, p < 0.001; d, p < 0.01; e, p < 0.05).
Mean OC concentrations were lower in fetal than in maternal rats in all groups. Fetal OC levels were lower in FDR than in control fetuses. Fetuses of rats fed the 1.5% Ca-3.5% Sr diet had 2.7-fold increased OC levels compared with control fetuses; one fetal OC concentration was actually higher than the respective maternal value.
Fetal mineralization. Mean Ca content was lower in FDR than in control fetuses and was not increased by 1,25(OH)2D3 treatment; Ca content/body weight was not significantly different, however (Table 6). Fetuses of semistarved rats had normal Ca content and content/weight. However, fetuses of rats fed the 1.5% Ca-3.5% Sr diet had decreased Ca content and Ca content/weight compared with control (26 and 34%, respectively) and diabetic rats.
Fetal bones of rats fed the 1.5% Ca-3.5% Sr diet were not included in the histomorphometric analysis because it proved difficult to retain sections for this purpose; many fetal tibiae contained little mineralized tissue (Fig. 4). There was no difference in the percentage of trabecular mineralized tissue in the diaphysis of the fetal tibiae in the other five groups (p = 0.15). The width occupied by hypertrophic chondrocytes was higher in FDR than in control fetuses (Fig. 4, Table 6) but was normal in fetuses of semistarved rats and rats fed the 1.5% Ca-1.5% Sr diet. On qualitative histology of Goldner-stained sections, tibiae from FDR contained less osteoid tissue than control tibiae (not shown).

Microphotographs of a typical tibial section stained according to von Kossa from an FDR (a), a control fetus (b), and a fetus from a rat fed the 1.5% Ca-3.5% Sr diet (c). Note the delayed bone maturation in the FDR, with a wider zone occupied by hypertrophic chondrocytes (arrow). Note the severe hypomineralization in fetuses fed the 1.5% Ca-3.5% Sr diet. Bar is 200 µm (control fetus).
In a combined analysis of all rats in the present study, five parameters were significantly correlated with fetal Ca content: fetal weight (r = 0.69), placental calbindin-D9K (r = 0.56), maternal 1,25(OH)2D3 (r = 0.43), maternal free 1,25(OH)2D3 (r = 0.33), and duodenal calbindin-D9K (r = 0.32). In a multiple regression analysis, the best prediction (R2 = 0.80) of fetal Ca content included four parameters as independent variables: fetal weight, maternal free 1,25(OH)2D3, placental calbindin-D9K, and fetal OC (Table 7).
DISCUSSION
The aim of the present study was to determine the role of maternal intestinal Ca absorption, fetal hypotrophy, and transplacental Ca transport in the fetal hypomineralization of diabetic rats and to study fetal bone morphology. FDR had a lower Ca content, but the Ca content/body weight (obtained as a single pooled value per litter in the present study) was not significantly different from that in control fetuses, unlike our previous finding (11). However, Ca content/weight was slightly lower in fetuses of diabetic rats treated with 1,25(OH)2D3 than in control fetuses (p = 0.09). In addition, we confirmed that FDR have lower OC concentrations than control fetuses (23), which is compatible with a delay in bone development: OC concentrations increase steadily during late fetal and neonatal life in rats (37). Histology of the tibia documented increased hypertrophic cartilage width, indicating a delay in endochondral mineralization. In addition, there appeared to be a decreased amount of osteoid tissue in the diaphysis on qualitative histology. Together with the finding of a lower number of ossification centers on Alizarin Red staining in FDR (10), these biochemical and histologic data indicate that bone maturation is retarded in FDR.
Diabetic dams treated with 1,25(OH)2D3 showed higher concentrations of 1,25(OH)2D3 and Ca in plasma and calbindin-D9K in duodenal mucosa, confirming our data in male diabetic BB rats (24). This treatment, however, did not increase fetal 1,25(OH)2D3 or placental calbindin-D9K concentrations; placental calbindin-D9K has previously been shown to be vitamin D-independent in nondiabetic rats (38). In addition, 1,25(OH)2D3 treatment did not stimulate fetal endochondral mineralization or Ca content, indicating that hypomineralization in FDR is not explained by disturbed maternal duodenal Ca absorption.
Fetuses from nondiabetic semistarved dams with a weight similar to FDR did not show decreased plasma OC levels, confirming our previous data (23), or increased width of hypertrophic cartilage width compared with control fetuses; thus, their bone maturation was normal for their gestational age. Because 92% of fetal Ca is derived from the maternal diet (14), other sources of Ca must be used in pregnant semistarved rats. PTH as well as OC concentrations were higher than in rats fed ad libitum, which is compatible with increased bone remodeling and Ca removal from bone. After only 7 d of semistarvation, the BMD at the distal femoral metaphysis was lowest in this group and was significantly lower than in diabetic rats. Regarding duodenal Ca absorption, we found increased free 1,25(OH)2D3 compared with rats fed ad libitum but no significant change in calbindin-D9K concentrations. We have previously reported higher free 1,25(OH)2D3 but lower duodenal calbindin-D9K values in semistarved than in freely fed male rats, whereas the active duodenal Ca absorption measured by the everted sac technique remained within the control range (28). If this holds true for the pregnant semistarved rats in the present study, then it would seem that activating maternal bone Ca removal is the premier mechanism to ensure normal fetal mineralization in case of lower dietary Ca intake in the last week of pregnancy.
Because the high cation diet containing both 1.5% Ca and Sr did not have an effect on placental calbindin-D9K concentration, we include a group fed a diet containing 1.5% Ca and 3.5% Sr. In the latter group, the mean placental calbindin-D9K concentration was only slightly above that in diabetic rats. However, their immunohistologic pattern was different. In diabetic rats, calbindin-D9K staining was homogeneous in the labyrinth zone but qualitatively less intense than in control rats; the intensity of the staining of the intraplacental yolk sac appeared to be comparable to that in control placentas (20). Placentas are larger in diabetic rats, which is caused by "cysts" in the junctional zone (spongiosa) but not by a larger labyrinth (39). By contrast, degenerative zones were present in the placental labyrinth of Sr-fed rats in which calbindin-D9K staining was absent. Placental calbindin-D9K concentrations correlate with maternal-fetal Ca transport in normal late-pregnant rats (21). Decreased presence of calbindin-D9K in placentas from diabetic and 3.5% Sr-fed rats is in good agreement with the lower maternal-fetal Ca flux measured in perfused placentas from diabetic rats (13) and in dually perfused placentas from normal rats with SrCl2 in the perfusate (27).
Pregnant rats fed the 3.5% Sr diet had decreased duodenal calbindin-D9K concentrations. This confirms data of lower active Ca absorption and duodenal calbindin-D9K in weanling male rats fed a 2.4% Sr diet (40). The high cation load also stimulated urinary Ca excretion 10-fold and PTH concentrations were decreased, compatible with lower bone Ca removal. BMD at the distal femoral metaphysis was indeed highest in this group, but the results must be interpreted with caution because it has been reported (41) that BMD values obtained by DXA are spuriously higher if hydroxyapatite contains Sr instead of Ca ions. Interestingly, OC concentrations were 63% of control levels. This relatively moderate decrease in circulating OC levels in "Sr rickets" is comparable with that observed in vitamin D-deficient rachitic rats (42,43).
Fetuses of 3.5% Sr-fed rats were severely hypomineralized. Clearly, there is a discrepancy between their hypomineralization and the placental calbindin-D9K concentrations compared with FDR. This discrepancy is probably explained by the fact that Sr impairs not only the cytosolic Ca transport but also the uptake of Ca at the maternal-facing microvillous membrane of the syncytiotrophoblast, as shown in the human placenta (44). OC concentrations were 2.7-fold higher than in control fetuses, indicating a state of Ca deficiency. Indeed, we have reported higher concentrations of OC in the fetuses and neonates of dams fed a Ca-P restricted diet (0.2% Ca and P instead of 0.85% Ca-0.7% P) (37). Rats weaned onto a Ca-deficient diet also have markedly increased serum OC (42). In neonatal and growing rats on a low Ca diet, there is typically a discrepancy between OC levels and the degree of bone mineralization (37,43).
Table 7 shows that a large part of the variance in fetal Ca content in these experiments is explained by factors that we postulated to be of importance at the outset of the present study: 1) fetal weight; 2) maternal duodenal Ca absorption as reflected by the free 1,25(OH)2D3 concentration; 3) active transplacental Ca transport as reflected by placental calbindin-D9K concentrations; and 4) fetal bone formation and maturation as reflected by their plasma OC concentrations. The analysis thus shows that the experiments were appropriately designed to unravel the pathogenesis of decreased Ca content in FDR.
We interpret the data from this and previous studies as follows. There is no mineralization defect caused by Ca deficiency in FDR but a delay in bone maturation as indicated by a lower number of ossification centers on Alizarin staining (10), a wider hypertrophic chondrocyte zone on histology (Table 6), and lower plasma OC levels (23) (Fig. 3). These changes are not explained by their lower body weight. The data concur with previous findings in growing or adult diabetic rats: endochondral bone growth and bone formation at the trabecular, endocortical, and periosteal surfaces are all markedly lower than in nondiabetic rats, but the mineralization rate is normal when adjusted for the bone matrix production rate (45). Decreased placental calbindin-D9K concentrations (16) (Table 4) and maternal-fetal Ca flux (13) are likely to be secondary to the retardation in fetal bone maturation. Pregnant diabetic rats have a higher food and Ca intake (29,46) and, consequently (17), passive intestinal Ca absorption (46). Because of this, and because of decreased maternal skeletal Ca incorporation (11,45) and transplacental Ca flux, PTH and 1,25(OH)2D3 concentrations are set lower (10,11) (Table 3) to minimize bone Ca removal and active duodenal Ca absorption, respectively. Urinary Ca excretion is increased many-fold (11,46) (Table 1), caused in part by higher intestinal Ca absorption ("hyperabsorption hypercalciuria"), and is further augmented by 1,25(OH)2D3 treatment, which stimulates active duodenal Ca absorption (24).
Abbreviations
- IDM:
-
infants of diabetic mothers
- FDR:
-
fetuses of diabetic rats
- 1,25(OH)2D3:
-
1,25-dihydroxyvitamin D3
- DBP:
-
vitamin D-binding protein
- OC:
-
osteocalcin
- DXA:
-
dual energy x-ray absorptiometry
- BMC:
-
bone mineral content
- BMD:
-
bone mineral density
References
Tsang RC, Kleinman LI, Sutherland JM, Light IJ 1972 Hypocalcemia in infants of diabetic mothers. J Pediatr 80: 384–395
Demarini S, Mimouni F, Tsang RC, Khoury J, Hertzberg V 1994 Impact of metabolic control of diabetes during pregnancy on neonatal hypocalcemia: a randomized study. Obstet Gynecol 83: 918–922
Cruikshank DP, Pitkin RM, Varner MW, Williams GA, Hargis GK 1983 Calcium metabolism in diabetic mother, fetus, and newborn infant. Am J Obstet Gynecol 145: 1010–1015
Pedersen J, Osler M 1958 Development of ossification centres in infants of diabetic mothers. Acta Endocrinol (Copenh) 29: 467–472
Mimouni F, Steichen JJ, Tsang RC, Hertzberg V, Miodovnik M 1988 Decreased bone mineral content in infants of diabetic mothers. Am J Perinatol 5: 339–343
Lapillonne A, Guerin S, Braillon P, Claris O, Delmas PD, Salle BL 1997 Diabetes during pregnancy does not alter whole body bone mineral content in infants. J Clin Endocrinol Metab 82: 3993–3997
Verhaeghe J, Van Herck E, Bouillon R 1995 Umbilical cord osteocalcin in normal pregnancies and pregnancies complicated by fetal growth retardation or diabetes mellitus. Biol Neonat 68: 377–383
Mora S, Prinster C, Bellini A, Weber G, Proverbio MC, Puzzovio M, Bianchi C, Chiumello G 1997 Bone turnover in neonates: changes of urinary excretion rate of collagen type I cross-linked peptides during the first days of life and influence of gestational age. Bone 20: 563–566
Demarini S, Specker BL, Sierra RI, Miodovnik M, Tsang RC 1995 Evidence of increased intrauterine bone resorption in term infants of mothers with insulin-dependent diabetes. J Pediatr 126: 796–798
Verhaeghe J, Bouillon R, Nyomba BL, Lissens W, Van Assche FA 1986 Vitamin D and bone mineral homeostasis during pregnancy in the diabetic BB rat. Endocrinology 118: 1019–1025
Verhaeghe J, Bouillon R, Lissens W, Visser WJ, Van Assche FA 1988 Diabetes and low Ca-P diets have opposite effects on adult and fetal bone and mineral metabolism. Am J Physiol 254: E496–E504
Demignon J, Rebut-Bonneton C 1988 Effects of experimental diabetes on the vitamin D metabolism of pregnant rats and their fetuses. Calcif Tissue Int 42: 127–135
Husain SM, Birdsey TJ, Glazier JD, Mughal MZ, Garland HO, Sibley CP 1994 Effect of diabetes mellitus on maternofetal flux of calcium and magnesium and calbindin9K mRNA expression in rat placenta. Pediatr Res 35: 376–381
Wasserman RH, Comar CL, Nold MM, Langemann FW 1957 Placental transfer of calcium and strontium in the rat and rabbit. Am J Physiol 189: 91–97
Mimouni F, Tsang RC, Hertzberg VS, Neumann V, Ellis K 1989 Parathyroid hormone and calcitriol changes in normal and insulin-dependent diabetic pregnancies. Obstet Gynecol 74: 49–54
Verhaeghe J, Thomasset M, Bréhier A, Van Assche FA, Bouillon R 1988 1,25(OH)2D3 and calcium-binding protein in fetal rats: relationship to the maternal vitamin D status. Am J Physiol 254: E505–E512
Bronner F, Pansu D, Stein WD 1986 Analysis of intestinal calcium transport across the rat intestine. Am J Physiol 250: G561–G569
Chartrel NC, Clabaut MT, Boismare FA, Schrub JC 1990 Uteroplacental hemodynamic disturbances in establishment of fetal growth retardation in streptozotocin-induced diabetic rats. Diabetes 39: 743–746
Symonds HW, Bubar RH, Crackel W, Twardock AR 1978 The effect of litter size on placental blood flow and placental calcium transfer in the multifoetate guinea pig. Br J Nutr 39: 347–356
Mathieu CL, Burnett SH, Mills SE, Overpeck JG, Bruns DE, Bruns ME 1989 Gestational changes in calbindin-D9K in rat uterus, yolk sac, and placenta: implications for maternal-fetal calcium transport and uterine muscle function. Proc Natl Acad Sci USA 86: 3433–3437
Glazier JD, Atkinson DE, Thornburg KL, Sharpe PT, Edwards D, Boyd RDH, Sibley CP 1992 Gestational changes in Ca2+ transport across rat placenta and mRNA for calbindin9K and Ca2+-ATPase. Am J Physiol 263: R930–R935
Heinze E, Vetter U 1987 Skeletal growth of fetuses from streptozotocin diabetic rat mothers: in vivo and in vitro studies. Diabetologia 30: 100–103
Verhaeghe J, Van Herck E, Van Bree R, Van Assche FA, Bouillon R 1989 Osteocalcin during the reproductive cycle in normal and diabetic rats. J Endocrinol 120: 143–151
Verhaeghe J, Suiker AMH, Van Bree R, Van Herck E, Jans I, Visser WJ, Thomasset M, Allewaert K, Bouillon R 1993 Increased clearance of 1,25(OH)2D3 and tissue-specific responsiveness to 1,25(OH)2D3 in diabetic rats. Am J Physiol 265: E215–E223
Rosso P, Kava R 1980 Effects of food restriction on cardiac output and blood flow to the uterus and placenta in the pregnant rat. J Nutr 110: 2350–2354
Bruns MEH, Wallshein V, Bruns DE 1982 Regulation of calcium-binding protein in mouse placenta and intestine. Am J Physiol 242: E47–E52
Stulc J, Stulcová B, Svihovec J 1990 Transport of calcium across the dually perfused placenta of the rat. J Physiol (Lond) 420: 295–311
Nyomba BL, Verhaeghe J, Thomasset M, Lissens W, Bouillon R 1989 Bone mineral homeostasis in spontaneously diabetic BB rats. I. Abnormal vitamin D metabolism and impaired active intestinal calcium absorption. Endocrinology 124: 565–572
Verhaeghe J 1988 Calcium metabolism during reproduction in the diabetic BB rat. Thesis, Katholieke Universiteit Leuven, Belgium, 98
Rucinski B, Mann GN, Epstein S 1995 A new rapid and reproducible homologous immunoradiometric assay for amino-terminal parathyroid hormone in the rat. Calcif Tissue Int 56: 83–87
Bouillon R, Van Assche FA, Van Baelen H, Heyns W, De Moor P 1981 Influence of the vitamin D-binding protein on the serum concentration of 1,25-dihydroxyvitamin D3 . J Clin Invest 67: 589–596
Thomasset M, Parkes CO, Cuisinier-Gleizes P 1982 Rat calcium-binding proteins: distribution, development, and vitamin D dependence. Am J Physiol 143: E483–E488
Verhaeghe J, Suiker AMH, Einhorn TA, Geusens P, Visser WJ, Van Herck E, Van Bree R, Magitsky S, Bouillon R 1994 Brittle bones in spontaneously diabetic BB rats cannot be predicted by bone mineral measurements: studies in diabetic and ovariectomized rats. J Bone Miner Res 9: 1657–1667
Baron R, Vignery A, Neff L, Silverglate A, Santa Maria A 1983 Processing of undecalcified bone specimens for bone histomorphometry. In: Recker RR (ed) Bone Histomorphometry: Techniques and Interpretation. CRC Press, Boca Raton, FL, 23–29.
Dupret JM, L'Horset F, Perret C, Bernaudin JF, Thomasset M 1992 Calbindin-D9K gene expression in the lung of the rat. Absence of regulation by 1,25-dihydroxyvitamin D3 and estrogen. Endocrinology 131: 2643–2648
Warembourg M, Perret C, Thomasset M 1986 Distribution of vitamin D-dependent calcium-binding protein messenger ribonucleic acid in rat placenta and duodenum. Endocrinology 119: 176–184
Verhaeghe J, Thomasset M, Van Assche FA, Bouillon R 1990 Osteocalcin is vitamin D-dependent during the perinatal period in the rat. J Dev Physiol 14: 311–317
Glazier JD, Mawer EB, Sibley CP 1995 Calbindin-D9K gene expression in rat chorioallantic placenta is not regulated by 1,25-dihydroxyvitamin D3 . Pediatr Res 37: 720–725
Prager R, Abramovici A, Liban E, Taron Z 1974 Histopathological changes in the placenta of streptozotocin-induced diabetic rats. Diabetologia 10: 89–91
Armbrecht HJ, Wasserman RH, Bruns MEH 1979 Effect of 1,25-dihydroxyvitamin D3 on intestinal calcium absorption in strontium-fed rats. Arch Biochem Biophys 192: 466–473
Christoffersen J, Christoffersen MR, Kolthoff N, Bärenholdt O 1997 Effects of strontium ions on growth and dissolution of hydroxyapatite and on bone mineral detection. Bone 20: 47–54
Price PA 1985 Vitamin K-dependent formation of bone G1a protein (osteocalcin) and its function. Vitam Horm 42: 65–108
Lian JB, Carnes DL, Glimcher MJ 1987 Bone and serum concentrations of osteocalcin as a function of 1,25-dihydroxyvitamin D3 circulating levels in bone disorders in rats. Endocrinology 120: 2123–2130
Kamath SG, Kelley LK, Friedman AF, Smith CH 1992 Transport and binding in calcium uptake by microvillous membrane of human placenta. Am J Physiol 262: C789–C794
Verhaeghe J, Bouillon R 1996 Effects of diabetes and insulin on bone metabolism In: Bilezikian JP, Raisz LG, Rodan GA (eds) Principles of Bone Biology. Academic Press, San Diego, 549–561.
Birdsey TJ, Husain SM, Garland HO, Sibley CP 1995 The effect of diabetes mellitus on urinary calcium excretion in pregnant rats and their offspring. J Endocrinol 145: 11–18
Acknowledgements
The authors thank Prof. F.P. Luyten, Rheumatology Service, K.U. Leuven, and Reviewer I for their critical comments, and A.-M. De Schutter, I. Jans, R. Janssens, J. Jolly, J. Nijs, and H. Peeters for their help. We also thank Novo Nordisk A/S (Bagsvaerd, Denmark) for the insulin vials and Boehringer (Mannheim, Germany) for Keto-Diabur stix.
Author information
Authors and Affiliations
Additional information
Supported by grant G.3C06.93 for Fundamental Clinical Research (J.V.) and financed by grant 3.0157.95, both from the Belgian Nationaal Fonds voor Wetenschappelijk Onderzoek. R.B. is supported by the “J. Servier Chair of Diabetes Research.”
Rights and permissions
About this article
Cite this article
Verhaeghe, J., van Bree, R., van Herck, E. et al. Pathogenesis of Fetal Hypomineralization in Diabetic Rats: Evidence for Delayed Bone Maturation. Pediatr Res 45, 209–217 (1999). https://doi.org/10.1203/00006450-199902000-00009
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199902000-00009
This article is cited by
-
Of mice and mutations: Phenotypic effects of the diabetic db/db and ob/ob mutations on the skull and teeth of mice
European Archives of Paediatric Dentistry (2008)
-
Enamel mineral concentration in diabetic rodents
European Archives of Paediatric Dentistry (2007)
-
Diabetes in rat pregnancy alters renal calcium and magnesium reabsorption and bone formation in adult offspring
Diabetologia (2005)
