Abstract
In the endeavor to close the anthropogenic carbon cycle and produce goods in a de-fossilized future, electrochemical carbon dioxide reduction constitutes a powerful tool.[1] Herein,3d transition metal complexes range among the most effective catalysts to overcome the kinetic barriers for CO2 activation.[2]
In a recent literature review, we analyzed the main reaction pathways of homogeneously catalyzed CO2 electroreduction from an organometallic perspective and classified them into two mechanisms: (1) direct coordination of carbon dioxide to the metal center prior to activation and (2) formation of a metal hydride as the intermediate reactive to CO2.[3] Although properties of the metal center such as hydricity, or substrate (CO2) and product (CO, HCO2H...) binding affinity, are crucial regarding the mechanism and the ability of the complex to mediate CO2 reduction, systematic variations of the metal within an identical, redox-innocent ligand backbone remain scarcely investigated.
In this study,[4] we report on the synthesis, structures, and electrochemistry of 3d transition metal complexes (M = Mn(I), Fe(II), Co(II), Ni(II), Cu(I) and Zn(II)) coordinated by a new redox-innocent PNP pincer ligand system (Figure 1). The ligand combines a large steric demand, partially shielding the metal center, with a high degree of aromaticity. With these features, the ligand only marginally interferes in the redox processes but still takes sufficient π-backbonding to stabilize low-valent metals.
The coordination compounds derived thereof were analyzed by spectroscopic methods (e.g., NMR, EPR, and Mössbauer) as well as crystal X-Ray diffraction to determine their spatial and electronic structures. These findings served as the starting point for a further exploration of the electrochemical reduction of the complexes via cyclic voltammetry. While the Mn, Cu, and Zn complexes solely exhibit ligand reduction or decomposition processes, the distinct electrochemical waves found for the Fe, Co and Ni coordination compounds could be assigned to metal-centered reduction events from M(II) down to M(0).
For both cobalt and nickel, the reductions appear to be accompanied by their chloride ligands being lost or exchanged with the acetonitrile solvent, a fact that we currently investigate[5] as these phenomena remain misjudged across many (CO2) electroreduction catalysts. In contrast to Co and Ni, Fe undergoes a more complex reduction pattern likely yielding a dimeric species.
Despite their unequal reduction pathways, the evolution of the voltammograms of the complexes under CO2 atmosphere indicates an interaction of each of the complexes with the substrate molecule in their reduced metal states. The d10 Ni(0) species putatively forms an Aresta-type Ni-η2-CO2 complex, in which the electron transfer to the substrate through backbonding is insufficient to enable electrocatalytic activity. At contrast, CO2 binding at the d9 Co(0) intermediate likely leads to additional electron uptake and formation of a formal Co(I) metallacarboxylate complex able to promote turnover (Figure 2).
Eventually, we related our findings to the few literature precedents that incorporate redox-innocent ligands. This assessment shows that beneficial characteristics in the electrochemical activation of CO2 by complexes based on redox-innocent ligands are an unsaturated coordination sphere (coordination number = 4 or 5) as well as a d7 to d9 configuration in the reduced oxidation state (+I or 0). The on-purpose design of complexes that simultaneously meet these three characteristics hence provides a promising strategy for catalyst development. In particular, dynamic structural and electronic changes under electrochemical conditions, such as the exchange of chlorido ligands with acetonitrile as faced in this study, must be controlled to ensure the primary operation of the metal centers in the desired catalytic manifold.
References
[1] P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo, E. H. Sargent, Science 2019, 364, eaav3506.
[2] R. Francke, B. Schille, M. Roemelt, Chem. Rev. 2018, 118, 4631.
[3] N. W. Kinzel, C. Werlé, W. Leitner, Angew. Chem. Int. Ed. 2021, 60, 11628.
[4] N. W. Kinzel,D. Demirbas, E. Bill, T. Weyhermüller, C. Werlé,N. Kaeffer, W. Leitner, Inorg. Chem. 2021, doi: 10.1021/acs.inorgchem.1c02909.
[5] N. W. Kinzel, N. Kaeffer, W. Leitner in preparation.
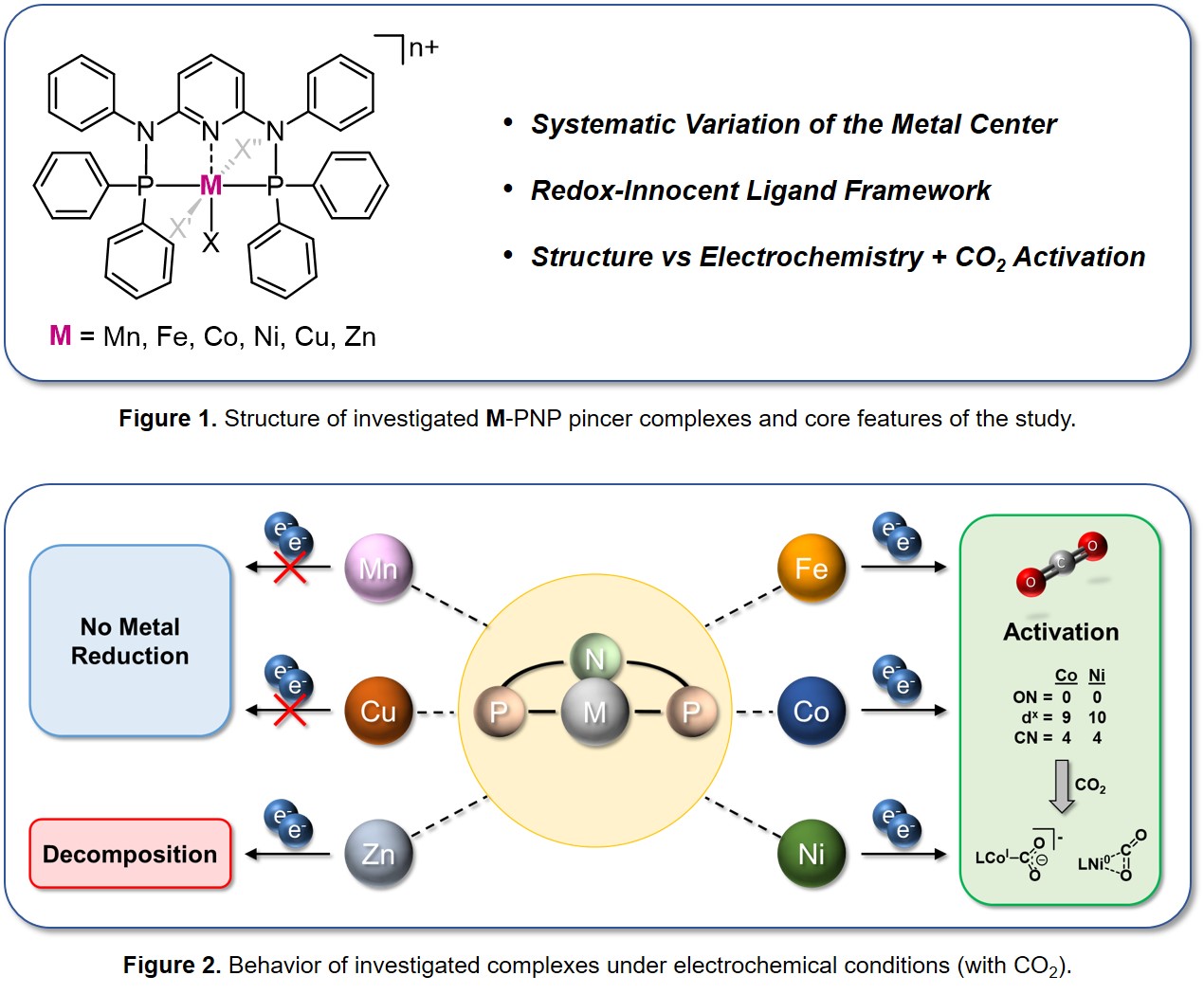
Figure 1