Abstract
Cyclic voltammetry and controlled-potential (bulk) electrolysis have been employed to investigate the electrochemical reduction of 4,4'-(2,2,2-trichloroethane-1,1-diyl)bis(methoxybenzene), commonly known as the pesticide methoxychlor, at glassy carbon and silver cathodes in dimethylformamide (DMF) containing 0.050 M tetra-n-butylammonium tetrafluoroborate (TBABF4). Reduction of methoxychlor at both glassy carbon and silver shows four voltammetric peaks, the first three of which are associated with cleavage of carbon–chlorine bonds; the fourth peak is assigned to reduction of 4,4'-(ethene-1,1-diyl)bis(methoxybenzene). Bulk electrolyses of methoxychlor at reticulated vitreous carbon and silver mesh cathodes at potentials corresponding to each of the first three voltammetric peaks were conducted; coulometric n values and product distributions (determined by means of GC and GC–MS techniques) depend on potential. In particular, two completely dechlorinated products, namely 4,4'-(ethane-1,1-diyl)bis(methoxybenzene) and 4,4'-(ethene-1,1-diyl)bis(methoxybenzene) have been identified and quantitated. A mechanistic scheme is proposed to account for the formation of the various products.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
When global use of the infamous pesticide 4,4'-(2,2,2-trichloroethane-1,1-diyl)bis(chlorobenzene) (1)—known more familiarly as DDT—was restricted, a close relative, namely 4,4'-(2,2,2-trichloroethane-1,1-diyl)bis(methoxybenzene) (2) or methoxychlor, became a temporarily popular replacement. Unfortunately, when concerns about the toxicity of 2 arose, it was banned in the European Union in 2002 and became unregistered in the United States in 2003. However, in other parts of the world, 2 is still employed as an agricultural pesticide and for home applications in the treatment of flea and fly infestations.
Like many members of the family of halogenated pesticides, methoxychlor degrades very slowly in the environment, which allows for its transport and subsequent detection in Arctic samples far from the original point of use.1 For hydrolytic degradation pathways in aquatic systems of pH 6–9, the half-life is approximately one year, and the resulting olefinic metabolite degrades even more slowly.2 When exposed to sunlight, this olefin derivative undergoes an interesting photoisomerization in water.3 Sunlight increases the photolysis rate, so that the half-life at the surface of distilled water is closer to 4.5 months, and even faster in natural waters where half-lives of only 2–5 h are seen.4 Biological degradation of methoxychlor results in the loss of one chlorine as well as demethylation of the hydroxylbenzene moieties.5 Uptake of methoxychlor in a model ecosystem has also been examined; whereas methoxychlor was found to be in a dynamic state in fish, it accumulated in snails. Furthermore, incubated mouse livers were unable to metabolize methoxychlor and 97% was recovered.6
Concerns about the accumulation of methoxychlor in humans prompted studies conducted in the agricultural region of Spain (decades after local regulations had restricted usage); methoxychlor was detected in the serum of 60.7% of young men tested.7 Extensive research has dealt with the estrogenetic activity of methoxychlor, which has adverse effects on fertility and early pregnancy.8 A study of 2,2-bis(hydroxyphenyl)-1,1,1-trichloroethane (a metabolite of methoxychlor) demonstrated that it decreases the production of testosterone in rats, linking methoxychlor to male infertility.9 Moreover, methoxychlor has been associated with human breast cancer and irregular cell development.10
Interest in the use of electrochemical reduction for detection, degradation, and remediation of halogenated pesticides has prompted a number of investigations, some of which are described in a recent review.11 In early work by Rosenthal and Lacoste,12 a polarographic study of a number of aromatic chloroethanes, including methoxychlor, established that electrochemical reduction resulted in cleavage of all three carbon–chlorine bonds. Tandon and co-workers extended this research in their own polarographic studies of the reduction of methoxychlor in aqueous media containing dimethylformamide (DMF) and ethanol,13 as well as in the presence of various surfactants;14 it was reported that uptake of six electrons is required for cleavage of the three carbon–chlorine bonds. There has been only one investigation of the electrochemical oxidation of methoxychlor at a platinum anode in acetonitrile; major products included 4,4'-dimethoxybenzophenone and dimethoxykelthane.15 To the best of our knowledge, bulk reduction of methoxychlor to identify the dechlorinated products has never been performed, despite the fact that electroreductive carbon–chlorine bond cleavages are well documented in the literature.
Two previous publications from our laboratory describe the electrochemical reduction of a related compound (DDT) at carbon16 and silver17 cathodes in DMF containing a tetraalkylammonium salt as supporting electrolyte. Both of these papers revealed that each of the aliphatic carbon–chlorine bonds undergoes reductive cleavage via a two-electron process, followed by subsequent protonation from residual water in the solvent–electrolyte, and accompanied by hydroxide-promoted elimination reactions to expel chloride ions from the substrate. For these just-mentioned reports,16,17 significant differences in potentials were needed to reduce certain carbon–chlorine bonds of DDT at carbon and silver cathodes. An increasing number of studies has demonstrated that organic chlorides are easier to reduce at silver cathodes than at carbon electrodes.18–20 Recently, it has been shown in our laboratory that the position and identity of the halogen atom affect the reduction potential and mechanism.21,22 Research from the groups of Isse23,24 and Amatore25 has provided further insight concerning the mechanisms of electroreductive carbon–chlorine bond cleavage at silver cathodes; it has been determined that activated species adsorb onto the silver surface, thereby promoting the observed catalytic effects.
In the present work, cyclic voltammetry and controlled-potential (bulk) electrolysis have been used to investigate the electrochemical reduction of methoxychlor at carbon and silver cathodes in DMF containing 0.050 M tetra-n-butylammonium tetrafluoroborate (TBABF4) as supporting electrolyte. Additionally, the cyclic voltammetric behavior of a related compound, 4,4'-(ethene-1,1-diyl)bis(methoxybenzene), was examined at both glassy carbon and silver cathodes to characterize the final stage of methoxychlor reduction. Gas chromatography (GC) and gas chromatography–mass spectrometry (GC–MS) have been employed to separate, identify, and quantitate all products arising from the electrolytic reduction of methoxychlor. A mechanistic scheme is proposed which indicates that the overall dechlorination of methoxychlor involves a combination of electrochemical cleavage of carbon–chlorine bonds and hydroxide-promoted elimination of chloride ions.
Experimental
Reagents
Methoxychlor (2) was obtained from Alfa Aesar (98.5% purity). Related compounds were also purchased for use as analytical standards: 4,4'-(2,2-dichloroethene-1,1-diyl)bis(methoxybenzene) (3, Chem Service, Inc., 94.9%); 4,4'-(ethene-1,1-diyl)bis(methoxybenzene) (7, Sigma Aldrich, purified), and 4,4'-(ethane-1,1-diyl)bis(methoxybenzene) (8, Enamine Ltd., 90%).
Deuterium oxide (D2O, 99.9 atom % D) and n-hexadecane (99%, used as an internal standard for gas chromatography) were acquired from Aldrich Chemical Company and used without further purification. Solvent used in each experiment was dimethylformamide (DMF, EMD Chemicals, 99.9%). Tetra-n-butylammonium tetrafluoroborate (TBABF4, TCI Chemicals, 98%), employed as supporting electrolyte, was recrystallized from diethyl ether–ethyl acetate and stored in a vacuum oven at 80ºC. Zero-grade argon (Air Products) was used to remove oxygen from solutions prior to each experiment.
Electrodes, cells, and instrumentation
Previous publications26,27 describe our electrochemical cells, the Princeton Applied Research Corporation (PARC) instrumentation, and experimental procedures utilized for cyclic voltammetry. Short rods of silver (99.9% purity, Alfa Aesar) and glassy carbon (Grade GC-20, Tokai Electrode Manufacturing Company) were press-fitted into machined Teflon tubes to provide working electrode disks, each with a geometric area of 0.071 cm2; electrical connection was obtained via a stainless-steel rod inserted into the opposite end of the Teflon tube. These cathodes were cleaned by polishing in a suspension of 0.05 μm aluminum oxide on a Master-Tex (Buehler) polishing pad, as indicated in an earlier paper.27
Controlled-potential electrolyses were conducted in a two-compartment cell described previously.28 A PARC model 173 potentiostat–galvanostat was interfaced with a data acquisition card; current–time curves were obtained via a customized LabView program and integrated with the aid of OriginPro 2015 software. Bulk electrolyses were carried out with two different electrode materials: (a) reticulated vitreous carbon (RVC) disks (4 mm thickness, 2.4 cm diameter, 200 cm2 geometric area, purchased from ERG Aerospace Corporation, Oakland, CA) and (b) silver gauze (99.9%, 20 mesh woven from 0.356 mm diameter wire, 20 cm2 geometric area, obtained from Alfa Aesar).
All potentials cited in this report were measured with respect to a cadmium-saturated mercury amalgam reference electrode in contact with DMF saturated with both cadmium chloride and sodium chloride; this reference electrode has a potential of –0.76 V vs. an aqueous saturated calomel electrode (SCE) at 25°C29–31
Separation, identification, and quantitation of electrolysis products
After each bulk electrolysis, the catholyte was partitioned between brine and diethyl ether (EMD Chemicals, 99%) and the ether layer was rinsed twice with distilled water; then the ether phase was dried over anhydrous sodium sulfate and concentrated via rotary evaporation to a volume of approximately 1 mL prior to analysis. Gas chromatography–mass spectrometry (Hewlett-Packard 6890N gas chromatograph, fitted with a 30 m × 0.25 mm i.d. Rxi-5-SilMS, 0.25 μm thick film capillary column and coupled to a Hewlett-Packard model 5973 inert mass-selective detector) was used to separate and acquire mass spectral information for all electrolysis products. By comparing experimentally acquired mass spectra with those found in the NIST 2007 mass-spectral database, we were able to identify six different products, as shown below:
To quantitate each of the products, a known amount of an internal standard (n-hexadecane) was added to each solution prior to the start of an electrolysis. Products were extracted as described above, and concentrated ether extracts were injected into an Agilent 7890A gas chromatograph equipped with a flame-ionization detector and a 30 m × 0.25 mm capillary column (J & W Scientific) with a DB-5 stationary phase consisting of 5% phenylpolysiloxane and 95% methylpolysiloxane. Known amounts of commercially obtained chemical standards for 2 and 3 and the completely dechlorinated analogues 7 and 8 were also injected into the chromatograph; their resulting chromatographic peak areas were then measured with respect to the internal standard (n-hexadecane) to determine a response factor used to calculate their respective abundances in electrochemical experiments, according to a procedure previously described.32 Because total dechlorination of 2 was the goal of this work, we were most interested in the yields of completely dechlorinated products; however, we also estimated the quantities of partially dechlorinated species. Response factors for 2 and dechlorinated 7 and 8 were similar (ranging from 0.82–0.96); therefore, an average response factor of 0.92 was used to calculate the yields of 4, 5, and 6 formed in each electrolysis. Product yields are reported as the absolute percentage of starting material that was converted into a particular species.
Results and Discussion
Cyclic voltammetric behavior of methoxychlor (2) and 4,4'-(ethene-1,1-diyl)bis(methoxybenzene) (7)
Shown in Figure 1 are cyclic voltammograms recorded at 100 mV s−1 for the reduction of 5.0 mM solutions of 2 at glassy carbon (dashed line) and silver (solid line) electrodes in DMF containing 0.050 M TBABF4. Cyclic voltammograms for reduction of 2 at glassy carbon exhibit a prominent cathodic peak at –1.14 V, followed by a small shoulder at –1.44 V, and two more irreversible cathodic peaks at –1.68 and –1.91 V. Similarly, voltammograms for reduction of 5.0 mM 2 at silver show an irreversible cathodic peak at –0.90 V, a shoulder at –1.40 V, and two additional prominent peaks at –1.66 and –1.94 V.
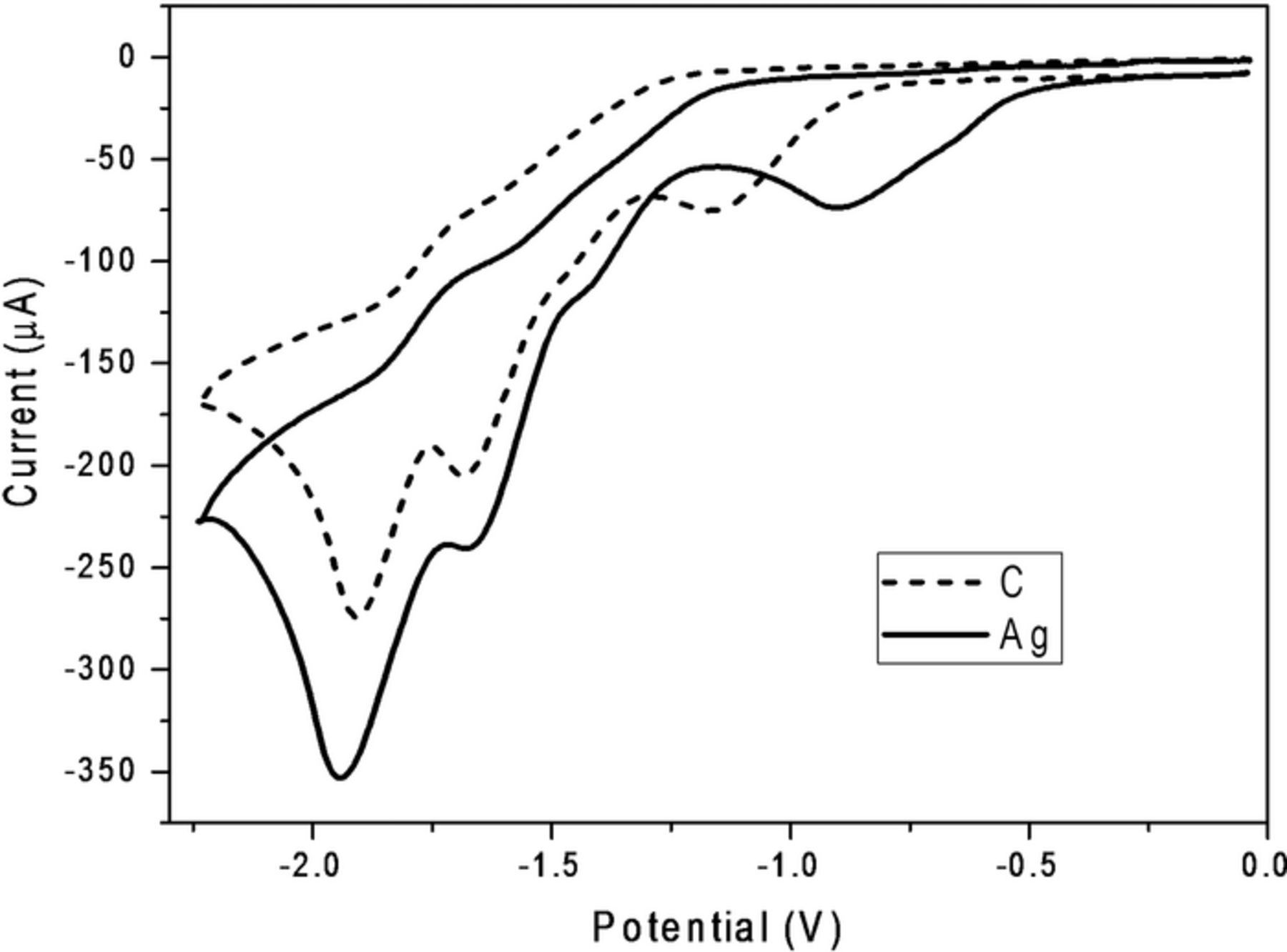
Figure 1. Cyclic voltammograms recorded at 100 mV s−1 for reduction of 5.0 mM methoxychlor (2) at a glassy carbon cathode (dotted curve) and at a silver cathode (solid curve) in oxygen-free DMF containing 0.050 M TBABF4; the area of each electrode was 0.071 cm2, and all scans go from ca. 0 to –2.2 to 0 V. Potentials are given with respect to a cadmium-saturated mercury amalgam reference electrode in contact with DMF saturated with both cadmium chloride and sodium chloride; this electrode has a potential of –0.76 V versus SCE at 25°C.
In comparing the two sets of voltammograms, we notice that the first stage of reduction results in a much broader peak at a silver cathode than at a glassy carbon electrode; in fact, the first peak recorded with silver seems to consist of two nearly merged waves. It is noteworthy that the first stage of reduction occurs at a significantly less negative potential for silver than for glassy carbon. We attribute this potential shift to the well-known catalytic effects of silver as a cathode material for carbon–chlorine bond cleavage, as reported in literature.18–25 It is interesting that there is little shift in the peak potential for the second stage of reduction when glassy carbon is compared with silver; furthermore, the third and fourth cathodic peak potentials are independent of cathode material.
Depicted in Figure 2 are cyclic voltammograms recorded at 50, 100, 250, and 500 mV s−1 for reduction of 5.0 mM 2 at the glassy carbon electrode. For each of the four cathodic peaks, we observed (a) an increase in current and (b) a shift in potential to significantly more negative values with increasing scan rate. In similar fashion, Figure 3 displays cyclic voltammograms recorded at 50, 100, 250, and 500 mV s−1 for reduction of 5.0 mM 2 at the silver cathode; again, as the scan rate was increased, we observed an increase in peak current and a shift toward more negative peak potentials. These observations are consistent with irreversible reduction, and the concerted dissociative electron transfer mechanism expected for cleavage of aliphatic halides.33
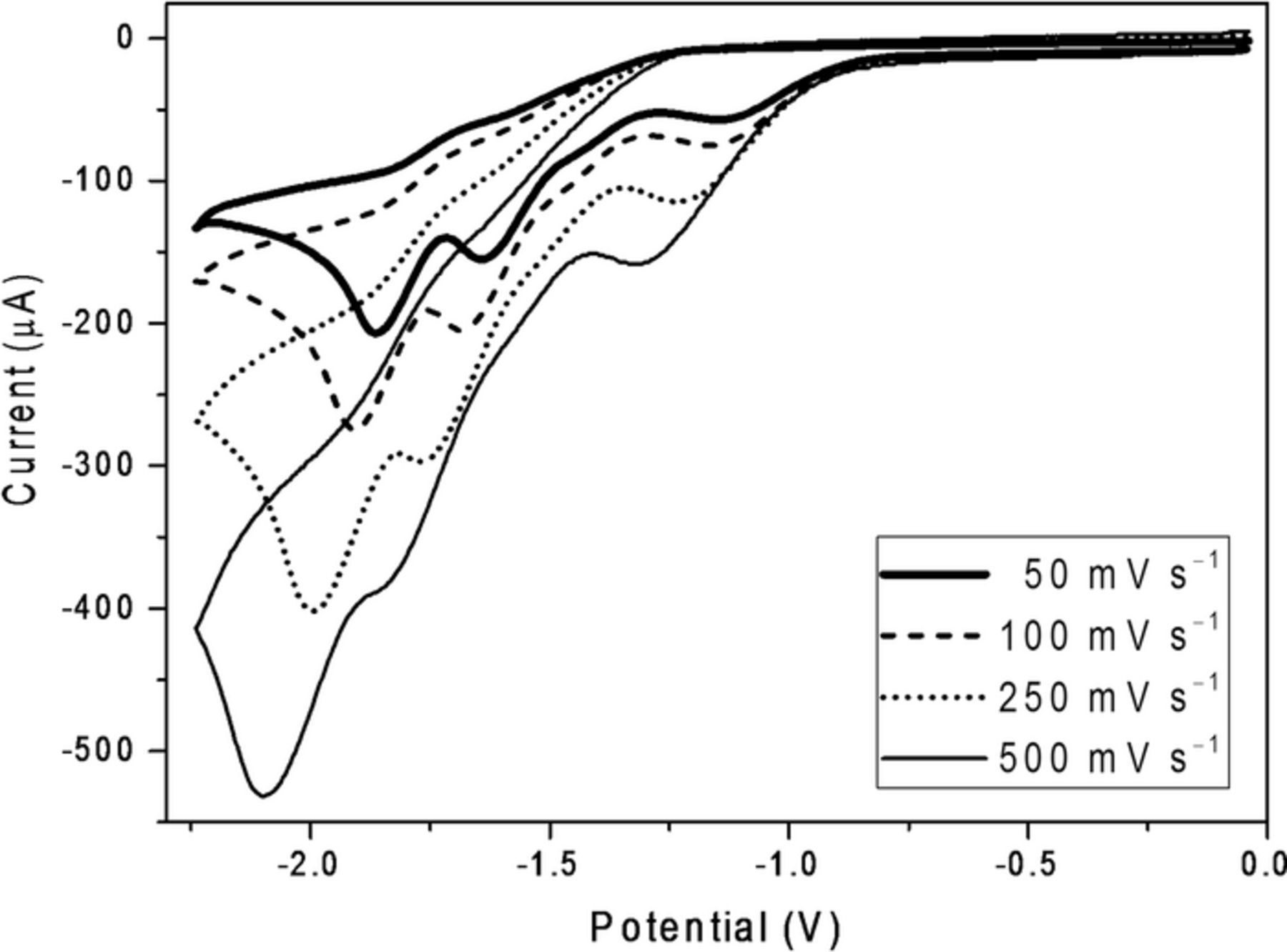
Figure 2. Cyclic voltammograms recorded at scan rates of 25, 100, 250, and 500 mV s−1 for reduction of 5.0 mM methoxychlor (2) at a glassy carbon cathode (area of 0.071 cm2) in oxygen-free DMF containing 0.050 M TBABF4. Scans go from ca. 0 to –2.2 to 0 V. Potentials are given with respect to a cadmium-saturated mercury amalgam reference electrode in contact with DMF saturated with both cadmium chloride and sodium chloride; this electrode has a potential of –0.76 V versus SCE at 25°C.
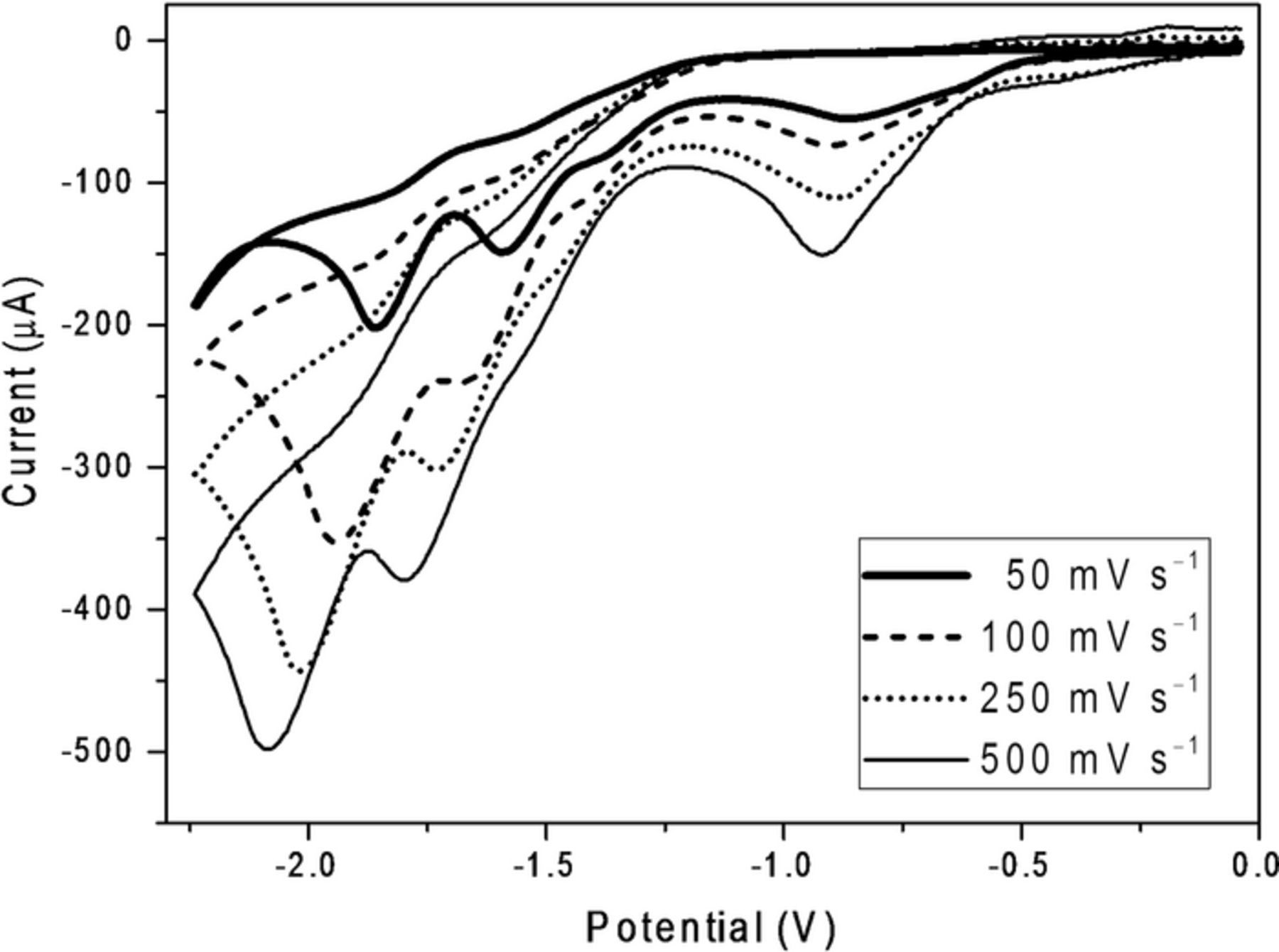
Figure 3. Cyclic voltammograms recorded at scan rates of 25, 100, 250, and 500 mV s−1 for reduction of 5.0 mM methoxychlor (2) at a silver cathode (area of 0.071 cm2) in oxygen-free DMF containing 0.050 M TBABF4. Scans go from ca. 0 to –2.2 to 0 V. Potentials are given with respect to a cadmium-saturated mercury amalgam reference electrode in contact with DMF saturated with both cadmium chloride and sodium chloride; this electrode has a potential of –0.76 V versus SCE at 25°C.
Shown in Figure 4 are cyclic voltammograms, recorded with both glassy carbon (dashed line) and silver (solid line) cathodes, for reduction of 7. For both cathode materials, a single irreversible peak is observed near –1.86 V, which value is very close to that for the most negative peaks depicted in cyclic voltammograms for reduction of 2 shown in Figure 1. We attribute this peak to the reduction of 7 to afford 8.
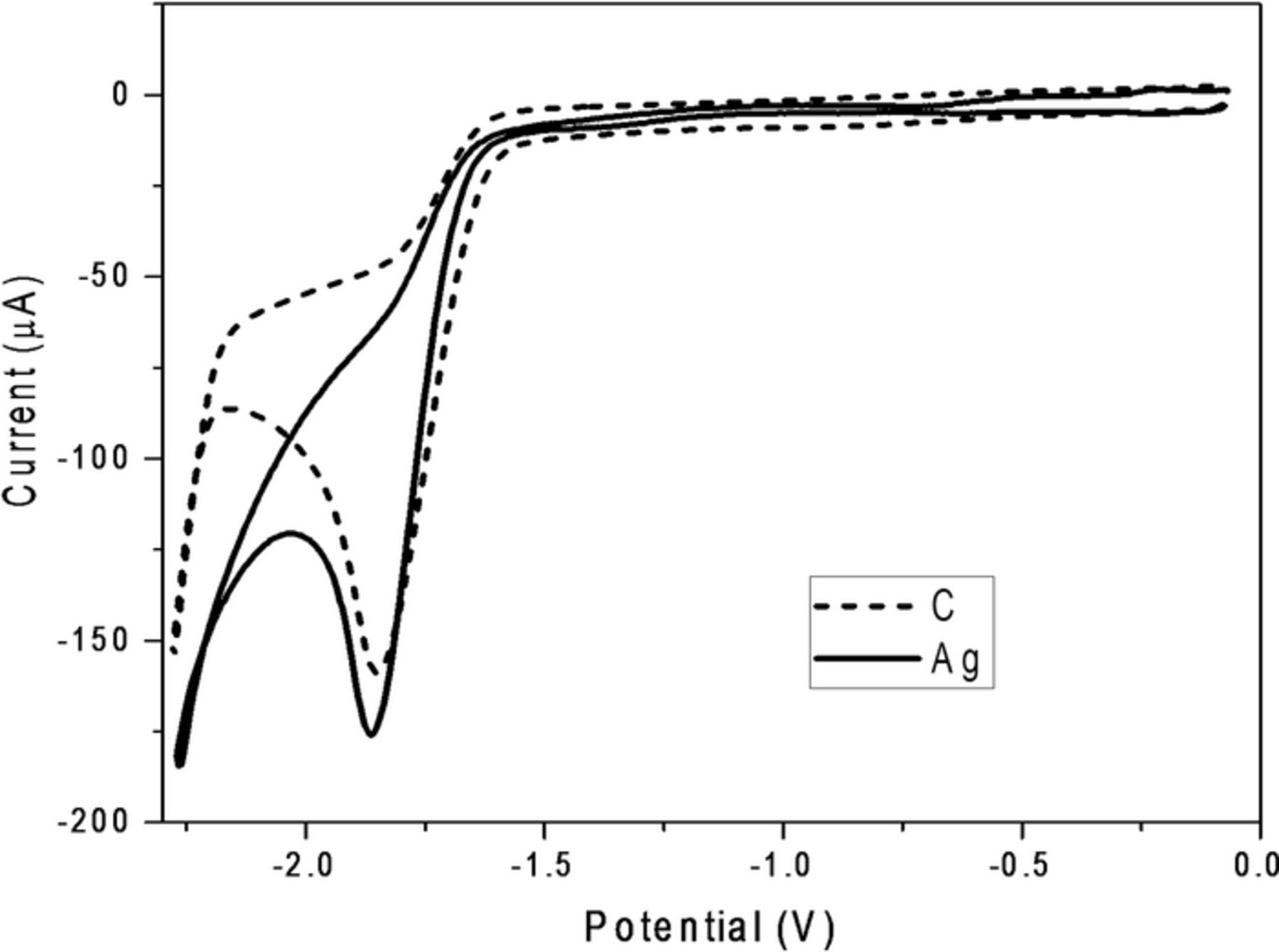
Figure 4. Cyclic voltammograms recorded at 100 mV s−1 for reduction of 5.0 mM 4,4'-(ethene-1,1-diyl)bis(methoxybenzene) (7) at a glassy carbon cathode (dotted curve) and at a silver cathode (solid curve) in oxygen-free DMF containing 0.050 M TBABF4. Areas of electrodes were 0.071 cm2, and scans go from ca. 0 to –2.2 to 0 V. Potentials are given with respect to a cadmium-saturated mercury amalgam reference electrode in contact with DMF saturated with both cadmium chloride and sodium chloride; this electrode has a potential of –0.76 V versus SCE at 25°C.
It is suggested in the literature that hydroxide ions, resulting from deprotonation of residual water in the solvent–electrolyte system, can promote the elimination of one chloride ion from the substrate to form an olefinic species.34 In fact, in our previous study17 of the electrochemical reduction of DDT (which is structurally similar to methoxychlor), we observed that such an elimination can occur within the duration of a cyclic voltammetry experiment. Furthermore, electrochemical reduction of activated olefins is well documented in the literature,17,35–37 a process occurring (a) via two-electron reduction that is followed by protonation (via the solvent–electrolyte) or (b) by dimerization. On the basis of that earlier research, together with evidence for the reduction of 7, as revealed in Figure 4, we assign the most negative peak displayed in the cyclic voltammograms for 2 (Figure 1) to reduction of an activated olefin that is generated chemically via hydroxide-promoted elimination of chloride.
Controlled-potential (bulk) electrolyses of methoxychlor (2)
Table I lists coulometric n values and product distributions obtained from a set of controlled-potential (bulk) electrolyses of 5.0 mM solutions of 2 in DMF–0.050 M TMABF4 at reticulated vitreous carbon (RVC) cathodes held at potentials corresponding to values more negative than the cathodic peak potentials seen in Figure 1. For electrolyses of 2 which exhibits four voltammetric peaks, some of which are close together, it is very difficult to employ a large cathode, because it will not truly be an equipotential surface. Therefore, as pointed out by a reviewer, the actual potential applied in many electrolytic reductions can be significantly more positive than the value set by the potentiostat.
Table I. Coulometric data and product distributions for controlled-potential (bulk) electrolyses of methoxychlor (2) at reticulated vitreous carbon cathodes in oxygen-free DMF containing 0.050 M TBABF4.
| Product distribution (%)b | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | E (V) | na | 2 | 3 | 4 | 5 | 6 | 7 | 8 | Total |
| 1 | –1.15 | 1.1 | 26 | 11 | 48 | 15 | 2 | NDc | ND | 102 |
| 2 | –1.55 | 2.3 | ND | 6 | 14 | ND | 70 | 5 | 17 | 112 |
| 3 | –1.75 | 4.7 | ND | TRd | ND | ND | ND | 56 | 47 | 103 |
aAverage number of electrons per molecule of 2. bYield expressed as the percentage of 2 incorporated into each product. cND = not detected. dTR = a trace amount (<1%) was detected.
Each entry in Table I is the average obtained from at least three separate electrolyses. Product yields reported for 4, 5, and 6 were estimated on the basis of an average gas chromatographic response factor determined experimentally from authentic samples of the other products (2, 3, 7, and 8); small errors in these estimated response factors are probably responsible for total product recoveries being greater than 100%.
Both the coulometric n values and the extent of dechlorination (reflected by higher yields of dehalogenated products) rise as a more negative electrolysis potential is applied. When methoxychlor 2 was reduced at –1.15 V, the measured n value was 1.1 and the major product was 4, along with smaller amounts of 3, 5, and 6, and a substantial amount (25%) of unreduced 2 was also found.
At –1.55 V, the measured n value was 2.3 and the major product was 6, with lesser amounts of 3, 4, 7, and 8. At the most negative potential (–1.75 V), the measured n value was 4.7, and 2 was converted entirely to completely dechlorinated products (7 and 8).
Similar trends can be ascertained from corresponding bulk electrolyses of 2 conducted at silver mesh cathodes, the results of which are listed in Table II. For electrolyses carried out at –0.95 V, the measured n value was 1.1 and the major product was 4; moreover, 3 was detected along with smaller amounts of unreduced 2, together with 5, 6, and 7. When the potential was set at –1.50 V, the n value increased to 2.0, and the resulting product mixture consisted of 3, 5, 6, 7, and 8. Finally, at the most negative potential (–1.75 V), a majority of the substrate was completely dechlorinated to give 7 and 8, although small amounts of 3 and 6 were detected. In each case, we accounted for essentially all of 2.
Table II. Coulometric data and product distributions for controlled-potential (bulk) electrolyses of methoxychlor (2) at silver mesh cathodes in oxygen-free DMF containing 0.050 M TBABF4.
| Product distribution (%)b | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | E (V) | na | 2 | 3 | 4 | 5 | 6 | 7 | 8 | Total |
| 1 | –0.95 | 1.1 | 7 | 27 | 42 | 4 | 14 | 4 | NDc | 98 |
| 2 | –1.50 | 2.0 | ND | 28 | ND | 22 | 20 | 17 | 19 | 106 |
| 3 | –1.75 | 6.1 | ND | 4 | ND | ND | 7 | 25 | 63 | 99 |
aAverage number of electrons per molecule of 2. bYield expressed as the percentage of 2 incorporated into each product. cND = not detected.
To confirm the involvement of carbanion intermediates in the reduction of 2, we conducted additional experiments at –1.75 V and at both carbon and silver cathodes in the presence of a 100-fold excess of D2O in the solvent–supporting electrolyte. Mass spectral evidence, corrected for the natural isotopic abundances of 2H and 13C,38 provided strong evidence for incorporation of deuterium; 7 and 8 were mainly dideuterated and trideuterated, respectively, a finding that clearly points to the intermediacy of carbanions. This result is consistent with observations made in our previous investigations of the direct reduction of DDT at carbon and silver cathodes.16,17 In that work, bulk electrolyses involving the use of DMF-d7 as solvent revealed no deuterated products, suggesting the absence of radical intermediates. On the other hand, electrolyses of DDT conducted in the presence of excess D2O showed evidence of deuterium incorporation, and thus the presence of carbanionic intermediates which can abstract a proton (deuteron) from water (D2O) in the solvent–electrolyte system.
Mechanistic aspects of the reduction of methoxychlor (2) and its products
In accord with earlier studies of halogenated environmental pollutants carried out in our laboratory,16,17,22,39 reductive dehalogenation of polychlorinated compounds can involve a combination of electrochemical steps and base-promoted chemical eliminations of halide ions. Scheme 1 depicts a set of proposed steps to account for the formation of all products detected after bulk electrolyses of 2 at both carbon and silver cathodes. To investigate the occurrence of non-electrochemical base-promoted elimination reactions, we set up cells that contained 5.0 mM 2 and 10.0 mM tetramethylammonium hydroxide in DMF–0.050 M TBABF4, with no potential being applied to a silver mesh electrode. After the solution was stirred for 1 h (the approximate duration of an electrolysis carried out at –0.95 V), the solution was partitioned between diethyl ether and brine; then the ether phase was dried over anhydrous sodium sulfate, concentrated by means of rotary evaporation, and subjected to GC–MS analysis. We found that a majority of 2 was converted to 3.
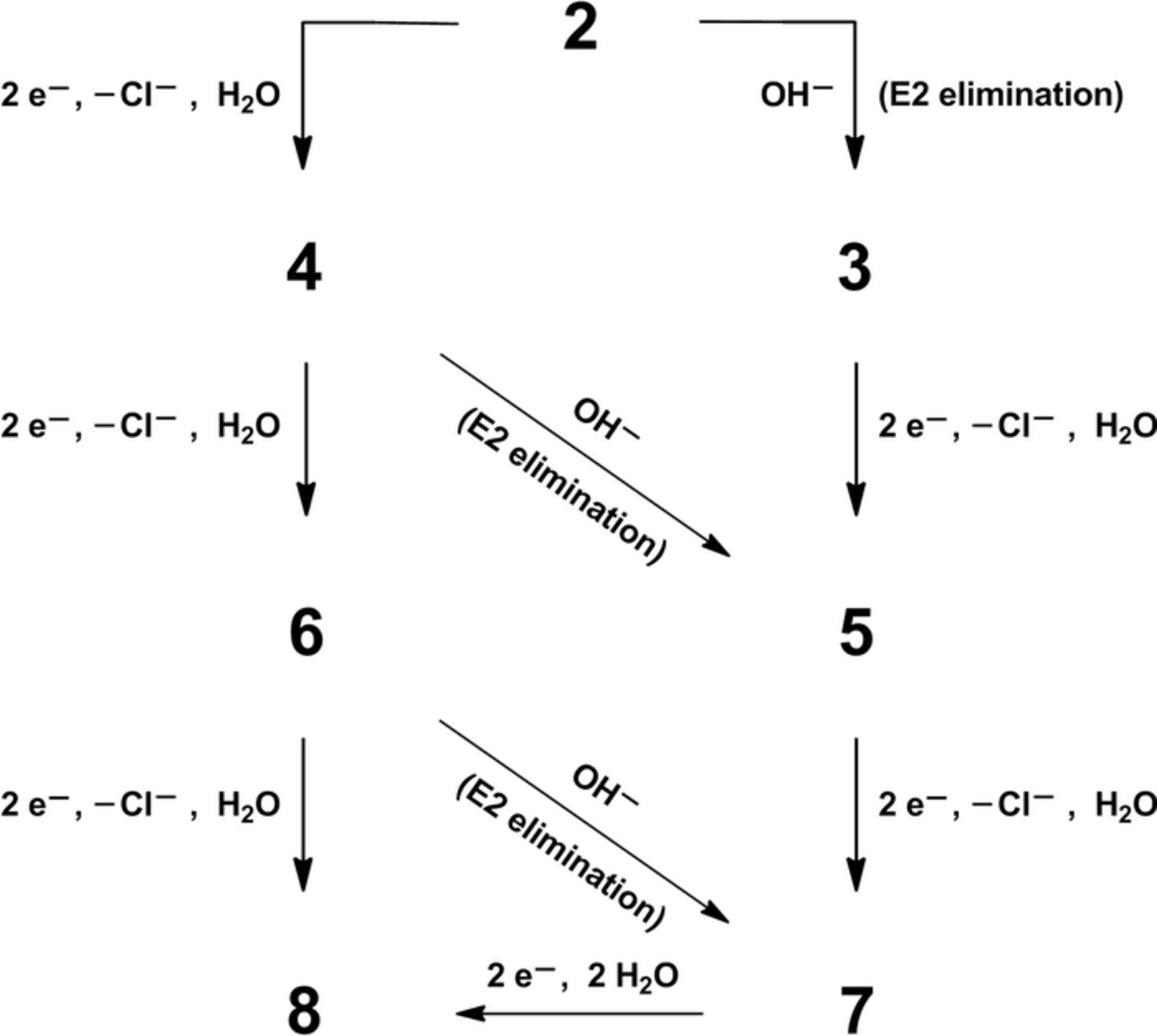
Scheme 1. Proposed mechanistic pathways for electrochemical and base-promoted degradation of methoxychlor (2).
As shown in Scheme 1, we suggest that, at the first stage of reduction (–0.95 V for a silver cathode and –1.15 V for a carbon cathode), 2 undergoes a two-electron reductive cleavage of a carbon–chlorine bond to yield a carbanion intermediate, which is subsequently protonated by residual water in the solvent–electrolyte to afford 4. Parenthetically, it should be noted that water is estimated to be present at a concentration of approximately 40 mM in a newly opened bottle of DMF.40 Therefore, hydroxide ions (arising from deprotonation of adventitious water) may promote elimination of a chloride ion from 2 to give 3; additionally, 4 could undergo an analogous process to afford 5.
During the second stage of reduction (–1.50 V at a silver cathode and –1.55 V at a carbon cathode), the above processes are followed by additional two-electron reduction of 4 to yield 6. Once formed, 6 is subject to a hydroxide-promoted elimination reaction to afford 7. Any 3, previously formed via base-promoted elimination, can be reduced in a two-electron process to give more 5.
Finally, a potential of –1.75 V is sufficient for both carbon and silver electrodes to convert 6 to 8, as well as any 5 to 7, each process involving a two-electron reduction. As a final reaction in Scheme 1, we propose that 7 can undergo a two-electron, two-proton reduction to yield 8; however, because of the onset of solvent–electrolyte breakdown, a bulk electrolysis of 7 was not feasible (Figure 4).
In our earlier investigation17 of the electrochemical reduction of DDT at silver cathodes, we observed that measured coulometric n values did not match well the theoretical n values calculated on the basis of the tabulated product distributions and on the proposition that reductive cleavage of each carbon–chlorine bond is strictly a net two-electron process. In fact, we found that some measured n values were larger than theoretical n values, which suggests the possibility of DISP1 processes;33 in other cases, some measured n values were smaller than theoretical ones, which may be indicative of one-electron reduction processes (in which a radical intermediate abstracts a hydrogen atom from the solvent). For the present study, the same phenomenon (n values being higher or lower than theoretically expected) has been encountered with respect to Tables I and II. Accordingly, the effect of scan rate on cathodic peak potentials for each of the first three stages of reduction of 2 at both carbon and silver electrodes was examined, and ∂Ep/∂log(v) significantly exceeded –29.6 mV per log unit, which is expected of dissociative electron-transfer processes. Thus, we conclude that DISP1 reactions play no important role in the reduction of 2 at either carbon or silver. Furthermore, we believe that incorporation of deuterium into products resulting from bulk electrolyses done in the presence of D2O indicates that two-electron carbon–chlorine bond cleavage is the dominant mechanistic pathway.
A reviewer of this work suggested that formation of various products could take place via self-protonation pathways (without direct involvement of adventitious water). For example, if 2 is electrolyzed at a reticulated vitreous carbon cathode held at –1.15 V, the major product is 4 (Table I), which could arise if its carbanion precursor accepts a proton from 2 (instead of from water present in the system). It is instructive to try to compare the acidities of water and 2. Using data tabulated by Bordwell,41 that provides pKa values for water and diphenylmethane in dimethyl sulfoxide, and work reported by Maran and co-workers42 that allows one to calculate corresponding pKa values in dimethylformamide, we arrived at pKaDMF = 31.5 and 32.5, respectively, for water and diphenylmethane. Moreover, the presence of three chlorine atoms on a molecule of 2 must assuredly render it significantly more acidic than diphenylmethane itself. Thus, 2 (as long as it remains incompletely electrolyzed) could be a significant source of protons. In analogous fashion, 4 and 6 might be viable proton donors for carbanion intermediates during later stages of the reduction of 2. Moreover, if 2, 4, and 6 do act as proton donors during intermediate stages of an electrolysis, their conjugate bases will ultimately be reprotonated by residual water in the solvent–electrolyte (as well as by water used during the workup procedure prior to final mass spectrometric measurements) to afford the observed final products (4, 6, and 8).
Conclusions
In this work, we have investigated the electrochemical reduction of methoxychlor (2) at carbon and silver cathodes in DMF containing 0.050 M TBABF4. This study was aimed at electrochemical dechlorination of 2 to afford less environmentally harmful species. Cyclic voltammograms displayed four cathodic peaks for the irreversible reduction of 2 at both glassy carbon and silver cathodes in the DMF–0.050 M TBABF4 medium, the first three peaks being due to reductive cleavage of carbon–chlorine bonds. Products arising from controlled-potential (bulk) electrolyses of 2 at reticulated vitreous carbon and silver gauze cathodes in DMF–0.050 M TBABF4 include a mixture of monochloro and dichloro derivatives of 2, along with their corresponding olefins. Bulk electrolyses conducted at a potential between the third and fourth cathodic peaks result in two completely dechlorinated products (7 and 8). These last two compounds are far less biologically and ecologically harmful than 2 and its less chlorinated derivatives. In consideration of the long lifetimes of the halogenated species (due to limited natural degradation pathways), electrochemical reduction should be further explored as a means of remediation.
Acknowledgments
We thank Dr. Jonathan A. Karty and Angela M. Hansen of the Indiana University Mass Spectrometry Facility, where all of the mass-spectral data were acquired. In addition, Dr. Karty assisted with the interpretation of results obtained from the deuterium-labeling experiments.