Abstract
An electrochemical sensor based on poly(3,4-ethylenedioxythiophene) functionalized reduced graphene oxide with palladium nanoparticles (Pd/PEDOT/rGO) has been constructed for electrochemical determination of dopamine (DA) in presence of ascorbic acid (AA) at high concentration. The structural features of the catalyst were characterized using several instrumental methods. The transmission electron microscopy (TEM) analysis suggests a well dispersed spherical PdNPs set successfully onto PEDOT/rGO film. Electrochemical determination of DA in presence of AA was investigated on modified glassy carbon electrode in 0.1 M phosphate buffer solution at pH 7.4 using cyclic voltammetry, differential pulse voltammetry (DPV) and amperometry techniques. A wider linear analytical range for DA detection (1–200 μM) and comparatively lower limit of detection (LOD) 0.14 μM were obtained using DPV technique, while LOD increases up to 0.16 μM in the presence of five times higher AA. Also, no significant interference was observed from similar biomolecule species such as norepinephrine, epinephrine, uric acid and AA. This DA-sensor retained 85.6% of its initial response up to 15 days.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
Graphene is a single atom thick and 2D network sheet of sp2 hybridized carbon has gained enormous importance over the past few years due to their unique properties. It has exceptional properties such as, high surface area, tunable bandgap and excellent electron conductivity.1,2 Among all the available support materials, reduced graphene oxide (rGO) consisting of chemically converted monolayer carbon atoms has emerged as one of the most promising supports for catalysts because the unique advantage of hydrophilicity and large specific surface area of its precursor, graphene oxide (GO), makes it possible to anchor well-dispersed metal nanoparticles (NPs) in the solution-based reduction.3 The addition of rGO into nanoelectrocatalysts cannot only maximize the specific surface area for electron transfer but also enhance mass transport of reactants to the electrocatalyst. However, it is still a major obstacle to obtain monodispersed metal NPs with very small sizes on unfunctionalized rGO.4 Therefore, it is important to functionalize the rGO with other guest molecules such as polymers.
GO contains numerous functional groups like hydroxyl and epoxy on the basal plane and carboxylic acid groups at the edges. Such a chemical structure for GO provides excellent opportunities for the functionalization with many different conductive materials.5,6 Chemical reduction of GO (rGO) is an efficient approach to large scale production which includes noncovalent and covalent functionalization of rGO.7 Many applications of graphene have been reported including catalysis.8–15 Palladium (Pd) is a good alternative of Pt and Au that has good catalytic activity. Its catalytic activity has been widely recognized in electroanalysis.13,16–19 Moreover, Pd is preferable to Pt or Au due to its lower price, better catalysis, tolerance to the CO poisoning.1,18 Poly(3,4-ethylenedioxythiophene) (PEDOT), one of the conducting polymers, has been extensively studied due to its easy synthesis, high electrochemical stability, high charge mobility, low energy bandgap and high electrical conductivity.19–21 PEDOT is also an excellent electrode modifying material which can promote electron transfer reactions and is useful for fabrication of electrochemical sensors.21 The comparatively smaller size of NPs have high surface-area-to volume ratio, a large number of edges and corner atoms which are significantly important for superior catalysis. Moreover, the smaller size of NPs leads to the better activity of electrocatalysis.1,14
Dopamine (DA) is one of the neurotransmitters which transmit messages from on neuron to the next in the mammalian central nervous system. DA contains a catecholamine group and plays an important role in the renal, hormonal, cardiovascular and central nervous systems. Its deficiency causes brain disorders such as schizophrenia and Parkinson's disease.22,23 Usually, DA and ascorbic acid (AA) are coexisting in our body fluid. Therefore, selective determination of DA is very important for analytical application and diagnostic research. Electrochemical methods have been used for detection of small biomolecules because of convenience, rapid response and high sensitivity. However, the oxidation potentials of DA and AA are very close, so their characteristic peaks in a voltammogram usually overlap resulting in poor sensitivity and selectivity.24 To resolve this problem, several materials, such as, polymer,25–29 graphene,29–32 carbon nanotube (CNT),33,34 metal complexes35–37 and NPs38–40 have been used.
In this work, the PdNPs decorated PEDOT functionalized rGO (Pd/PEDOT/rGO) has been synthesized via a chemical process, which has the following merits: (a) the Pd/PEDOT/rGO is easily synthesized by a wet-chemical process; (b) the rGO can be easily functionalized with PEDOT into rGO via noncovalent approach; and (c) the PdNPs in Pd/PEDOT/rGO can be electrochemically utilized in maximum due to well dispersion with smaller in size. Pd/PEDOT/rGO's electrocatalytic activity toward DA and AA was studied using cyclic voltammetry (CV), differential pulse voltammetry (DPV) and chronoamperometry (CA) techniques in 0.1 M phosphate buffer solution (PBS) at pH 7.4. Interference experiments were also carried out using common interfering substances such as norepinephrine (NP), epinephrine (EP), uric acid (UA) and AA.
Experimental
Characterization
The field emission transmission electron microscopy (FE-TEM) and energy-dispersive X-ray spectroscopy (EDX) observations were carried out in a JEM-2100F microscope at 200 kV. XPS measurements were performed on a MultiLab 2000 (Thermo Electron Corporation, England) with a 14.9 keV Al Kα X-ray source. The pH was measured by a pH glass electrode with a JENCO meter. A three-electrode potentiostat (CHI 700C Electrochemical Workstation, USA) in a grounded Faraday cage was used for voltammetric measurements. A Pt wire was used as an auxiliary electrode. A calibrated Ag/AgCl electrode from Bioanalytical Systems Inc. (BAS) in 3 M NaCl solution was used as a reference electrode.
Materials and chemicals
The graphite powder (325-mesh), EDOT, NaBH4 and K2PdCl4, NP, EP, UA, AA and DA were purchased from Aldrich, South Korea. The ethanol and acetonitrile (MeCN) were purchased from OCI Co., Ltd. All other reagents were of analytical grade and were used without further purification. A PBS was prepared with 0.1 M NaH2PO4 and its pH was adjusted with 0.1 M NaOH and 0.1 M H3PO4. Double-distilled water (DW) was used in the preparation of the aqueous electrolyte solutions.
Preparation of catalyst and electrode
GO was obtained by oxidizing graphite according to the improved Hummer's method.41 20 mg GO powder was dispersed in 30 mL MeCN under ultrasonic agitation after which 20 μL EDOT was added to the mix. The mixture was then stirred for 15 h, centrifuged and then washed several times with DW and ethanol. Finally, the resulting product (EDOT/GO) was dried at 50 °C in a vacuum oven for 24 h.
The GCE surface was coated with a 10 μL EDOT/GO suspension (1 mg/mL DW). After drying at room temperature (RT), EDOT/GO/GCE was electrochemically polymerized and reduced by ten successive cycles of CVs in an electrochemical cell containing 1 M KCl solution over a potential range of ±1.5 V at a 100 mV s−1 scan rate. The PEDOT/rGO/GCE was washed with DW before and after each experiment.
To make Pd/PEDOT/rGO, 5 mg PEDOT/rGO powder (collected from GC plate by smooth scratching) was dispersed in 10 mL ethanol-water (1:1, v/v ratio) solution by ultrasonication, followed by the addition of a metal precursor (10 mM K2PdCl4). Subsequently, 0.1% NaBH4 (2 mL) solution was added in a drop wise manner with stirring. The resulting mixture was stirred for 3 h at RT after which the mixture was filtered and then washed several times with DW. Finally, the resulting product was dried at 70 °C in a vacuum oven for 12 h. For comparison the Pd/rGO was prepared in the same way without EDOT and rGO and PEDOT/rGO was electrochemically prepared as mentioned above.
For the electrode preparation, Pd/rGO, PEDOT/rGO or Pd/PEDOT/rGO suspensions in DW (1 mg/mL) were sonicated in a water bath for 1 h. A 10 μL portion of each suspension was used to coat the surface of a glassy carbon electrode (GCE, 0.5 cm in diameter) that was prepolished with a 0.05 μm alumina suspension on a polishing cloth (BAS, USA). The resulting catalyst-coated GCE was washed with DW before and after each experiment. All the experiments were performed at RT in 0.1 M PBS solution, which was purged in an argon atmosphere for 5 min prior to each measurement.
Results and Discussion
Instrumental characterization
The surface morphology of rGO and PEDOT/rGO were characterized by SEM. Figure 1a shows the SEM image of rGO before treatment with EDOT. The 2D rGO exhibits a typically smooth sheet-like structure. The PEDOT/rGO has a coated surface after electrochemical polymerization compared to rGO. The PEDOT/rGO also showed a more crumpled surface compared to rGO which suggests attachment of the PEDOT layer onto rGO (Figure 1b).
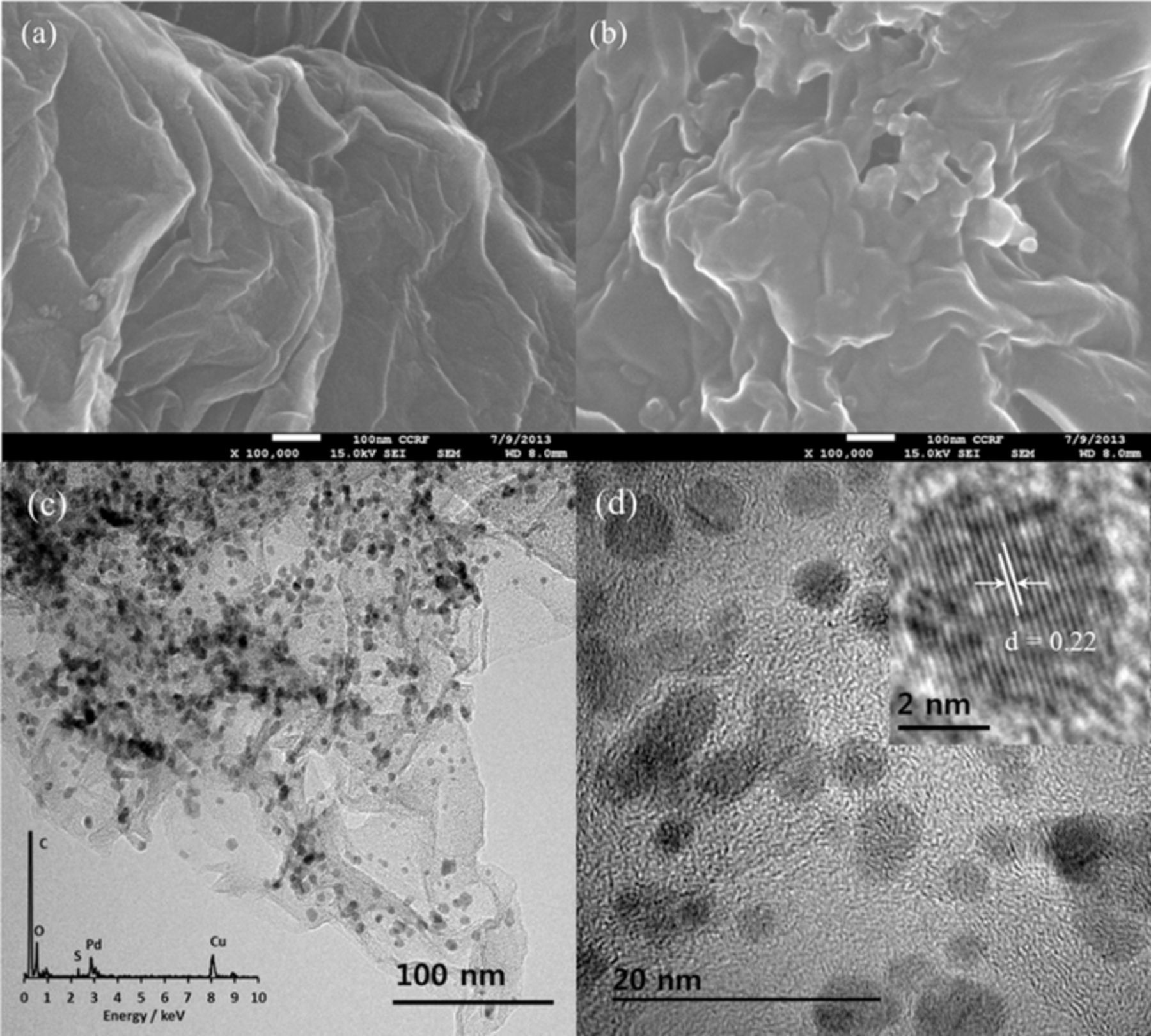
Figure 1. The SEM images of rGO (a) and PEDOT/rGO (b); TEM images (c and d) and corresponding EDX spectrum and HRTEM image of Pd/PEDOT/rGO in insets.
The TEM image of Pd/PEDOT/rGO has shown in Figures 1c and 1d. The TEM image of Pd/PEDOT/rGO shows black spherical spots representing PdNPs on the rGO demonstrating that PdNPs were set successfully onto the PEDOT functionalized rGO. The particle size range was estimated to be 2∼5 nm. The EDX spectrum of Pd/PEDOT/rGO shows signals from C, O, S, Pd and Cu (Figure 1c inset). The peaks of elemental Cu were attributed to the TEM grid. The HRTEM image in Figure 1d inset shows the Pd lattice lines in a selected PdNP and the d-spacing between two lines was calculated as 0.22 nm which is similar to the other reports.1,42
The XPS spectrum of as prepared Pd/PEDOT/rGO has shown in Figure 2. The XPS spectrum shows distinct C1s, O1s, S2p, Pd3d and Pd3p peaks at around 286, 534, 163.1, 340.9, 335.9 and 560 eV, respectively (Figure 2a).43–45 The high-resolution C1s XPS spectrum (Figure 2b) shows the three peaks at binding energy of 285, 286.4 and 288.8 eV which are attributed to the C=C, C=O and O=C=O bonds, respectively, of Pd/PEDOT/rGO. The predominant intensity of C=C indicates the reduction of the oxygen-containing functional groups in rGO. According to the previous report, the C=O peak at 286.4 eV has the influence of the C=S bond.14,15,45,46 The core level Pd3d spectrum of Pd/PEDOT/rGO shows doublet signals (Figure 2c) at binding energies of 335.9 (Pd3d5/2) and 340.9 eV (Pd3d3/2) which are attributed to the Pd0 species. Other tiny two peaks at 337.7 (Pd3d5/2) and 342.6 eV (Pd3d3/2) can be assigned to Pd2+ ion.47 It indicates that the maximum ionic-Pd had reduced to the metallic form (Pd0) by NaBH4. The Pd and PEDOT were calculated as 17.3 and 18.6 wt% (derived from S, 4.2 wt%), respectively, for Pd/PEDOT/rGO.

Figure 2. The XPS survey spectrum (a), the core level of C1s spectrum (b) and the core level of Pd3d spectrum (c) of Pd/PEDOT/rGO.
Electrochemical application of catalysts
The voltammetric behavior of DA and AA at various modified electrodes was investigated via CV. Figure 3 shows the CVs at the Pd/rGO, PEDOT/rGO and Pd/PEDOT/rGO modified GCE in 0.1 M PBS at pH 7.4 without and with 300 μM AA and/or DA at a scan rate of 100 mV s−1. The CVs on Pd/rGO in Figure 3a, AA shows broader oxidation peaks with the peak potentials at 206 mV. For DA, the redox peaks appear at 237 and 3 mV and the separation of peaks potential is about 234 mV. It is clear that the electrochemical reactions of these species at the Pd/rGO modified electrode are irreversible. Figure 3b shows the CVs of DA and AA at the PEDOT/rGO modified GCE. The oxidation peak potentials of DA (138 mV) and AA (118 mV) were negatively shifted compared to those at the Pd/rGO. In case of AA, because AA at pH 7.4 is negatively charged (pKa = 4.1), the peak potential shift may be due to the formation of hydrogen bonds between the ascorbate and positively charged backbone of the polymer in PEDOT/rGO. For DA, the redox peaks were observed at 138 and 65 mV with a peak to peak separation of 73 mV indicating that the redox reaction of DA is better reversible.
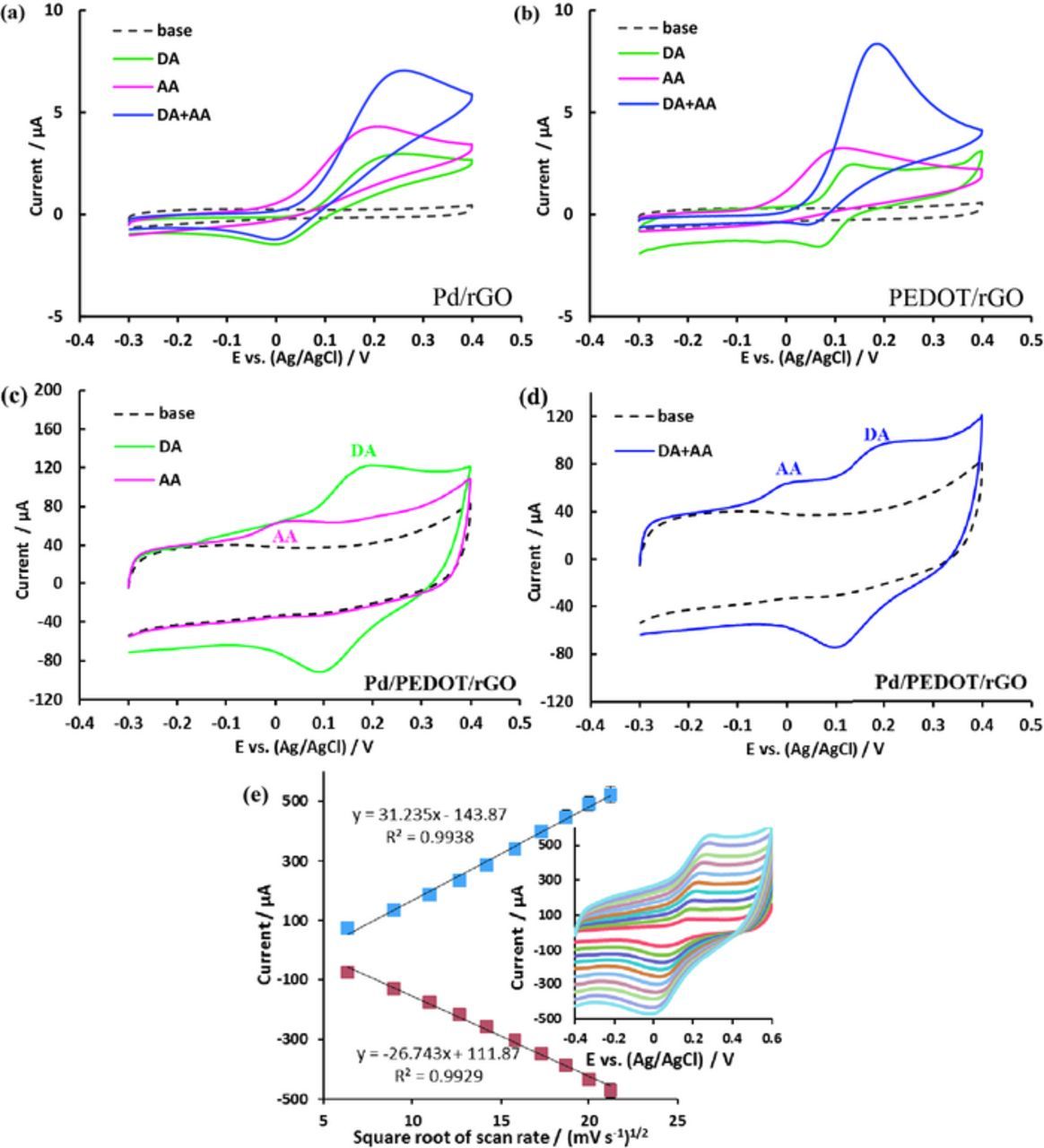
Figure 3. CVs at Pd/rGO (a) and PEDOT/rGO (b) Pd/PEDOT/rGO (c and d) in 0.1 M PBS at pH 7.4 without (dotted line) and with (colored lines) 300 μM DA, 300 μM AA and their mixture at 100 mV s−1 scan rate; the plot of peak current vs. square root of scan rate (e) and scan rate dependent CVs in 0.1 M PBS with 300 μM DA in inset.
It is, however, the CVs of DA and AA at the Pd/PEDOT/rGO modified electrode have shown in Figure 3. For AA in Figure 3c, the peak potential for the oxidation has shifted negatively to around 19 mV and the oxidation peak current (12.5 μA) is much higher compared to Pd/rGO (3.2 μA) or PEDOT/rGO (2.8 μA). For DA, the redox peaks have observed at 190 and 100 mV. The separation between the redox peaks is about 90 mV and the oxidation peak current (38 μA) is much higher than that obtained at Pd/rGO (3.05 μA) and PEDOT/rGO (1.96 μA), suggesting the significant electrocatalytic activities of Pd/PEDOT/rGO. The large difference in the anodic peak potentials between DA and AA is suggesting a selective determination of DA in the presence of AA while the oxidation peaks of DA and AA were overlapped at Pd/rGO and PEDOT/rGO modified electrodes (Figures 3a and 3b). This peak separation also has seen in the previous report.39 Thus, at Pd/PEDOT/rGO, two well defined oxidation peaks are observed with larger peak separation and higher peak current in the mixture of AA and DA, respectively. Therefore, the Pd/PEDOT/rGO electrode exhibited high electrocatalytic activity toward DA oxidation and leading to improve the reversibility; while, the irreversibility of DA's electrochemistry at conventional electrodes is a great challenge for DA detection.26 However, the incorporation of PdNPs into the PEDOT/rGO film has effective to improve the electrocatalytic activity toward selective determination of DA in presence of AA. The enhanced electrocatalytic determination of DA at the Pd/PEDOT/rGO compared to the other electrodes probably due to intermolecular charge-transfer in between graphene sheets and PdNPs via a conductive and low bandgap polymer, PEDOT12,48 synergic effect of and well attachment and dispersion of PdNPs.
The kinetics of electrode reaction was investigated by evaluating the effect of scan rate on the redox peak current in Figure 3e. The scan rate dependent CVs exhibit a profound effect on the redox peak current of 300 μM DA in 0.1 M PBS at Pd/PEDOT/rGO. For the scan rates in the range of 40–450 mV s−1, relationship is established between the redox peak current and the square root of scan rate (Figure 3e), indicating the surface controlled mechanism is significant at the low scan rate. The linear relationship is founded between the redox peak current and the square root of scan rate, suggesting the diffusion controlled behavior is predominated at the high scan rate.49 Such a variation on the reaction mechanism from the surface controlled to diffusion controlled at high sweeping rates demonstrates that the SPGNE possesses a faster electron transfer kinetics that could follow higher scan rates.
Figure 4a shows the DPV recorded at various concentrations of DA at Pd/PEDOT/rGO at 50 mV pulse amplitude. The concentration range of DA was 1–200 μM. The results shows that the oxidation peak currents increased with increasing concentration of DA, following the linear regression equation ip (μA) = 0.145 × [CDA] (μM) + 2.012. The plots showed good linearity, with a 0.997 correlation coefficient (Figure 4a inset). Figure 4b shows the DPV recorded at different concentrations of DA (1–200 μM) in presence of 1 mM AA at the Pd/PEDOT/rGO modified electrode. These concentration ranges are nicely fitted to the normal blood concentration range.50,51 The peak current is increasing with the increasing of DA concentration but the peak current for AA is almost constant, indicating that the addition of DA does not affect to the determination of AA. As we observed in CVs experiment, however, the both peaks are not overlapping each other. The peak current of DA is linear to the DA concentrations in the range of 1–200 μM, following the linear regression equation ip (μA) = 0.1274 × [CDA] (μM) + 1.4387. The plots showed good linearity, with a correlation coefficient of 0.998 (Figure 4b inset).
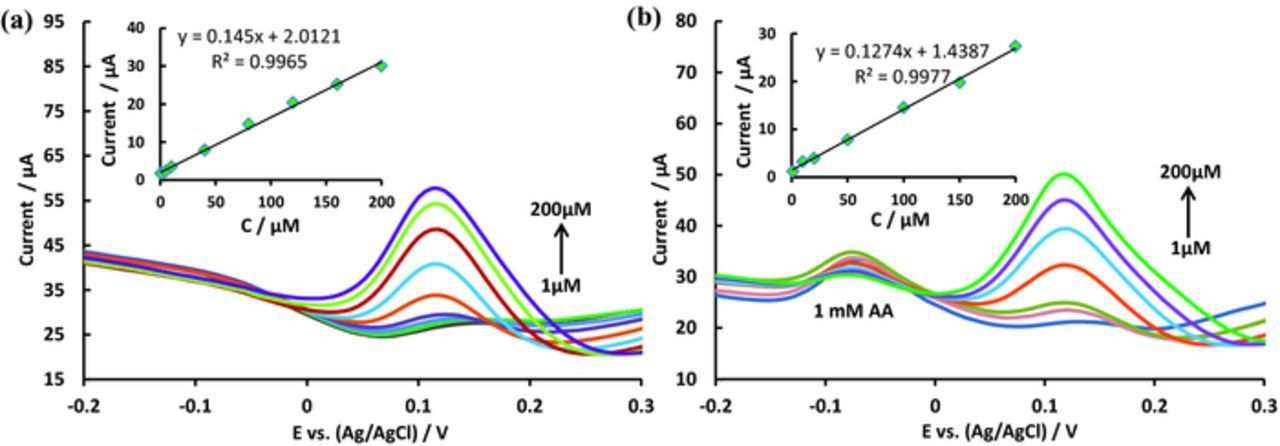
Figure 4. DPVs of Pd/PEDOT/rGO at various concentrations (1.0–200 μM) of DA (a) and 1.0–200 μM of DA in 1 mM AA (b) in a 0.1 M PBS solution (pH 7.4) at 50 mV pulse amplitude. Insert: Plots of oxidation peak currents vs. concentration.
The amperometric method was used to examine the sensitivity of Pd/PEDOT/rGO modified electrode toward the detection of DA at RT. Based on the above experimental results, we selected a potential of 0.25 V for DA detection. Figure 5a shows the amperometric response for DA at the applied potential of 0.25 V. The amperometric current response increased linearly from 2 to 200 μM with a correlation coefficient of 0.993. The linear regression equation is ipc (μA) = 0.0213 × [CDA] (μM) + 0.077. The detection limit was 0.14 μM (S/N = 3) at a signal-to-noise ratio (S/N) of 3. Figure 5b displays the amperometric response for DA in the presence of 100 μM AA at the applied potential of 0.25 V. The amperometric current response increased linearly from 2 to 200 μM concentration of DA. The linear regression equation is ipc (μA) = 0.0193 × [CDA] (μM) - 0.0011, with a correlation coefficient of 0.998. The detection limit was found to be 0.16 μM (S/N = 3). The analytical parameters which include the detection limit and linear range using Pd/PEDOT/rGO modified GCE are better or comparable to the other previous results at different modified electrode for DA sensing, as displayed in Table I. The comparison with the listed data suggests that the proposed sensor has improved significantly in terms of detection limit and linear range. The superior catalytic activity afforded by the attachment of smaller size of PdNPs onto the PEDOT functionalized rGO.
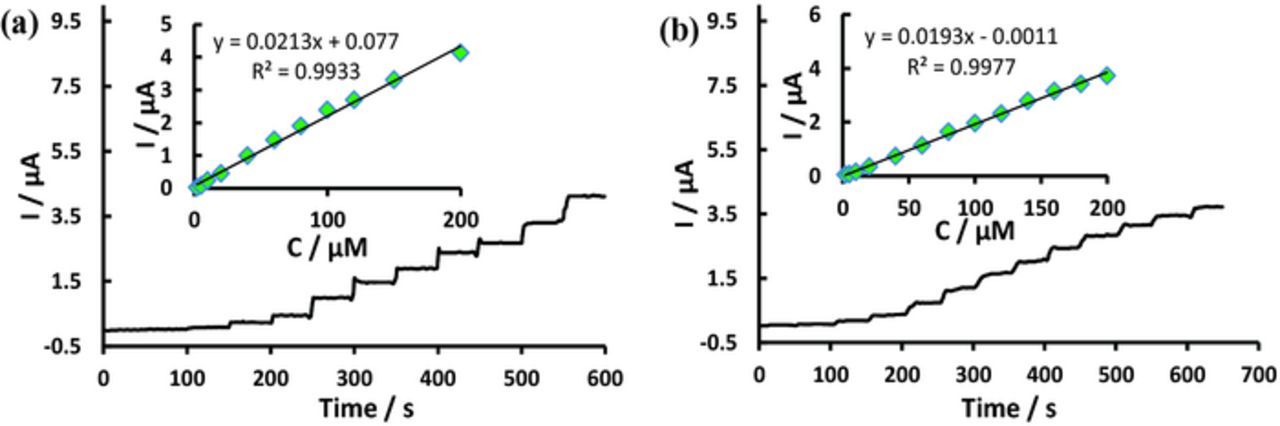
Figure 5. CA response of Pd/PEDOT/rGO at an applied potential of 0.25 V at various concentrations 2–200 μM DA (a) and 2–200 μM DA in presence of 100 μM AA (b).
Table I. Comparison of different-modified electrodes for the detection of DA.
| Electrode materials | Method | Detection limit (μM) | Linear range (μM) | Reference |
|---|---|---|---|---|
| Pd/PEDOT/rGO | DPV | 0.14 | 1–200 | This work |
| PEDOT/PNMPy/PEDOT/AuNP | CV | 2–3 | 1–100 | 26 |
| poly-ACBK | DPV | 0.5 | 1–200 | 27 |
| PEDOT-modified Ni/Si | DPV | 1.5 | 12–48 | 28 |
| PANI-GO | DPV | 0.5 | 2–18 | 29 |
| ERGO-DA | DPV | 0.04 | 0.5–100 | 30 |
| GO | DPV | 0.27 | 1.0–15 | 31 |
| ERGO | DPV | 0.5 | 0.5–60 | 32 |
| CuLB | DPV | 1.4 | 2–120 | 37 |
| Pd/CNF | DPV | 0.2 | 0.5–160 | 39 |
| Chitosan-graphene | DPV | 1 | 1–24 | 52 |
| RGO-AuNPs-CSHMs | DPV | 0.3 | 1.0–200 | 53 |
| Fe3O4@AuNPs-GO | DPV | 0.1 | 0.5–50 | 54 |
| RGO-Pd-NPs | LSV | 0.233 | 1.0–150 | 55 |
| CoTPP-CRGO | DPV | 0.03 | 0.1–12 | 56 |
| GO/SnO2 | DPV | 0.08 | 0.1–10 | 57 |
The sensor's ability to function in the presence of interferences was investigated at the Pd/PEDOT/rGO modified electrode and the results are shown in Figure 6. The interfering effects of common and similar interfering electroactive substances were assessed. Figure 6 shows the amperometric response for the sensor with injections of 30 μM of DA, NP, EP, UA, and AA at an applied potential of 0.25 V. As can be seen, the current responses of NP, EP, UA, and AA were insignificant compared to DA. The current responses for these interfering substances were approximately below 5% compared to the current response of 30 μM DA. The proposed sensor demonstrated good selectivity and sensitivity toward DA.
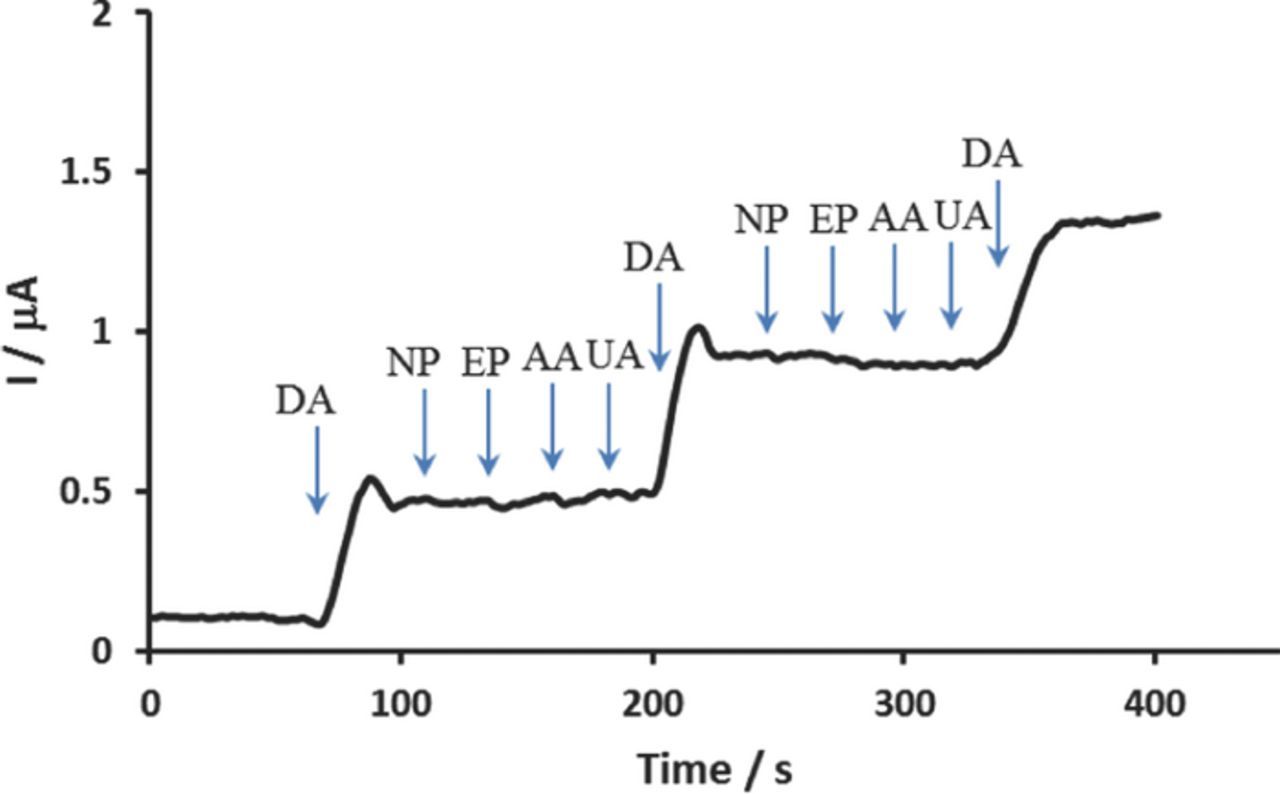
Figure 6. CA response of Pd/PEDOT/rGO with the sequential addition of 30 μM of DA, NP, EP, AA, and UA at an applied potential of 0.25 V.
The stability and reproducibility of the sensor were also investigated by measuring its f-peak current response in 0.1 M PBS at pH 7.4 containing 300 μM DA. The long term stability of the sensor was evaluated by CV experiment after storage at RT for 15 days. The sensor retained 85.6% of its initial current response. In addition, the relative standard deviation was 3.9% for five successive assays using five different electrodes. These results indicated that the proposed sensor possessed good stability and acceptable reproducibility.
Conclusions
In this paper, we have synthesized Pd/PEDOT/rGO by adding PdNPs on PEDOT functionalized rGO and applied as an electrochemical DA-sensor. The clear oxidation peak separation was appeared for DA and AA at different potentials on the Pd/PEDOT/rGO modified GCE. The Pd/PEDOT/rGO exhibited high electrooxidation of DA and AA by significantly decreasing their oxidation overpotentials and enhancing the peak currents compared to Pd/rGO and PEDOT/rGO. This electrochemical DA-sensor showed excellent selectivity and high sensitivity for DA detection without interference of excess amount of AA. The linear concentration range and detection limit were determined as 1–200 μM and 0.14 μM for DA, respectively, using DPV. Also, the presence of NP, EP, UA, and AA showed negligible effects on the determination of DA. This sensor retained 85.6% of its initial response up to 15 days.
Acknowledgment
This research has supported by the Science Research Program through a National Research Foundation of Korea (NRF), grant funded by the Ministry of Education, Science and Technology (2010-0007864).