

Abstract
Polymer-electrolyte fuel cells (PEFCs) utilize porous catalyst layers (CLs) formed of carbon supports on which Pt particles are deposited and ionomer films are distributed. Carbon supports themselves have varying degrees of porosity, where high-surface-area carbon (HSC) supports possess nanometer-sized interior pores that are suitable for Pt nanoparticle deposition but prevent deleterious ionomer penetration. However, this requires protons to transport through water pathways inside the pores. To understand the generation of such pathways, we examine the various mechanisms of water uptake by PEFC CLs, and the subsequent impact of water uptake on Pt utilization through developing a multiphysics model of the water wetting phenomena as a function of relative humidity. The model details water uptake via ionomer absorption, capillary condensation in the hydrophilic pores, and surface adsorption using molecular potential that account for various water and surface dipole interactions. The results quantify how mesoporous carbons with highly hydrophilic pores increase Pt utilization through the development of wetted layers, which at the same time enable optimized gas-transport pathways. It also demonstrates the impact of pore-size distribution (PSD) and physical and chemical parameters on the water uptake phenomena, allowing for future CL particle and structure optimization.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
List of Symbols

| Pore radius |

| Total volume from a given pore-type |
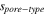
| Adjustable parameter relating to the width of the PSD for a given pore-type |

| Adjustable parameter relating to the peak position of the PSD for a given pore-type |
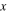
| Distance from wall |

| Local Lennard-Jones (LJ) interaction potential |
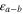
|
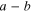 LJ interaction potential well-depth LJ interaction potential well-depth |

| Distance of closes approach between 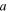 and and 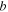
|
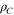
| Carbon number density in pore walls |
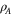
| Surface density of surface oxides on pore walls |
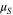
| Dipole moment of surface oxide group |
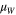
| Dipole moment of water |
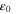
| Dielectric constant of gas phase |
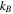
| Boltzmann constant |

| Temperature in Kelvin |
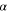
| Angle formed by 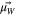 with the plane z = 0 (where carbon wall is represented by x = 0 plane, see SI for details) with the plane z = 0 (where carbon wall is represented by x = 0 plane, see SI for details) |
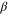
| Angle formed between the x-axis and the projection of 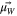 on the z = 0 plane on the z = 0 plane |
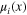
| Local chemical potential of component 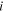
|
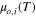
| Standard state chemical potential of component 
|
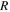
| Gas constant |
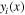
| Local mole fraction of component 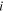
|
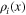
| Local molar density of component 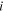
|
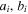
| Characteristic constants of component 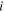 in Peng-Robinson EOS in Peng-Robinson EOS |
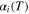
| Function to modulate temperature dependence of molecular attractive forces of component i |
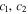
| Constant in 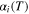 function function |
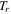
| Reduced temperature |
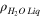
| Molar density of liquid water at temperature considered for model calculations |
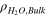
| Molar density of water in bulk gas-phase |
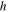
| Thickness of adsorbed water film |
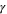
| Surface tension of water at temperature considered for model calculations |
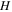
| Mean curvature of the condensed meniscus in pores |
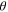
| Water contact angle on carbon walls |
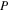
| Total operating pressure |
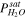
| Saturation water vapor pressure at temperature considered for model calculations |
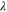
| Moles of water absorbed per mole of sulfonic acid groups |
High current-density performance of polymer-electrolyte fuel-cells (PEFCs) is limited by significant gas-transport resistances, particularly at the oxygen electrode. Transport losses increase with a reduction in Pt content, necessitating high Pt loadings to achieve desired performance, thus increasing costs. 1–4 Consequently, recent research has focused on optimizing CL structure and material properties to allow facile species transport to/from reaction sites. 5–10
The large transport resistance observed in PEFC catalyst layers (CLs) is due to the presence of a local gas-transport resistance close to the Pt particles. 1,3,11,12 In our prior studies, we demonstrated that the local transport resistance consists of (i) diffusion resistance through the ionomer thin film, and (ii) an interfacial resistance at the Pt/ionomer interface due to interactions between the ionomer acid moieties and the Pt surface. 13–19 These findings also agree with prior literature suggesting low permeability of ionomer thin films compared to bulk ionomer due to confinement effects, 19–24 and specific interactions between the acid groups and the Pt surface. 25–28 Consequently, novel ionomer chemistries are being examined such as ionomers with multiple-acid side chains, high oxygen permeability, 5,10,29 and lower equivalent weight (EW), where EW is defined as grams of polymer per mol of acid group. 5,30 However, while lower EW or a higher side-chain density provide greater water uptake, thereby resulting in enhanced gas permeability and H+ conductivity, they also increase the deleterious interactions of ionomer with the Pt. To ameliorate this effect, different catalyst supports are being investigated. 31–35
The catalyst support upon which the Pt nanoparticles are dispersed and on which ionomer thin films are distributed is the other key component of CLs. Carbon blacks are often used as the catalyst support. Common carbon-black types are: (i) solid non-porous compact carbon, such as Vulcan, and (ii) porous high-surface area carbon (HSC) supports such as Ketjen Black (KB) with interior micropores.
33
Vulcan carbon results in almost all of the Pt particles deposited on the exterior surface which are then in direct contact with ionomer.
31,34,36
Conversely, HSC supports house a significant fraction of Pt particles in the interior micropores that do not allow ionomer penetration due to size restrictions.
31,34,36
The Pt particles are hence not in direct contact with ionomer, thereby reducing the interfacial resistance and interaction of the ionomer acid moieties with the Pt surface.
8,35
Deposition of Pt particles in the micropores also limits their dissolution and sintering during operation.
32,37
Pt-supported HSC demonstrates higher mass activity because of better Pt dispersion and reduced poisoning. However, the transport resistance worsens due to longer and more resistive reactant pathways,
8,35
where the reactant gas and ions must diffuse/transport through small tortuous micropores to reach the Pt reaction sites. Furthermore, absence of ionomer in HSC interior pores necessitates  transport through adsorbed and/or condensed water pathways to the Pt sites (within the electrical double layer).
38
As a result, CLs with HSC supports exhibit a relative-humidity (RH)-dependent Pt utilization, defined here as the electrochemically active surface area (ECSA) measured at a given RH normalized to ECSA at 100% RH.
31,36,39
In the absence of continuous water pathways in HSC micropores, Pt particles cannot access H+ ions and become electrochemically inactive, subsequently resulting in low Pt utilization. Several recent studies demonstrate improved performance with novel carbon supports that have comparatively wider micropores, short micropore depth, and/or low micropore tortuosity (relative to HSC).
8,9
A carbon support with such tailored morphology is believed to allow facile reactant transport to the Pt sites, while also preventing ionomer from directly contacting the Pt.
transport through adsorbed and/or condensed water pathways to the Pt sites (within the electrical double layer).
38
As a result, CLs with HSC supports exhibit a relative-humidity (RH)-dependent Pt utilization, defined here as the electrochemically active surface area (ECSA) measured at a given RH normalized to ECSA at 100% RH.
31,36,39
In the absence of continuous water pathways in HSC micropores, Pt particles cannot access H+ ions and become electrochemically inactive, subsequently resulting in low Pt utilization. Several recent studies demonstrate improved performance with novel carbon supports that have comparatively wider micropores, short micropore depth, and/or low micropore tortuosity (relative to HSC).
8,9
A carbon support with such tailored morphology is believed to allow facile reactant transport to the Pt sites, while also preventing ionomer from directly contacting the Pt.
Focusing on PEFCs, Soboleva et al. experimentally studied water uptake in CLs with different carbon supports and Pt loadings.
36
CLs with HSC supportsdemonstrated significantly higher water uptake than did Vulcan, with both Pt utilization and CL double-layer capacitance increasing with increasing RH.
36
Eikerling et al. developed a macrohomogeneous model of water uptake in CLs with varying pore-size distributions (PSDs).
40
CLs with optimal wetting i.e., with co-existing liquid and gas phases in the primary pores (i.e., pores between carbon particles in the agglomerate) and only gas phase in the secondary pores (pores between agglomerates) provided efficient gas transport as well as high Pt utilization.
41
This study, however, examined CLs with Vulcan carbon that do not possess micropores. In an extensively detailed study of water uptake in PEFC CLs and subsequent impact on  conductivity, Iden et al. modeled water uptake in HSC using the semi-empirical Do-Do model that assumes initial water adsorption due to hydrogen bonding of water molecules with the functional groups on the carbon surface, followed by additional adsorption on top of the adsorbed water molecules via water-water hydrogen bonding.
42
Although the model results did not match experimental data at higher RHs due to neglect of capillary condensation, the model highlighted various water-uptake mechanisms, such as surface adsorption and micropore filling.
43
Iden et al. examined Ketjen Black carbon without Pt deposition and concluded that adsorbed water in micropores does not contribute to
conductivity, Iden et al. modeled water uptake in HSC using the semi-empirical Do-Do model that assumes initial water adsorption due to hydrogen bonding of water molecules with the functional groups on the carbon surface, followed by additional adsorption on top of the adsorbed water molecules via water-water hydrogen bonding.
42
Although the model results did not match experimental data at higher RHs due to neglect of capillary condensation, the model highlighted various water-uptake mechanisms, such as surface adsorption and micropore filling.
43
Iden et al. examined Ketjen Black carbon without Pt deposition and concluded that adsorbed water in micropores does not contribute to  conduction. However, Soboleva et al. demonstrated that water uptake by bare Ketjen Black carbon without Pt nanoparticles is significantly different from Pt-deposited Ketjen Black and hence results cannot be directly translated to PEFC CLs.
conduction. However, Soboleva et al. demonstrated that water uptake by bare Ketjen Black carbon without Pt nanoparticles is significantly different from Pt-deposited Ketjen Black and hence results cannot be directly translated to PEFC CLs.
These studies illuminate the critical role of water in PEFC CLs. However, they do not establish quantitative correlations between water uptake and PSD, RH, or performance metrics, specifically Pt utilization. In this study, we develop a self-consistent model to predict water uptake and Pt utilization quantitatively as a function of RH for a given pore-size and Pt-particle distribution. Using literature-reported PSD of CLs and a Pt particle distribution proportional to the micropore surface area (SA), the micropores within carbon particles are modeled without any ionomer, where the macro and mesopores that form outside the carbon particles include ionomer deposition on carbon-particle external surfaces. Water uptake is modeled via surface adsorption, capillary condensation, and ionomer absorption. Lastly, Pt utilization is determined by accounting for Pt particles in contact with water or ionomer, and the impact of RH and micropore-PSDs on calculated Pt utilization is examined.
Theory
CL pore-size and Pt-particle distribution
PEFC CLs are heterogeneous porous structures formed of carbon agglomerates held together by ionomer binder. The agglomerates are composed of several carbon particles, as illustrated in Fig. 1. The pores formed between carbon agglomerates are called macropores and are often the largest pores in the CL structure with a size range of 15 to 130 nm. Pores also form inside the agglomerates, between individual carbon particles. These are referred to as mesopores ranging from 2 to 35 nm. Lastly, a third category of pores i.e., micropores, exist inside porous carbon particles with sizes of 0.5 to 6 nm. Pores in CLs are irregularly shaped, especially the micropores which are known to have constricted openings that prevent ionomer penetration. The reported size range for each pore type is derived utilizing several techniques such as  adsorption,
9,34
quenched solid-density functional theory (QSDFT),
9
and electron tomography.
31
To model CL behavior as a function of RH, it is paramount to utilize a PSD representative of HSC supports because pore size and shape determine the critical radius for capillary condensation, interfacial surface area available for water adsorption and ionomer distribution, and Pt-particle distribution on the catalyst particles (i.e., interior versus exterior surface area). To simplify the modeled system, we assume all pores are cylindrical in shape.
adsorption,
9,34
quenched solid-density functional theory (QSDFT),
9
and electron tomography.
31
To model CL behavior as a function of RH, it is paramount to utilize a PSD representative of HSC supports because pore size and shape determine the critical radius for capillary condensation, interfacial surface area available for water adsorption and ionomer distribution, and Pt-particle distribution on the catalyst particles (i.e., interior versus exterior surface area). To simplify the modeled system, we assume all pores are cylindrical in shape.

Figure 1. Schematic of pore types in HSC-based CL. Carbon particles are in grey, Pt particles in yellow, and ionomer in blue. Macropores form in-between carbon agglomerates, mesopores form inside the carbon agglomerates, and the micropores exist in the interior of individual carbon particles. A typical pore-size range for each pore type is listed.
Download figure:
Standard image High-resolution imageBecause of their physical relevance, log-normal distributions are often utilized to model PSDs in PEFC CLs, 40,44,45 where each pore type has its own distribution.
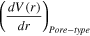
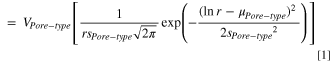
where  is the pore radius,
is the pore radius,  is the volume of pores between
is the volume of pores between  and
and 
 is the total volume of corresponding pore type and
is the total volume of corresponding pore type and  and
and  are parameters that determine the overall shape, i.e., peak-position and width of the distribution. The three parameters in Eq. 1 (
are parameters that determine the overall shape, i.e., peak-position and width of the distribution. The three parameters in Eq. 1 ( and
and  ) are adjustable. Model parameters were manually adjusted to fit the overall PSD to match volume and surface-area (SA) experimental data, as shown in Tables I and II. Because pores are modeled as straight cylinders, pore length
) are adjustable. Model parameters were manually adjusted to fit the overall PSD to match volume and surface-area (SA) experimental data, as shown in Tables I and II. Because pores are modeled as straight cylinders, pore length  was determined using the pore radius, pore volume, and cylinder volume equation
was determined using the pore radius, pore volume, and cylinder volume equation  while pore SA was calculated using the lateral surface area
while pore SA was calculated using the lateral surface area  Values of the fitting parameters are provided in Table S1. The overall PSD is a summation of the individual PSD corresponding to each pore type as shown in Fig. 2.
Values of the fitting parameters are provided in Table S1. The overall PSD is a summation of the individual PSD corresponding to each pore type as shown in Fig. 2.
Table I. Volumes used to scale PSD in model and experimental results.
| Pore type | Model (cm3/gcarbon) | Experiment 34 (cm3/gcarbon) |
|---|---|---|
| Micropores (0.5–2nm) | 0.100 | 0.11 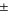 0.02 0.02 |
| Micropores (2–6nm) | 0.076 | |
| Mesopores (2–35 nm) | 0.224 | 0.59 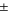 0.07 0.07 |
| Macropores (15–130 nm) | 0.290 | |
Table II. Surface area (SA) comparison of model and experiments.
| Model | Experiment 34 | |
|---|---|---|
| SA (<2nm) | 285.24 m2/gcarbon | 292.3 m2/gcarbon |
| SA (>2nm) | 233.30 m2/gcarbon | 209.3 m2/gcarbon |
| Total | 518.54 m2/gcarbon | 501.6 m2/gcarbon |

Figure 2. Overall PSD and individual sub-PSDs for the three pore-types. Each PSD is a piece-wise log-normal distribution. The overall PSD is the sum of three individual PSDs.
Download figure:
Standard image High-resolution imageFigure 3 compares the predicted PSD to experimental data on Pt/HSC CL with 5 wt% ionomer (0.1 ionomer to carbon weight ratio, denoted as I:C ratio) as the reference system. 34 Loose HSC or Pt/HSC PSDs (not bound via ionomer) overestimate the pore volume of larger pores, whereas high-ionomer-content CL PSDs underestimate small pore volume due to pore blockage by ionomer. Hereafter PSD from CLs with low ionomer content (5 wt%) are used. The experimentally reported pore volumes presented in Fig. 3 were measured over unequal pore-interval ranges, resulting in sharp peaks/irregularities in the theoretical curve. To perform a 1:1 comparison, the model-predicted volumes (shown in Fig. 3) were also summed over similar pore-size intervals as in the experimental data. Comparison of model and experimentally reported SA for the various pore types is presented in Table II, with good overall agreement in trends. Typically, the pore volume and SAs vary widely between the different types of HSC supports. Hence, observed deviations between model and experimental measurements are acceptable to predict general trends in HSC CL systems. Further, BET-BJH theory used in the experiments by Soboleva et al., is not necessarily suitable for heterogeneous microporous systems 46 ; utilizing density-functional theory for estimating the PSD of such complex systems may provide better agreement between model and experimental data, but necessitates substantial computational effort. 46,47

Figure 3. Comparison of model PSD with experimental data for Pt/HSC with an I:C ratio of 0.1. Experimental data reported from  adsorption isotherms and interpreted via BET-BJH analysis, reproduced from Soboleva et al.
36
adsorption isotherms and interpreted via BET-BJH analysis, reproduced from Soboleva et al.
36
Download figure:
Standard image High-resolution imageApart from the CL PSD, the Pt-particle size and physical distribution (on interior versus exterior SA of carbon particles) also influence Pt accessibility as a function of RH. For example, Pt particles on the exterior carbon surface that are in contact with ionomer remain electrochemically active even at low RHs, whereas reactant accessibility to Pt particles in interior micropores depends on water pathways and thus is highly RH sensitive. Moreover, Pt particles located inside the micropores and those located on the particle exterior surface are exposed to very different local microenvironments, resulting in different performance. In the model, we reference the data reported by Park et al. (see Fig. 4), 9 wherein the authors reported the frequency of different sized Pt particles and their distribution between interior and exterior pores. About 73% of the Pt particles, contributing ∼65% of the total Pt area, were observed to be deposited in the micropores. These data are in good agreement with those of Padgett et al. 31 We thus assign 35% of the total Pt area to exterior pores with ionomer access that remain electrochemically active even at low RH. The distribution of interior Pt particles accounting for the remaining 65% of the total Pt area across the micropores of different pore size is calculated using two criteria. First, the pore size must be larger than Pt particle size since pores cannot house Pt particles larger than the pore size. Second, the fraction of Pt particles in a micropore is equal to the corresponding micropore SA normalized to the total SA of all the micropores that have pore size equal or greater than a given Pt particle size.

Figure 4. Pt particle size distribution in the interior micropores and external surface of carbon particles. Data from Park et al. 9
Download figure:
Standard image High-resolution imageCL water uptake
Each component of a CL: carbon support, Pt nanoparticles, and ionomer, has a different affinity for water and different modes of water uptake. Hydrophilic pores in PEFC CLs take up water either via surface adsorption or via capillary condensation, whereas ionomer absorbs water. Each mechanism in the different components is modeled separately as detailed in the following sections.
Surface adsorption
Surface adsorbed water on hydrophilic and oxidized surfaces plays a critical role in many fields such as biology, tribology, etc. 48 Within HSC supports, surface oxygen groups render the surface hydrophilic resulting in water adsorption. Focusing on the more critical cathode PEFC, where air is fed to the cell, the gas molecules are simplified to water vapor and N2 and their interactions with the carbon pore walls and subsequent adsorption modeled using Lennard-Jones (LJ) and dipole-dipole interaction potentials as detailed below. Extensions to other gases (e.g, H2) is straightforward although the assumption of only N2 and water vapor does not induce significant error due to the ideal nature of the gas at typical PEFC operating conditions (see SI).
Both nitrogen and water molecules in the gas phase exhibit intermolecular interactions with the carbon wall that can be described using LJ potentials. Because we consider only pores with pore sizes greater than 0.5 nm (water molecular size ∼0.2 nm 49,50 ), we approximate the carbon pore walls as planar infinite and use the additive 9–3 LJ interaction potential for the gas-wall interaction potential, 51,52

where  is the normal distance from the wall surface,
is the normal distance from the wall surface,  is the 12–6 LJ potential well depth for gas (N2 or water)/carbon interaction,
is the 12–6 LJ potential well depth for gas (N2 or water)/carbon interaction,  is related to the corresponding 12–6 LJ potential collision diameter, and
is related to the corresponding 12–6 LJ potential collision diameter, and  is the solid carbon density. The first term on the right in the above equation represents repulsive forces, whereas the second term represents attractive forces. Oxygen groups are only present on the surface of pore walls and present a planar 2-D surface for intermolecular interactions, thus the interaction potential due to surface oxygen groups is modeled with a 10–4 potential,
53
is the solid carbon density. The first term on the right in the above equation represents repulsive forces, whereas the second term represents attractive forces. Oxygen groups are only present on the surface of pore walls and present a planar 2-D surface for intermolecular interactions, thus the interaction potential due to surface oxygen groups is modeled with a 10–4 potential,
53
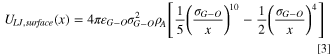
where  is the 12–6 LJ potential well-depth for gas (N2 or water)/oxygen group interaction,
is the 12–6 LJ potential well-depth for gas (N2 or water)/oxygen group interaction,  is the corresponding 12–6 LJ potential collision diameter, and
is the corresponding 12–6 LJ potential collision diameter, and  is the surface density of oxygen groups. The micropore walls exhibit varying
is the surface density of oxygen groups. The micropore walls exhibit varying  depending on exposure to
depending on exposure to  processing conditions, etc
processing conditions, etc  on each pore wall is thus assigned a random value from a gaussian distribution around a mean value instead of a constant fixed value. Pores with higher
on each pore wall is thus assigned a random value from a gaussian distribution around a mean value instead of a constant fixed value. Pores with higher  have greater adsorbed water thickness due to stronger interactions.
have greater adsorbed water thickness due to stronger interactions.
Here, we treat the interacting molecules such as H2O, N2 and surface oxygen groups as LJ spheres. 54–56 Properties of a hydroxyl group are used to model the surface oxygen groups since OH is the most abundant type of oxygen group in carbonaceous materials. 55 Values of various LJ parameters are listed in Table S2. 12–6 LJ potential parameters for N2/carbon interactions were obtained from the study by Kim et al. 54 Lorentz-Berthelot mixing rules were employed to calculate other cross-species 12–6 LJ potential parameters from pure species 12–6 LJ potential parameters, 57
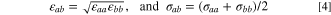
In addition to intermolecular interactions, water molecules and surface oxygen groups exert strong dipole-dipole interactions due to their polar nature, which play a critical role in enhancing surface adsorption of water. Dipole-dipole interactions depend strongly on the orientation of the interacting molecules and have been extensively studied by Molecular Dynamics and Monte Carlo samplings.
56,58–60
By assuming that surface dipoles are distributed uniformly and oriented perpendicular to the surface and by approximating the pore walls as planar surfaces, the potential energy of water ( defined per mole) due to dipole-dipole interactions with the surface oxygen group is obtained from the expression (see SI for derivation)
defined per mole) due to dipole-dipole interactions with the surface oxygen group is obtained from the expression (see SI for derivation)
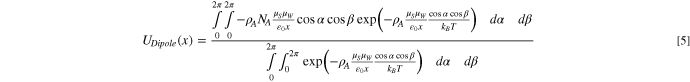
where  and
and  are the dipole moments of water and surface oxygen group, respectively,
are the dipole moments of water and surface oxygen group, respectively,  is the dielectric permittivity of the gas phase, and
is the dielectric permittivity of the gas phase, and  and
and  are Boltzmann's constant and Avogadro's number, respectively. In 3-D space, the water dipole vector is formed by three component vectors along the three principal axis directions. Only the component dipole vector aligned parallel to the surface-oxygen-group dipoles contributes to
are Boltzmann's constant and Avogadro's number, respectively. In 3-D space, the water dipole vector is formed by three component vectors along the three principal axis directions. Only the component dipole vector aligned parallel to the surface-oxygen-group dipoles contributes to  while the other component vectors perpendicular to the surface-oxygen-group dipoles have net zero interaction potential (see SI). Equation 5 is derived by summing the interaction potential over the various orientations of the gaseous water dipole. At each
while the other component vectors perpendicular to the surface-oxygen-group dipoles have net zero interaction potential (see SI). Equation 5 is derived by summing the interaction potential over the various orientations of the gaseous water dipole. At each  Eq. 5 is evaluated by converting the integral to a Riemann sum (See SI for details).
Eq. 5 is evaluated by converting the integral to a Riemann sum (See SI for details).
Note that the above formulation of  assumes that the distance
assumes that the distance  is greater than the size of the molecule (i.e., the distance over which the dipole charges are separated). At small
is greater than the size of the molecule (i.e., the distance over which the dipole charges are separated). At small  the overall dipole interaction potential is a result of Coulombic interactions between the individual poles. However, the model employs Eq. 5 for all
the overall dipole interaction potential is a result of Coulombic interactions between the individual poles. However, the model employs Eq. 5 for all  as an approximation, thus over-predicting adsorbed-film thickness. To the best of our knowledge, dipole-dipole interactions for molecules separated by small distances cannot be expressed analytically and are typically estimated from molecular dynamics.
54,56
as an approximation, thus over-predicting adsorbed-film thickness. To the best of our knowledge, dipole-dipole interactions for molecules separated by small distances cannot be expressed analytically and are typically estimated from molecular dynamics.
54,56
As noted above, the gas-molecule wall-interaction potentials result in adsorption of water molecules on the carbon surface. To estimate the thickness of the adsorbed water layer, we examine the local chemical potential of H2O and  molecules close to the wall. The LJ and dipole interaction potentials with the carbon surface, in addition to size and LJ interactions between the adsorbate molecules, allinfluence the local chemical potential of water and nitrogen molecules close to the carbon surface. A local density approximation along with Amagat's ideal mixing approximation (i.e., the Lewis fugacity rule)
61,62
and the Peng-Robinson equation of state
61–63
yields
64
(see SI for details)
molecules close to the wall. The LJ and dipole interaction potentials with the carbon surface, in addition to size and LJ interactions between the adsorbate molecules, allinfluence the local chemical potential of water and nitrogen molecules close to the carbon surface. A local density approximation along with Amagat's ideal mixing approximation (i.e., the Lewis fugacity rule)
61,62
and the Peng-Robinson equation of state
61–63
yields
64
(see SI for details)
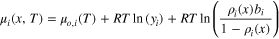
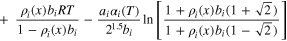

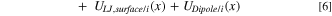
where  is a reference temperature-dependent standard-state chemical potential of component i,
is a reference temperature-dependent standard-state chemical potential of component i,  is the local molar density of component i, and
is the local molar density of component i, and  is the mole fraction of component i expressed in terms of molar densities
is the mole fraction of component i expressed in terms of molar densities

Characteristic constants  and
and  of component
of component  in Eq. 6 are calculated using the corresponding critical-state parameters,
63
as outlined in the SI.
in Eq. 6 are calculated using the corresponding critical-state parameters,
63
as outlined in the SI.  modulates the temperature dependence of attractive forces between molecules,
63
modulates the temperature dependence of attractive forces between molecules,
63

where  is the reduced temperature (gas temperature divided by the its critical temperature). Peng and Robinson initially defined the constants
is the reduced temperature (gas temperature divided by the its critical temperature). Peng and Robinson initially defined the constants  and
and  using the acentric factor for nonpolar hydrocarbon compounds.
63
Since then, over 100 modifications for
using the acentric factor for nonpolar hydrocarbon compounds.
63
Since then, over 100 modifications for  have been proposed to accommodate different types of compounds, operating conditions, and mixtures.
65
We utilize the initial definitions of
have been proposed to accommodate different types of compounds, operating conditions, and mixtures.
65
We utilize the initial definitions of  and
and  in terms of the acentric factor for N2. For water, we use the values
in terms of the acentric factor for N2. For water, we use the values  and
and  reported by Peng and Robinson in a later publication
66
(see Table S2).
reported by Peng and Robinson in a later publication
66
(see Table S2).
Upon equating the local chemical potential at distance  from the wall to the bulk chemical potential for each component gas (i.e.,
from the wall to the bulk chemical potential for each component gas (i.e.,  and
and  ), Eqs. 6–8 yield two coupled nonlinear algebraic relations to describe
), Eqs. 6–8 yield two coupled nonlinear algebraic relations to describe  Once solved, the adsorbed water film thickness
Once solved, the adsorbed water film thickness  from the calculated density profile is given by
from the calculated density profile is given by
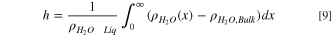
where  is the molar density of gas-phase water far from the wall (where LJ and dipole surface interaction potentials are non-existent), and
is the molar density of gas-phase water far from the wall (where LJ and dipole surface interaction potentials are non-existent), and  is the liquid-phase pure-water molar density. Due to the strong dipole-dipole interactions between water molecules and the surface oxygen groups, the adsorbed film is primarily composed of water. Hence, water bulk density is used as the normalizing factor in Eq. 9. Density profiles at various RHs are shown in Fig. 5 for x ≫ 0.15 nm. Below this distance, the potential energy increases sharply due to strong repulsive intermolecular forces. The steep slope of the potential function in this region coupled with system accuracy limits causes convergence issues of the iteration algorithm. Additionally, 0.15 nm is less than the distance of closest approach
is the liquid-phase pure-water molar density. Due to the strong dipole-dipole interactions between water molecules and the surface oxygen groups, the adsorbed film is primarily composed of water. Hence, water bulk density is used as the normalizing factor in Eq. 9. Density profiles at various RHs are shown in Fig. 5 for x ≫ 0.15 nm. Below this distance, the potential energy increases sharply due to strong repulsive intermolecular forces. The steep slope of the potential function in this region coupled with system accuracy limits causes convergence issues of the iteration algorithm. Additionally, 0.15 nm is less than the distance of closest approach  At this distance, the molar density calculated is ∼
At this distance, the molar density calculated is ∼ hence, the adsorbed film thickness at distance
hence, the adsorbed film thickness at distance  < 0.15 nm is neglected. Even at 99% RH, the thickness of the adsorbed film is ∼0.15 nm, which is somewhat less than a continuous water monolayer thickness. The thickness of adsorbed layer is highly sensitive to the value of
< 0.15 nm is neglected. Even at 99% RH, the thickness of the adsorbed film is ∼0.15 nm, which is somewhat less than a continuous water monolayer thickness. The thickness of adsorbed layer is highly sensitive to the value of  and multilayer adsorption is observed just by doubling the value of
and multilayer adsorption is observed just by doubling the value of  . Note that
. Note that  can vary widely between carbon types based on processing condition, air exposure during storage, etc.
can vary widely between carbon types based on processing condition, air exposure during storage, etc.

Figure 5. Density profiles of gas-phase mixture as a function of distance from the wall for various RHs. With higher RH, the thickness of the adsorbed film increases.  Even at 99% RH, the thickness of adsorbed film is ∼0.15 nm, which is less than continuous water monolayer thickness, indicating conduction of
Even at 99% RH, the thickness of adsorbed film is ∼0.15 nm, which is less than continuous water monolayer thickness, indicating conduction of  between patches of adsorbed water film via surface oxides. On the resolution of the ordinate scale, the increasing density of water in the gas phase does not register.
between patches of adsorbed water film via surface oxides. On the resolution of the ordinate scale, the increasing density of water in the gas phase does not register.
Download figure:
Standard image High-resolution imageIn the model, only micropores adsorb water. The mesopores and macropores are modeled with ionomer deposition on the pore walls, preventing water from adsorbing directly on the bare carbon surface. In an actual CL system with heterogeneous ionomer distributions, bare carbon surfaces in meso and macropores can also adsorb water. This contribution is, however, minimal given the small surface area of meso and macropores compared to that of the micropores, and thus is neglected. Water adsorption on Pt surfaces is similarly neglected due to their relatively small SA compared to carbon SA in the micropores. 67
At typical PEFC operating temperatures (60 to 80 °C), there are multiple roots of the Peng-Robinson EOS. 64 The smallest and largest roots correspond to gas and liquid phase respectively, while the middle root is considered aphysical. Hence, different adsorbed film thicknesses can be calculated depending on the initial guess. Since we are modeling the adsorption curve of the isotherm here, the bulk gas-phase density of water and nitrogen are used as initial guesses for the densities of various components for solving Eq.6 via Newton-Raphson iterative method.
Capillary condensation
Capillary condensation describes liquid filling of small capillary pores (2 to 50 nm) below the planar saturation pressure due to surface tension and concave meniscus curvature lowering the chemical potential. 68,69 Kelvin's equation for gas mixtures governs the condensation conditions. 69 By assuming that the condensed liquid consists of only water, and the gas phase includes both carrier gas (inert N2) and water vapor, the Kelvin equation is written as
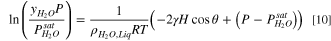
where  is the water/air surface tension,
is the water/air surface tension,  is the total mean curvature of the meniscus and depends on pore radius/size and shape,
is the total mean curvature of the meniscus and depends on pore radius/size and shape,  is the saturation water vapor pressure at temperature T, and
is the saturation water vapor pressure at temperature T, and  is the mole fraction of water vapor in the bulk gas phase, and
is the mole fraction of water vapor in the bulk gas phase, and  is the contact angle of water with the solid surface measured in the water phase (assumed to be 0 due to hydrophilic oxygen-groups and adsorbed water on the pore walls). Contact angle is not an independent parameter and can be calculated from adsorption isotherm.
70
Here we use an assumed value of
is the contact angle of water with the solid surface measured in the water phase (assumed to be 0 due to hydrophilic oxygen-groups and adsorbed water on the pore walls). Contact angle is not an independent parameter and can be calculated from adsorption isotherm.
70
Here we use an assumed value of  to limit the scope of the paper. The constricted cylindrical pores in CLs have a mean curvature equal to the inverse of pore radius
to limit the scope of the paper. The constricted cylindrical pores in CLs have a mean curvature equal to the inverse of pore radius  different from that for straight cylindrical capillaries with mean curvature
different from that for straight cylindrical capillaries with mean curvature  to account mainly for presence of narrow constrictions in the CL pores. Water uptake due to capillary condensation at a given RH thus equals the total volume of pores with pore radius less than the critical radius for capillary condensation
to account mainly for presence of narrow constrictions in the CL pores. Water uptake due to capillary condensation at a given RH thus equals the total volume of pores with pore radius less than the critical radius for capillary condensation  which equals
which equals 
Ionomer absorption
Ionomer water uptake is estimated from CL water content, λ, defined as the moles of water absorbed per mole of sulfonic acid groups. Based on experiments, Kusoglu and Weber report a polynomial fit for i as a function of RH, 19,71
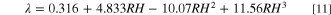
Equation 11 is obtained from bulk ionomer data that should be representative of the response of the heterogeneous CL due to the presence of ionomer aggregates that behave similar to bulk ionomer and dominate water uptake (compared to ionomer thin films that are reported to exhibit reduced water uptake).
19
The water volume absorbed by ionomer is equal to the calculated  value multiplied by water molar volume
value multiplied by water molar volume  and the total moles of sulfonic groups, given by the ionomer mass divided by ionomer EW (1000 g mol−1).
and the total moles of sulfonic groups, given by the ionomer mass divided by ionomer EW (1000 g mol−1).
The above sections quantify water uptake via different modes in the CL. The total water uptake at a given RH, hence, is the sum of water uptake from the individual modes i.e., surface adsorption, capillary condensation, and ionomer absorption, as represented by each term on the right side of the following expression, respectively.
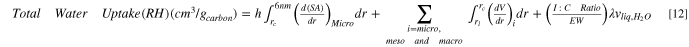
where  is the critical radius for capillary condensation, and
is the critical radius for capillary condensation, and  is the smallest pore radius in respective pore type. SA represents pore surface area and
is the smallest pore radius in respective pore type. SA represents pore surface area and  is the adsorbed film thickness calculated from Eq. 9. The first term on the right in Eq. 12 represents water adsorbed on carbon walls and includes the larger micropores above
is the adsorbed film thickness calculated from Eq. 9. The first term on the right in Eq. 12 represents water adsorbed on carbon walls and includes the larger micropores above  because only non-liquid-filled micropores exhibit bare carbon walls. The second term represents capillary condensation and includes all pore-types. The last term representing ionomer water uptake is calculated using
because only non-liquid-filled micropores exhibit bare carbon walls. The second term represents capillary condensation and includes all pore-types. The last term representing ionomer water uptake is calculated using  from Eq. 11 using CL parameters i.e., I:C ratio, and ionomer EW. The relative contribution by each mode depends on several factors such as CL PSD, carbon surface properties, and I:C ratio. These findings are examined below.
from Eq. 11 using CL parameters i.e., I:C ratio, and ionomer EW. The relative contribution by each mode depends on several factors such as CL PSD, carbon surface properties, and I:C ratio. These findings are examined below.
Pt accessibility
In PEFCs, Pt particles in the interior micropores of HSC are accessed by  through liquid-water pathways, i.e., adsorbed and/or capillary-condensed water.
31,38,39
In one of the early studies examining
through liquid-water pathways, i.e., adsorbed and/or capillary-condensed water.
31,38,39
In one of the early studies examining  conduction via adsorbed water layers, Khanna et al. suggested the dominance of a
conduction via adsorbed water layers, Khanna et al. suggested the dominance of a  tunneling mechanism at low humidity and vehicular transport at high humidity.
72
Furthermore, hydroxyl terminated surface defects are known to facilitate
tunneling mechanism at low humidity and vehicular transport at high humidity.
72
Furthermore, hydroxyl terminated surface defects are known to facilitate  transport.
73–75
Therefore we define a threshold adsorbed film thickness
transport.
73–75
Therefore we define a threshold adsorbed film thickness  at which the adsorbed water film can conduct water, thus rendering the Pt particles in electrochemical contact and leading to the electrochemically active surface area (ECSA). At adsorbed film thickness lower than
at which the adsorbed water film can conduct water, thus rendering the Pt particles in electrochemical contact and leading to the electrochemically active surface area (ECSA). At adsorbed film thickness lower than  the adsorbed films are not appreciably conductive and hence the Pt particles in contact do not contribute to ECSA. The conductivity of the adsorbed films is expressed as a step-function with the films exhibiting bulk liquid conductive at thickness
the adsorbed films are not appreciably conductive and hence the Pt particles in contact do not contribute to ECSA. The conductivity of the adsorbed films is expressed as a step-function with the films exhibiting bulk liquid conductive at thickness  and above. The total ECSA at any RH is determined by summing the Pt area in (i) meso and macropores pores since they are in contact with ionomer, and (ii) micropores that are either flooded due to capillary condensation or have adsorbed film thickness greater than
and above. The total ECSA at any RH is determined by summing the Pt area in (i) meso and macropores pores since they are in contact with ionomer, and (ii) micropores that are either flooded due to capillary condensation or have adsorbed film thickness greater than  It is assumed that all micropores are in contact with meso/macropores and can thus conduct
It is assumed that all micropores are in contact with meso/macropores and can thus conduct  when they have sufficient adsorbed film thickness or capillary-condensed water. In the model,
when they have sufficient adsorbed film thickness or capillary-condensed water. In the model,  is fit within reason (fractions of a monolayer) to reproduce experimental data.
is fit within reason (fractions of a monolayer) to reproduce experimental data.
Results and Discussion
Water uptake
Overall CL water uptake, as described in Eq. 12, is estimated by summing over (i) water absorption into the ionomer, estimated from λ, (ii) surface adsorption, given by product of water-film thickness and micropore carbon surface area, and (iii) capillary condensation approximated from the total volume of water-filled pores based on the given HSC PSD. Model results are compared to experimental data in Fig. 6. At low RH, all three modes of water uptake have non-negligible contributions. At ∼50% RH, all micropores (up to the size of 6 nm) are water-filled due to capillary condensation, and there is no further contribution from surface adsorbed water. At higher RH, additional water uptake is only due to capillary condensation in meso and macropores, and ionomer absorption. Capillary condensation mostly dominates the water uptake over the whole RH range.

Figure 6. Water uptake of CL with Pt/HSC catalyst and I:C ratio 0.8 – model predictions (dashed lines) versus experimental data (filled symbols). Uptake contributions are highlighted in different colors. Experimental data from Ref. 36.
Download figure:
Standard image High-resolution imageFigure 7a plots total water uptake for different oxide adsorption site densities ( ) on pore walls. Oxide defects are primarily responsible for water adsorption on pore walls due to dipole-dipole interactions. Thus, water uptake due to surface adsorption increases with
) on pore walls. Oxide defects are primarily responsible for water adsorption on pore walls due to dipole-dipole interactions. Thus, water uptake due to surface adsorption increases with  at low RHs. At high RH, where capillary condensation and ionomer adsorption dominate, no impact of varying
at low RHs. At high RH, where capillary condensation and ionomer adsorption dominate, no impact of varying  is observed. In the current model (including Fig. 6),
is observed. In the current model (including Fig. 6),  was set as
was set as  sites/m2 to best reproduce experimental data. Figure 7b compares the present model to experimental data collected by Soboleva et al. on CLs with different I:C ratios.
36
In the model, the same PSD was used for all I:C ratios, and only the ionomer content was increased. For CLs with I:C ratios of 0.1 and 0.8, model predictions provide good agreement with experimental data, particularly at low RH. At high RH, the model underpredicts the water uptake of CL with I:C ratio 0.1. One likely explanation is that the PSD itself changes with I:C ratio due to ink-based interactions from which CLs are fabricated, which in turn impacts capillary condensation. Additionally, the experimental water uptake of CLs with I:C of 0.1 and 0.8 converge at high RH suggesting flooding due to capillary condensation in all the samples and highlights the difficulty in accurately controlling high RH in experimental investigations. Interestingly, the water uptake of bare HSC (Ketjen black in powder form) without ionomer and Pt is significantly lower than that of the CLs. To match this set of data, the model was modified to increase water/carbon contact angle to 75°
76
and surface adsorption was neglected, which suggests that HSC surface properties are significantly different from HSC with Pt deposited. These differences can potentially arise during the Pt deposition process in which the carbon is subjected to several chemical steps including acid treatment.
77
Note that in Figs. 6 and 7, we plot only the adsorption curve of the isotherm. The desorption isotherm exhibits hysteresis with a distinctly larger desorption isotherm due to pore morphology resulting in trapping of condensed water in larger pores due to throat constrictions and is beyond the scope of the current study as it requires detailed nanoscopic tomographic imaging.
31,34,36
sites/m2 to best reproduce experimental data. Figure 7b compares the present model to experimental data collected by Soboleva et al. on CLs with different I:C ratios.
36
In the model, the same PSD was used for all I:C ratios, and only the ionomer content was increased. For CLs with I:C ratios of 0.1 and 0.8, model predictions provide good agreement with experimental data, particularly at low RH. At high RH, the model underpredicts the water uptake of CL with I:C ratio 0.1. One likely explanation is that the PSD itself changes with I:C ratio due to ink-based interactions from which CLs are fabricated, which in turn impacts capillary condensation. Additionally, the experimental water uptake of CLs with I:C of 0.1 and 0.8 converge at high RH suggesting flooding due to capillary condensation in all the samples and highlights the difficulty in accurately controlling high RH in experimental investigations. Interestingly, the water uptake of bare HSC (Ketjen black in powder form) without ionomer and Pt is significantly lower than that of the CLs. To match this set of data, the model was modified to increase water/carbon contact angle to 75°
76
and surface adsorption was neglected, which suggests that HSC surface properties are significantly different from HSC with Pt deposited. These differences can potentially arise during the Pt deposition process in which the carbon is subjected to several chemical steps including acid treatment.
77
Note that in Figs. 6 and 7, we plot only the adsorption curve of the isotherm. The desorption isotherm exhibits hysteresis with a distinctly larger desorption isotherm due to pore morphology resulting in trapping of condensed water in larger pores due to throat constrictions and is beyond the scope of the current study as it requires detailed nanoscopic tomographic imaging.
31,34,36

Figure 7. (a) Water uptake for varying oxide defect density on pore walls  (b) Model results (lines) and experimental data (filled symbols) comparison of CLs with different I:C ratios and Pt/HSC catalyst and HSC powder. Experimental data taken from Refs. 36, 43.
(b) Model results (lines) and experimental data (filled symbols) comparison of CLs with different I:C ratios and Pt/HSC catalyst and HSC powder. Experimental data taken from Refs. 36, 43.
Download figure:
Standard image High-resolution imageElectrochemical active surface area (ECSA)
The primary role of water in PEFC CLs is to facilitate  conduction through water pathways and ionomer to/from the Pt reaction sites and the ionomer membrane. Hence, ECSA consists of only those Pt particles that are in contact with a
conduction through water pathways and ionomer to/from the Pt reaction sites and the ionomer membrane. Hence, ECSA consists of only those Pt particles that are in contact with a  conducting pathway. Pt particles on the exterior carbon surfaces are in contact with ionomer and can access
conducting pathway. Pt particles on the exterior carbon surfaces are in contact with ionomer and can access  even at low RHs. Pt particles in the interior micropores, however, lose
even at low RHs. Pt particles in the interior micropores, however, lose  access at low RH due to insufficient or disconnected water pathways. At higher RH, where most of the pores are filled with capillary-condensed water, extremely high Pt accessibility is observed.
31,36,39
access at low RH due to insufficient or disconnected water pathways. At higher RH, where most of the pores are filled with capillary-condensed water, extremely high Pt accessibility is observed.
31,36,39
Figure 8a displays the model-predicted fraction of Pt particles with  access (hence electrochemically active) as a function of RH when
access (hence electrochemically active) as a function of RH when  was set to 0.5 times water monolayer thickness
was set to 0.5 times water monolayer thickness  49,50
As described earlier, the fraction of electrochemically active Pt particles is estimated by including contributions due to surface adsorbed films, capillary condensation in pores, and ionomer. In agreement with prior studies, ECSA increases with increasing RH is observed. By accounting for only Pt particles in water-filled pores with capillary condensation and exterior Pt particles in contact with ionomer, model results do not agree with ECSA experimental data, shown by the dotted line. However, when the Pt particles in wetted pores with surface adsorbed water are included, model results are in good agreement with experimental data. This result demonstrates the vital role of adsorbed films: despite their small volume, adsorbed films aid
49,50
As described earlier, the fraction of electrochemically active Pt particles is estimated by including contributions due to surface adsorbed films, capillary condensation in pores, and ionomer. In agreement with prior studies, ECSA increases with increasing RH is observed. By accounting for only Pt particles in water-filled pores with capillary condensation and exterior Pt particles in contact with ionomer, model results do not agree with ECSA experimental data, shown by the dotted line. However, when the Pt particles in wetted pores with surface adsorbed water are included, model results are in good agreement with experimental data. This result demonstrates the vital role of adsorbed films: despite their small volume, adsorbed films aid  conduction and allow high Pt accessibility in CLs with HSC supports, especially at low RHs. Hence, carbon supports with high surface adsorption can enable high Pt access even at low RH. Alternatively, increasing the conductivity of adsorbed films to enable
conduction and allow high Pt accessibility in CLs with HSC supports, especially at low RHs. Hence, carbon supports with high surface adsorption can enable high Pt access even at low RH. Alternatively, increasing the conductivity of adsorbed films to enable  conduction at low adsorbed film thickness can also provide larger Pt access. This result is shown in Fig. 8b, where the minimum threshold thickness of adsorbed film to enable
conduction at low adsorbed film thickness can also provide larger Pt access. This result is shown in Fig. 8b, where the minimum threshold thickness of adsorbed film to enable  conduction is varied. With low
conduction is varied. With low  higher Pt accessibility is obtained. Although the conductivity of adsorbed water cannot be modified, one can provide alternate pathways by adding additional supporting electrolytes such as ionic liquids to enhance
higher Pt accessibility is obtained. Although the conductivity of adsorbed water cannot be modified, one can provide alternate pathways by adding additional supporting electrolytes such as ionic liquids to enhance  conductivity.
78,79
It is interesting to note that the best fit to experimental data is obtained at
conductivity.
78,79
It is interesting to note that the best fit to experimental data is obtained at  less than monolayer thickness.
less than monolayer thickness.  conductivity through adsorbed water films of thickness as low as 0.5 nm have been reported in literature.
80
Below monolayer coverage, water molecules can be expected to adsorb in isolated patches.
81
conductivity through adsorbed water films of thickness as low as 0.5 nm have been reported in literature.
80
Below monolayer coverage, water molecules can be expected to adsorb in isolated patches.
81
 conduction will thus have to rely on surface defects to transport between these patches. Similar observations have been reported earlier in literature in other systems and are likely at play in PEFC CLs also.
73,74
conduction will thus have to rely on surface defects to transport between these patches. Similar observations have been reported earlier in literature in other systems and are likely at play in PEFC CLs also.
73,74

Figure 8. (a) Fraction of Pt particles electrochemically active versus RH. Solid line represents total fraction of electrochemically active Pt particles, including both water-filled and wetted pores, while the dashed line considers only water-filled pores with capillary condensed water. (b) Impact of threshold adsorbed film thickness  to allow
to allow  conduction on active ECSA fraction. Value of
conduction on active ECSA fraction. Value of  is expressed in terms of water monolayer thickness
is expressed in terms of water monolayer thickness  49,50
Data taken from Refs. 31, 36, 39.
49,50
Data taken from Refs. 31, 36, 39.
Download figure:
Standard image High-resolution imageThe water-filled and surface-wetted pores present very different local environments close to the Pt sites and can result in significantly different performances. Wetted pores allow high gas transport in comparison to water-filled pores where the reactant gas molecules must transport though the pore in the dissolved state. However,  transport can become limiting in wetted pores. The impact of PSD on Pt accessibility is demonstrated in Fig. 9 which plots the total fraction of electrochemically active Pt for carbon supports with micropore PSDs limited to larger pores (from 3–6 nm compared to 0.5–6 nm used in model). At low RH (<0.4), both PSDs demonstrate similar ECSA since the ECSA is primarily contributed by exterior Pt particles and interior Pt particles that are in contact with adsorbed water films. Under these operating conditions, CLs with larger PSDs provide enhanced performance due to less gas transport resistance to Pt reaction sites. At mid-RHs (0.4 to 0.7), however, we find that the active ECSA fraction in case of a larger PSD lags behind. This is because capillary condensation also lags behind in the case of a larger PSD, thus resulting in a lower active ECSA fraction at a given RH. The active ECSA fraction calculated from flooded pores with capillary condensation and exterior Pt particles (in contact with ionomer) is shown in the inset of Fig. 9. This plot shows a clear difference in the active ECSA fraction of the two different PSDs at mid-RH ranges. Hence, even though the larger PSD provides enhanced gas transport, it might be limited by restricting
transport can become limiting in wetted pores. The impact of PSD on Pt accessibility is demonstrated in Fig. 9 which plots the total fraction of electrochemically active Pt for carbon supports with micropore PSDs limited to larger pores (from 3–6 nm compared to 0.5–6 nm used in model). At low RH (<0.4), both PSDs demonstrate similar ECSA since the ECSA is primarily contributed by exterior Pt particles and interior Pt particles that are in contact with adsorbed water films. Under these operating conditions, CLs with larger PSDs provide enhanced performance due to less gas transport resistance to Pt reaction sites. At mid-RHs (0.4 to 0.7), however, we find that the active ECSA fraction in case of a larger PSD lags behind. This is because capillary condensation also lags behind in the case of a larger PSD, thus resulting in a lower active ECSA fraction at a given RH. The active ECSA fraction calculated from flooded pores with capillary condensation and exterior Pt particles (in contact with ionomer) is shown in the inset of Fig. 9. This plot shows a clear difference in the active ECSA fraction of the two different PSDs at mid-RH ranges. Hence, even though the larger PSD provides enhanced gas transport, it might be limited by restricting  transport to Pt reaction sites. Because adsorbed-film pores allow better reactant gas transport, carbon supports with larger micropore pore-size range may enhance gas-transport to the Pt particles and provide better performance in the high-current density region (where gas transport is cited as the limiting factor).
14,18
Conversely, smaller micropores may allow better performance if
transport to Pt reaction sites. Because adsorbed-film pores allow better reactant gas transport, carbon supports with larger micropore pore-size range may enhance gas-transport to the Pt particles and provide better performance in the high-current density region (where gas transport is cited as the limiting factor).
14,18
Conversely, smaller micropores may allow better performance if  transport is the limiting factor. This result agrees with recent studies that demonstrate superior performance of mesoporous carbons, particularly in the high current-density region.
8,9,35
Identification of the desired PSD to optimize both
transport is the limiting factor. This result agrees with recent studies that demonstrate superior performance of mesoporous carbons, particularly in the high current-density region.
8,9,35
Identification of the desired PSD to optimize both  and gas-transport requires further investigation of
and gas-transport requires further investigation of  -conductivity dependence on adsorbed water-film thickness, micropore geometry, and water adsorption mechanisms pertinent in HSC supports. Finally, it should be noted that ECSA measurements are at low currents and thus accessibility is only one metric that may not translate directly to higher operating current densities as the conductivity of the wetted films is expected to be much lower than that in the ionomer, and is an area of current research. Also, this analysis does not consider water consumption or production, both of which can occur during fule-cell operation.
-conductivity dependence on adsorbed water-film thickness, micropore geometry, and water adsorption mechanisms pertinent in HSC supports. Finally, it should be noted that ECSA measurements are at low currents and thus accessibility is only one metric that may not translate directly to higher operating current densities as the conductivity of the wetted films is expected to be much lower than that in the ionomer, and is an area of current research. Also, this analysis does not consider water consumption or production, both of which can occur during fule-cell operation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 9. ECSA fraction: total (solid lines) and from water-filled pores (dashed lines) for different micropore size ranges. The inset figure plots active ECSA fraction while neglecting the contribution of wetted pores with adsorbed water films and without capillary condensation versus RH for different PSDs.
Download figure:
Standard image High-resolution image{kind=link}
Conclusions
Catalyst-layer (CL) performance with high-surface area carbon (HSC) critically depends on water uptake because water provides the pathway for  conduction to interior Pt particles that have no direct ionomer contact. Predicting such uptake is critical for understanding overall device performance and optimizing CL design. In this paper, water uptake in CLs with HSC supports is modeled via three mechanisms: (i) surface adsorption via gas/solid molecular interactions, (ii) capillary condensation, and (iii) ionomer absorption using detailed physics-based descriptions, with good agreement compared to experimental data. Although capillary condensation dominates overall water uptake, adsorbed water films are important contributors to
conduction to interior Pt particles that have no direct ionomer contact. Predicting such uptake is critical for understanding overall device performance and optimizing CL design. In this paper, water uptake in CLs with HSC supports is modeled via three mechanisms: (i) surface adsorption via gas/solid molecular interactions, (ii) capillary condensation, and (iii) ionomer absorption using detailed physics-based descriptions, with good agreement compared to experimental data. Although capillary condensation dominates overall water uptake, adsorbed water films are important contributors to  conduction in micropores that make the Pt particles electrochemically accessible. Both capillary-condensed and adsorbed-water pathways contribute to Pt accessibility, with adsorbed water pathways becoming increasingly important at low RHs. The two pore types present different local microenvironments and reactant species transport. Designing carbon supports that optimize both reactant gas and ion transport can significantly improve overall CL performance. This conclusion agrees with recent studies that demonstrate superior high-current density performance of mesoporous carbons. Larger pore sizes prevent capillary condensation, while the surface adsorbed water films continue to conduct
conduction in micropores that make the Pt particles electrochemically accessible. Both capillary-condensed and adsorbed-water pathways contribute to Pt accessibility, with adsorbed water pathways becoming increasingly important at low RHs. The two pore types present different local microenvironments and reactant species transport. Designing carbon supports that optimize both reactant gas and ion transport can significantly improve overall CL performance. This conclusion agrees with recent studies that demonstrate superior high-current density performance of mesoporous carbons. Larger pore sizes prevent capillary condensation, while the surface adsorbed water films continue to conduct  thus enabling efficient transport of both reactant species. Overall, the model provides a quantitative methodology to explore these phenomena.
thus enabling efficient transport of both reactant species. Overall, the model provides a quantitative methodology to explore these phenomena.
Acknowledgments
This material is based on work performed by the Million Mile Fuel Cell Truck (M2FCT) Consortium (https://millionmilefuelcelltruck.org) (technology manager: Greg Kleen), which is supported by the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy, Hydrogen and Fuel Cell Technologies Office, under contract number DE-AC02–05CH1123, with initial funding from the Fuel Cell Performance and Durability Consortium (FC-PAD) under the same contract.
Supplementary data (0.5 MB PDF)