
Abstract
Composite-solid electrolytes, in which ion-conducting polymers are combined with superionic ceramics, could revolutionize electrochemical-energy-storage devices enabling higher energy density, providing greater stability during operation and enhanced safety. However, the interfacial resistance between the ceramic and polymer phases strongly suppresses the ionic conductivity and presents the main obstacle to the use of these materials. Here, we emphasize the need for a distinct focus on reducing energy barriers to interfacial ion transport and improving the cation transference number. To achieve this goal, it is essential to develop a fundamental understanding of the parameters that influence the interfacial barriers to ion transport in composite electrolytes, and to understand the effect of the type of ceramic ("active" and "inert") and its content on ion-transport phenomena. We suggest that adapting the polymer chemistry, mainly directed on polymerized ionic liquids, (PolyILs), and combined with functionalization of the surface of ceramic nanoparticles is a promising route for overcoming the high-energy-barrier challenge. Owing to high content of ion-conducting ceramics and high t+ of PolyILs, the fractional contribution of the migrating cationic species to the total ionic conductivity of polymer-in-ceramic electrolytes via an interfacial percolation path, will be close to unity, thus eliminating complications that might arise from emerging concentration gradients during the operation of solid-state batteries.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
Devices for the storage of electrical energy play an important role in our current life, and their importance for the world is recognized by the 2019 Nobel Prize in Chemistry awarded for the development of lithium-ion batteries. 1 The future use of electrical-energy storage in electrical vehicles, renewable energy and portable electronics requires rather revolutionary changes in current battery and supercapacitor technology. 2–4 The development of solid electrolytes might provide the anticipated transformative technological leap enabling the production of solid-state alkali-ion or alkali-metal batteries with a significantly higher energy density and improved safety. 2,4,5 At present, no single candidate material can satisfy all the prerequisites for such a transformative solid electrolyte—high ionic conductivity, combined with good mechanical properties and flexibility, wide electrochemical stability window and good adhesion to the electrodes.
Today, two types of solid electrolytes are explored: inorganic ceramic electrolytes, and polymer electrolytes. Ceramic electrolytes typically have both high ionic conductivities and high lithium transference number (tLi+ ∼ 1). Unfortunately, they are severely brittle and have poor contact with the electrodes. As a result, most of these batteries often fail after a few cycles as a result of dimensional changes accompanying charge and discharge. 5–7 Nevertheless, specific coatings and other modifications 8 to the ceramic surface yield much better results. 9
Polymer electrolytes obviously differ from ceramic electrolytes in their properties. Polymer electrolytes in general have high flexibility and good adhesion to the electrodes, but show low ionic conductivity at room temperature, and in most cases low tLi+ ∼ 0.2–0.5. Considering the vast knowledge of the pros and cons of the two electrolyte families, the obvious route is to disperse ceramic particles in a polymer matrix and thus produce a composite electrolyte that will satisfy most of the requirements of solid electrolytes. 2,4 Indeed, this "the whole is greater than the sum of its parts" approach offers new routes towards safer electrolytes with high ionic conductivity and good mechanical properties. 10
A basic question arises—how compatible structurally and electrochemically are the ion-conducting ceramics with the polymers?
Unfortunately, not much. The poor matching of the two phases that differ in structure and differ in properties leads to high interfacial-energy barriers to ion transport in composite materials, consequently rendering them ineffective as electrolytes. 11–13 Despite several promising solid electrolyte composites, 14–18 there is no clear understanding of what controls ion transport through the polymer-ceramic interface, and how to increase the overall ion transport. 19 In addition, the cation transference number in composite materials is also low. 17,20–22 The studies addressing anode/electrolyte and cathode/electrolyte interfaces are beyond the scope of this manuscript.
Background and Discussion
Solid electrolytes
Ionic conductivity in inorganic solids is caused by the existence of microscopic defects or disorder. 23 In other words, a perfect crystal of an ionic compound would be an insulator. The crystalline structures that permit fast ion transport are generally disordered, channeled, or layered, 24 all having reduced ion-hopping-activation barriers. 25–27 Despite the fact that cations, as well as anions, can move in the solid lattice, the mobility of cations is generally favored because of their small size. Many of the known superionic solids are cationic (e.g., Li+, Na+, and K+) conductors. Therefore, inorganic solid-state electrolytes are considered as classical single-ion conductors. The apparent activation energy (Ea) for ion conductivity contains contributions from both the defect-formation energy, Ef, and the migration energy, Em. 28 For example, the main mechanism of ionic conductivity in crystalline lithium germanium phosphate (LGP) is the Li+ ion migrating through crystallites and jumping between them, thus creating a percolation path (Fig. 1).
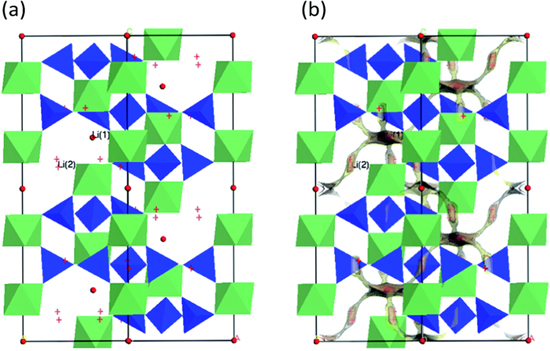
Figure 1. Schematic presentation (a) of the LiGe2(PO4)3 structure (GeO6, green; PO4, blue, Li(1): red spheres, and Li(2): red crosses), and (b) bond-valence site energy-pathway model for the migration of Li+ ion in crystalline LGP. Yellow iso-surfaces represent structural regions that Li+ ions can reach with an activation barrier of 0.35 eV. Reproduced from Ref. 10 with permission of The Royal Society of Chemistry.
Download figure:
Standard image High-resolution imageSince solid electrolytes are typically polycrystalline, the presence of grain boundaries syphons the ion-conduction path to either "through" the bulk (when the surface resistance to ion conduction is greater than that of the bulk) or "on-top" of the crystallites, i.e., the surface (when the surface resistance is reduced as a result of high concentration of defects). Therefore, controlling the grain-boundary morphology, in the hope of achieving low Ea for cation hopping is important in the study of practical solid-state electrolytes and remains an area of intensive research. 7 Naturally, there is a connection between the material's crystalline order and its ion conductivity; therefore, considerable efforts have been focused on several candidate solid lithium electrolytes having different crystal structures, mainly from the following families: LISICON-like (lithium superionic conductor), argyrodites, garnets, NASICON-like (sodium superionic conductor), lithium nitrides, lithium hydrides, perovskites, and lithium halides. 5,28–32 In addition, researchers have been trying to increase the overall ion conductivity of each solid-electrolyte family by inducing structural defects and compositional doping to the bulk or by designing coatings 8,33 for the crystallites. These engineered interfaces have led to considerable success 9 and their study, or rather lack of it, is the focus of this review.
Contrary to common beliefs on the poor ionic conductivity of solid-state electrolytes, the argyrodites, thio-LISICON and Li10MP2S12 (LMPS, M = Si, Ge, Sn) almost reach the values of ubiquitous liquid electrolytes (e.g., ethylene carbonate/dimethyl carbonate with 1 M LiPF6 ≈ 10−2 S cm−1).
Lithium argyrodites Li6PS5X (with X = Cl, Br, or I) are newly discovered fast lithium-ion conductors. This family of solid electrolytes, as reported in Ref. 10, can reach ionic-conductivity values as high as 7 × 10−3 S cm−1. The electrochemical stability windows of Li6PS5X (X = Cl, Br, I) argyrodite compounds are very wide (0−7 V vs Li/Li+). 11 Li1.3Al0.3Ti1.7(PO4)3 has the highest bulk conductivity (σ ≈ 3 × 10−3 S cm−1) reported to date for NASICON lithium-ion conductors at room temperature. In addition, NASICON-like conductors are typically stable in air and water and are stable at high potentials. However, similar to perovskites, titanium-containing compounds can be reduced at low potentials. 13 The highest lithium-ion conductivity in the perovskite family was found for Li0.34La0.56TiO3 with a total lithium-ion conductivity of 7 × 10−5 S cm−1 and bulk ionic conductivity of 10−3 S cm−1. 7,13 Despite their high ionic conductivity and stability at high potentials, lithium lanthanum titanates (LLT) are unsuitable for use with lithium and graphite negative electrodes, since the LLT electrolyte is reduced at potentials ∼1.5 V vs Li/Li+.
Perovskite-based solid electrolytes have the lowest ion conductivities (in both bulk and grain boundaries) compared with other families. Moreover, the perovskite materials present further challenges as they require high-temperature sintering (where Li2O loss is an issue) and they are unstable with respect to metallic lithium as the Ti4+ cations are reduced upon contact, and therefore filler layers (thin polymer films) are placed between the lithium and solid electrolyte. 12,16,21,34 Recently, Li et al. suggested inducing periodic dislocations between the solid electrolyte and the electrodes in order to reconcile any intrinsic structural differences at the interface of the two. 35 Growing thin films, with a designed lattice mismatch (via epitaxial-growth method) leads to better contact between the solid electrolyte and the electrode since it can tolerate a large lattice mismatch. This paradigm paves the way to multilayered electrodes and perovskite solid electrolytes design. Moreover, researchers noted that thin-film ceramic electrolytes that are typically produced by vacuum evaporation or sputtering, provide a different structure, morphology and composition compared to thermally annealed bulk electrolytes. 36
Whatever the growth method, researchers strive to increase and ensure the contact between the solid phases (electrolyte and electrode) upon cycling. Bad contact between the two phases typically leads to sluggish charge transfer as a result of high and unstable interfacial resistance across the solid electrolyte/electrode interface, and low currents (if any) are measured. The origins of high interfacial resistance 22 can be traced to one or more of the following factors: poor physical interfacial contact, mechanical failure of the contact with volume variations, formation of lithium- or sodium-depleted space-charge layers as a result of the large chemical-potential difference between ceramics and electrode materials and degradation at the interface that is caused by mutual diffusion of elements and/or reactivity. Though it was expected that solid electrolytes would be impervious to lithium-dendrite-induced failure by virtue of their mechanical rigidity, recent reports have demonstrated the ability of metallic Li to penetrate solid materials and call for the reexamination of the dendrite-formation mechanism in solid electrolytes. 22,33 Two comments here: (i) one must make a solid electrode/electrolyte composite to "ionically wire" the electrode (in an ordinary LIB, the liquid electrolyte soaks the electrode). Sometimes this leads to unwanted side reactions when the composite is formed at high temperature; (ii) dendrites have been reported to grow along and around grain boundaries.
Polymer electrolytes
Polymer electrolytes are also promising candidate materials for solid-state batteries. One of the first polymers studied as electrolyte was poly(ethylene oxide) (PEO). Wright and co-workers were the first to measure the ionic conductivity of PEO-salt complexes in the early '70 s. 37 To date, PEO-based polymer electrolytes have been regarded as one of the most suitable electrolytes for lithium batteries, as shown by the intensive research of Armand and co-workers 38–41 and Bruce and co-workers. 42 Still, cationic conductivity at room temperature in polymer electrolytes remains far below the required level, and the cation transference number in most candidate materials remains low. 43–46 Polymer electrolytes (PE) have multiphase structures at microscopic and/or macroscopic levels. They consist of intricately distributed amorphous and various crystalline complexes of PEO and Li+, which makes ion transport very complex. A variety of relevant transport mechanisms such as the intrachain-hopping motion of cations through the formation of a weak coordination shell between Li+ ions and ether oxygens (EO), free ion motion along percolating channels in PEO melt, interchain ion hopping (between different molecular chains or segments) of PEO, etc. (Fig. 2). Most researchers agree that high ionic conductivity requires an ultra-fast segmental relaxation 47 and, according to some estimates, the required rate of segmental relaxation is not achievable in dry polymer electrolytes at ambient temperature. 48 Although it is possible to decouple the ion transport from the segmental dynamics, 49 the ionic conductivity achieved at ambient temperature remains far below the required ∼1 mS cm−1. 50–55
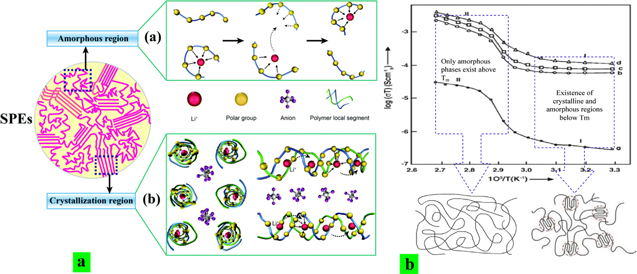
Figure 2. Schematic representation of ion-transport mechanism in amorphous and semicrystalline polymer electrolytes (2a). Reproduced from Ref. 56 with permission of The Royal Society of Chemistry. (2b) Temperature dependence of dc conductivity of (a) pure PEO, (b) (PEO:NaClO3) (90:10), (c) (PEO: NaClO3) (80:20), and (d) (PEO: NaClO3) (70:30). 57
Download figure:
Standard image High-resolution imageIn Ref. 58 blending of plasticized poly(vinylidenefluoride-co-hexafluoropropylene) (PVdF-HFP) with poly(ethyl methaacrylate) (PEMA) was suggested for the enhancement of conductivity of polymer electrolytes. To improve the conductivity of lithium in polystyrene-TFSI (PS-TFSI) polyIL, Bouchet et al. synthesized copolymers of PS-TFSI with PEO and obtained a single-ion BAB triblock copolymer. This combination yielded a copolymer with a low Tg resulting in a polymer electrolyte with improved Li-ion conductivity, albeit only at high temperatures, 1.3 × 10−5 S.cm−1 at 60 °C. The copolymer paradigm, slowly but steadily increases the room-temperature polymer-electrolyte conductivity to the essential ∼1 mS cm−1 threshold. 59–62
Polymerized ionic-liquid electrolytes
Recently, a relatively new class of polymer electrolytes - polymerized ionic liquids - (polyILs), has attracted significant attention. PolyILs are essentially single-ion conductors the counterion of which is attached to the polymer chain, providing a significant advantage in electrochemical devices. 41,63–65 Usually polyILs with Li+ as the mobile ion have a transference number close to unity, although their high glass-transition temperature (Tg) leads to a low conductivity under ambient conditions. 41,64 Recently, we succeeded in synthesizing PolyILs with a very flexible siloxane backbone, attached the TFSI anion and short PEO side chains. 66 This PolyIL reached a Li+ conductivity of about 10−5 S cm−1 at room temperature. 67 Poly(ethylene-co-acrylic lithium (fluoro sulfonyl imide) (PEALiFSI), with acrylic (fluoro sulfonyl)imide anion (AFSI) single-ion conducting polymer electrolyte (SICPE) synthesized in Ref. 61, exhibited remarkably high Li+ conductivity (5.84 × 10–4 S.cm–1 at 25 °C). Detailed studies of the mechanism of ionic conductivity in PolyILs 68,69 revealed that electrostatic and elastic forces affect the transport of mobile ions. However, electrostatic interactions dominate the energy barrier in the case of small cations, such as Li+ or Na+. Thus, in order to increase their conductivity, a significant decrease of electrostatic interactions is required. 48,68,69 This can be achieved by either a strong increase in the dielectric constant of the polymer or stronger delocalization of charge on the anion. 41,48 It has been also discovered that ion-ion correlations lower, by almost an order of magnitude, the overall conductivity in PolyILs. 68,69 This is in stark contrast to the superionic systems discussed above, in which ionic correlations enhance conductivity by a factor ∼3–5. The mechanism of ionic correlations in PolyILs remains a puzzle, 48,69 but reversing these correlations can additionally increase their conductivity by a factor of more than ten.
Composite electrolytes
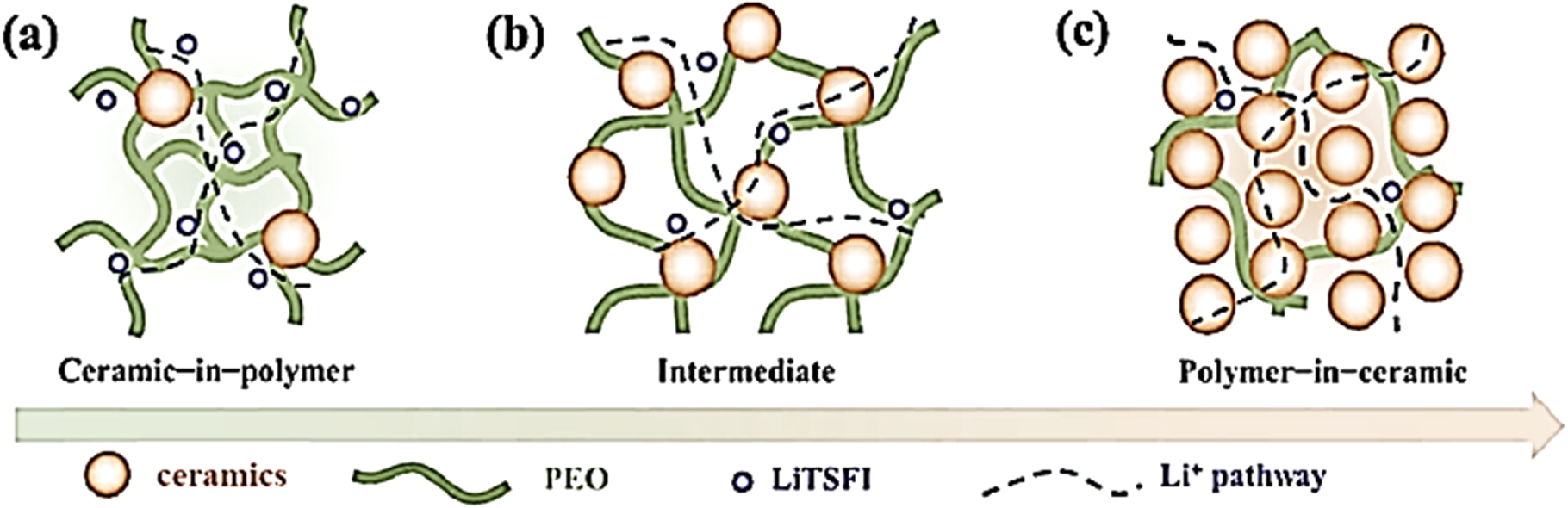
A typical design of a solid composite-electrolyte utilizes the salt with a polymer as the flexible binder or host. Two types of ceramic fillers can be considered for composite electrolytes—inert and active. The former have room-temperature (RT) conductivity much lower than that of polymer electrolytes. Examples are alumina, silica, lithium aluminate, barium titanate, etc. The latter are the ceramic fillers, which have RT ionic conductivities much higher than that of polymers, like ultra-thin layers of LIPON, argyrodites, LISICON, NASICON, etc. The composite electrolytes with host polymer as the main conducting medium show temperature-dependent lithium-ion-conductivity behavior, similar to that of polymer electrolytes. In composites with ceramic fillers as the main conducting medium and polymer as the binder, different high- and low-activation-energy conduction paths are distinguished. 13,36,70–74
The ceramic-in-polymer system is the main trend in composite-electrolyte studies because of its easier film processability. Ceramic-in-polymer electrolytes (CPE) with relatively low ceramic loading have higher room-temperature conductivities (10−5 to 10−6 S cm−1) compared to polymer electrolytes without ceramic filler (10−7 to 10−8 S cm−1). The common explanation of this phenomenon is that the addition of a few percent of ceramic particle fillers hinders polymer crystallization, enabling segmental relaxation in amorphous parts and therefore higher ionic conductivities. Kumar and Scanlon 75 underlined the need for careful matching of the amorphous phase volume and change of glass transition point (Tg) in CPEs. Scrosati et al. 34,76,77 and other research groups, including ours, 78,79 found improved conductivity, decreased degree of crystallinity and low and stable Li/CPE interface (SEI) resistance for the PEO-based electrolytes containing high-surface-area, inert ceramic fillers, such as SiO2, Al2O3, MgO, TiO2 montmorillonite, and metal-organic frameworks (MOFs). 80–82 We have demonstrated that ion-transport mechanisms differ in CPEs containing low and high content of lithium salt. 15 Two different Arrhenius dependencies were identified. The first one is characterized by the inflection point and the second by a conductivity jump. Both occur at temperatures close to the melting point of the PEO-salt eutectic. Noticeable contribution of crystalline phase and interfacial conduction was mentioned in concentrated CPEs with EO-to-Li salt ratio lower than 6:1. However, the prevailing concentration of the polymer medium in CPEs with inert ceramic fillers governs slow room-temperature migration and diffusion of lithium cations.
Active ceramic fillers used in polymer electrolytes include single-ion conductors of LISICON-type (Li14Zn(GeO4)4), NASICON-type (such as Li1.5Al0.5Ge1.5(PO4)3 [LAGP] and Li1.3Al0.3Ti1.7(PO4)3 [LATP]), perovskite-type (Li3La2/3−xTiO3 [LLTO]), garnet-type (Li7La3Zr2O12 [LLZO] and others. While active ceramic fillers are also expected to prevent formation of crystalline phases in composite electrolytes, to facilitate the dissociation of lithium salt and provide highly efficient pathway for lithium ion transport in CPEs with low-content of active ceramic component the major conduction pathway is via the host polymer, similarly to the inert-ceramic-filler electrolytes. Consequently, the conductivity of ceramic-in-polymer composite electrolytes is still orders of magnitude lower than that of high-temperature sintered superionic ceramics. In addition to the drop in ion conductivity at low temperatures, such composite electrolytes have low lithium transference number, again controlled by the polymer electrolytes. Zagórsky et al. showed that for 10 vol% LLZO microparticles the total lithium-ion conductivity of the PEO-LiTFSI system is 4.5 × 10−4 S.cm−1 at 70 °C, close to that of filler-free electrolyte. 83 This is caused by the high interfacial resistance (∼104 Ω cm2) between the garnet particles and the PEO(LiTFSI) matrix.
Naturally, the structural incompatibility and adherence of the polymer to the ceramic filler (and vice versa) affects the overall conductivity and performance, thus the engineering of the polymer/ceramic interface to promote stronger interactions between these two components 18 should be one of the focuses of future studies.
The polymer-in-ceramic approach utilizes a predominant ceramic scaffold to which a conductive polymer is introduced. These composite electrolytes contain high concentrations of ceramics (ion-charge carriers) with minimal quantities of polymer. The major ion-conduction path is assumed to be through the ceramic material and its interface, yielding high values of conduction. The question arises if both inert and active ceramics are capable of creating low-energy conduction paths, or this is a property of the active ceramics alone ? In addition, researchers are now concentrating on how to reduce the volume fraction of the polymer in the composite electrolyte to further increase its ionic conductivity without losing its flexibility and efficient electrode/electrolyte contacts. In Fig. 3 we illustrate the cation conduction paths in composite electrolytes with (a) low loading of ceramic-in-polymer, (b) intermediate loading and (c) high loading of ceramic material (polymer-in-ceramic). The second, (b), and the third case, (c), account for the active ceramic filler.
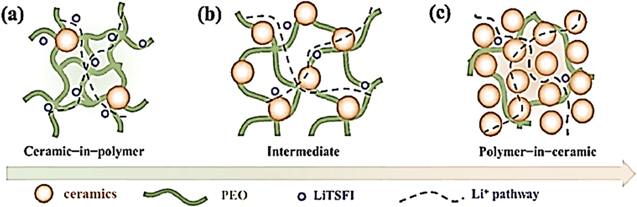
Figure 3. Schematic illustration for PEO-ceramic composite solid electrolyte. Reproduced with permission of Ref. 72 Copyright (2018) Elsevier.
Download figure:
Standard image High-resolution imageWe call the reader's attention to the fact that the use of a smaller volume fraction of polymer electrolyte raises the importance of the interfacial-energy barrier for cations crossing from superionic particles (ceramic fillers) to the polymer. Unfortunately, the polymer/ceramic interface remains barely studied and warrants intensive investigative efforts. Therefore, we are still puzzled on how one can reduce the interfacial barrier for Li+ and Na+ transport. Despite rigorous studies, we are still practicing screening methodologies rather than studying pre-designed material, leading to the somewhat sporadic choice of the polymer electrolyte and surface functionalization of the ceramic particles. It is clear that the volume fraction of the ceramic phase in the polymer phase significantly affects the overall performance of the composite solid electrolyte. 34 For example, Goodenough et al. varied the wt% of a garnet, Li6.4La3Zr1.4Ta0.6O12 (LLZTO), as the ceramic in PEO (and additional short PEG as plasticizer). They reported that the highest conductivity of about 10–4 S cm−1 at 55 °C was obtained for an 80 wt% ceramic blend, i.e., a polymer-in-ceramic electrolyte. 72
Table I presents the compositions and bulk conductivity values of some solid electrolytes described above.
Table I. Composition, structure and bulk ionic conductivity at ambient temperature of exemplary ceramic and composite electrolytes.
| Type of Electrolyte | Electrolyte Composition | Conductivity [mS cm−1] | References |
|---|---|---|---|
| Inorganic solid electrolytes | |||
| LISICON-like | Li10GeP2S12 | 12 | Kamaya et al. 84 |
| The framework is composed of (Ge0.5P0.5)S4 tetrahedra and LiS6 octahedra | |||
| LISICON-like/LGPS | Li10GeP2S12 | 10 | Kuhn et al. 85 |
| Tetragonal | |||
| Sulphide lithium super ion conductor | Li2S–P2S5 | 17 | Seino et al. 86 |
| Sulphide lithium super ion conductor | Li10Si0.3Sn0.7P2S12 | 8 | Bron et al. 87 |
| Sulphide lithium super ion conductor/LGPS like | Li9.6P3S12 | 12 | Kato et al. 88 |
| Sulphide lithium super ion conductor/LGPS like | Li9.54Si1.74P1.44S11.7Cl0.3 | 25 | Kato et al. 88 |
| Ceramic-in-polymer electrolytes | |||
| 20 wt% γ-LiAlO2 (inert ceramics) | PEO-LiCF3SO3/γ-LiAlO2 | 3 × 10−3 | Croce et al. 89 |
| 20 wt% γ-LiAlO2 (inert ceramics) | PEO-LiBF4/γ-LiAlO2 | 3 × 10−4 | Croce et al. 89 |
| 10 wt% TiO2 (inert ceramics) | PEO-LiClO4/TiO2 | 0.02 | Croce et al. 89 |
| 10 wt% (inert ceramics) | PEO-LiClO4/Al2O3 | 0.01 | Croce et al. 89 |
| 10 wt% BaTiO3 (inert ceramics + (poly[bis(triethylene glycol)benzoate] capped with an acetyl group) as a plasticizer | PEO-LiN(CF3CF2SO2)2/BaTiO3 | 0.13 | Itoh et al. 90 |
| 10 wt%MOF | PEO-LiClO4/Zn4O (1,4-benzenedi-carboxylate)3 MOF | 0.03 | Yuan et al. 80 |
| 12% v/v LLZTO (active ceramics) | PEO/Li6.4La3Zr1.4Ta0.6O12 | 0.2 | Zhang et al. 91 |
| 40 nm particles | |||
| 5 wt% nanosize LLZTO (active ceramics) | PPC-LiN(CF3CF2SO2/Li6.4La3Zr1.4Ta0.6O12 | 0.52 | Zhang et al. 92 |
| 15 wt%LLTO nanowires (active ceramics) | PAN- LiClO4/Li0.33La0.557TiO3 | 0.24 | Liu et al. 16 |
| Polymer-in-ceramic electrolytes | |||
| 40%v/v Al2O3 (inert ceramics) | PEODME500 1 M lithium perchlorate/alumina porous substrate | 0.01 | Syzdek et al. 93 |
| 75%v/v LiAlO2 (inert ceramics) | LiI-PEO/LiAlO2 | 0.5 | Blanga et al. 13 |
| 75–80 wt% LiISPS (active ceramics) | Li10+xIxSnP2S12-P(EO)-LiI composite with PEO polymer | 0.1–0.3 | Menkin et al. 74 |
| LLTO nanofiber mats (active ceramics) | PEO-LiTFSI/Li0.33La0.557TiO3 | 0.16 | Liu et al. 94 |
| 60 wt% LLZTO (active ceramics) | PEO-PEG-LiTFSI/LLZTO | 0.1 | Chen et al. 72 |
Ion-diffusion pathways
Researchers are still debating on how the ceramic phase governs the properties of the composite electrolytes. For example, is it the shape of the ceramic, its porosity, the acidity of the ceramic's surface or none of the above? Some researchers suggest that lithium-ion hopping occurs in a sequential manner on the surface of the ceramic fillers. 95–97 Wieczorek et al., 98 suggest that Lewis acid-base interactions are the reason for the enhanced conductivity. Cui et al. found that the shape of the ceramic filler, here in the form of high-aspect-ratio nanowires, creates long-range Li-ion channels that improve the conductivity of the composites. 16 In a recent work, Cui et al., showed that they can further enhance the ionic conductivity to 6.05 × 10−5 S.cm−1 at 30 °C in composite polymer electrolytes by aligning the ceramic nanowires. 99 Goodenough et al. suggested that the continuity of the ceramic phase is important as it forms a continuous percolating medium. They produced a composite electrolyte from a 3D nanostructured hydrogel‐derived Li0.35La0.55TiO3 (LLTO) as the ceramic framework and PEO‐LiTFSI. They achieved a Li-ion conductivity close to 10−4 S cm−1 at room temperature which they ascribed to percolation. 73 Wachsman et al. suggested the importance of controlling the pore size of the ceramic framework (scaffold) according to the desired function. Porous outer layer for electrode contact, dense inner layer for Li+ conduction will serve multiple functions, resulting in a low-resistance and mechanically strong structure capable of high-rate lithium cycling. Indeed, they demonstrated the use of microstructuring by a roll-to-roll technique to produce a porous-dense-porous trilayer composite electrolyte structure of doped Li7La3Zr2O12 (LLZ) ceramics with enlarged surface area (×40 of planar) and enhanced mechanical strength (×9 of planar) and tested it in a solid-state Li–S battery. 100 They reported cycling up to 10 mA cm−2 with low area-specific resistance values between 2 and 10 Ω-cm2 that are comparable to ∼22 Ω-cm2 reported for the total cell resistance of a commercial Li-ion battery. 101
The "enigmatic" interface
We are confident that an understanding of the mechanisms controlling the interfacial ion dynamics 33 is critical for the development of all-solid-state batteries 5 with metallic lithium anodes and composite electrolytes having a high ionic conductivity, to compete with current state-of-the-art liquid-electrolyte Li-ion batteries. 9 In either polymer-in-ceramic or ceramic-in-polymer electrolyte, the interfacial properties can enhance the conductivity of the composite electrolyte, while in most studied cases they suppress the ion transport between the ceramic phase and polymer matrix. 19 Therefore, it is imperative to comprehend the mechanisms governing interfacial ion dynamics, if we are to develop composite electrolytes with high ionic conductivities under ambient conditions.
Researchers are debating as to the fastest ion-diffusion pathways: on the ceramic surface, at the polymer-ceramic interface, through the ceramic bulk or via the polymer. Zheng et al. reported that Li+ ions favor the pathway through the LLZO phase instead of the PEO‐LLZO interface or PEO. 102 They reached this conclusion after they applied solid‐state Li NMR to monitor the replacement of 7Li in the composite electrolyte by 6Li from a 6Li metal electrode while cycling. On the other hand, Mustarelli et al. suggested that the major/fastest ion pathway is through the amorphous polymer. They studied poly(ethylene oxide)/Li1.3Al0.3Ti1.7(PO4)3 (PEO/LATP) electrolytes with LiTFSI containing up to 70 wt% ceramics and reported 4 × 10−5 ohm−1.cm−1 at room temperature. They concluded that increasing the polymer's amorphous fraction at room temperature will result in an overall higher composite electrolyte conductivity at low temperatures. 36 They did not detect any contribution from the polymer-ceramic interface.
Money et al. 103 suggested that the interface can enhance the ionic conductivity of the composite PEO4:LiClO4 polymer electrolyte by lowering the relaxation time of the polymer phase. They showed that the ionic conductivity of the electrolyte containing 4 wt% δ-Al2O3 is coupled with the structural relaxation time of the polymer. Fullerton-Shirey and Maranas examined the structure and mobility of PEO/LiClO4 solid polymer electrolytes embedded with Al2O3 nanoparticles at certain eutectic compositions. The acidic filler (α-Al2O3) was found to be more effective at increasing the conductivity at non-eutectic compositions. However, once at the eutectic composition, the surface chemistry has no apparent effect. 104,105 A mechanism was proposed in which the alumina nanoparticles stabilize the PEO6 structure at their surface. The different surface chemistries govern the extent of stabilized PEO6 resulting in either enhanced or decreased Li+ movement in the structure.
The surface chemistry of the ceramic filler was also studied by Ganesan et al. who applied simulations of atomistic molecular dynamics to examine ionic diffusivities and segmental dynamics of a PEO-LiBF4 system containing either dispersed TiO2 105 or Al2O3 106 nanoparticles. They discovered that the segmental dynamics of the PEO chains near the particle surface are significantly hindered as a result of the preferential interactions between the polymer segments and the surface. This applies to both "salt-free" and "salt-doped" systems. Therefore, increasing the loading of the nanoparticles will reduce the ionic mobilities and conductivities compared to pure PEO electrolyte. The conduction mechanism was explained according to the Lewis acid–base theory. Wieczorek et al. proposed that the acidic sites on the alumina surface attract the anion ClO4 − in turn freeing more Li+ from the ion pairs, thus enhancing the ionic conductivity. 98 The immobilization of the polymer chains near the ceramic surface was also noted by Dudney et al. 19 In the presence of "active" Ohara ceramic (Li1+x+yAlxTi2−xSiyP3−yO12), both the segmental mobility of the PEO polymer and the intrinsic ionic conductivity of the polymer phase (σPE) decreased by 60% and 30%, respectively, compared to that in the neat polymer electrolyte. The authors hypothesize that the affinity between the Ohara surface and Li+ causes the PEO chains near the Ohara ceramic surface to become less mobile because of coordination bonds of the PEO chains with the surface-bound Li.
We have recently conducted a comparative study of the inert and active ceramic matrix with imbedded LiI:P(EO)n electrolytes 74 (Fig. 4).

Figure 4. Arrhenius plots of ion conductivity of LSPS:LiI:P(EO) and LAO:LiI:P(EO) electrolytes, containing 75%–80% of ceramics. Reproduced with permission of Ref. 74 σ1-the conductivity of solid electrolyte calculated from the resistance of high-frequency intercept of the arc with the X-axis in typical Nyquist plot, and σ2 is calculated that from the resistance of the arc. The interpretation of the impedance spectrum, based on equivalent-circuit-type models, is discussed in detail in Ref. 74.
Download figure:
Standard image High-resolution imageWe suggest that the high-ionic-conductivity (0.5 mS cm−1) and low-activation-energy (2.3 kJ mol−1) ion−1 paths are created by the grain boundaries between the excess of LiI and inert LiAlO2 ceramic nanoparticles. Both confined-in-ceramic polymer electrolyte and ceramic LiAlO2 grains impede the total ion mobility. The fast ion transport in polymer-in-ceramic electrolytes composed of high-conductivity active Li10SnP2S12, goes through lithium-iodide-rich glass ceramics, and is restricted by slow ion transport via the imbedded polymer electrolyte. Unexpectedly, it was found that at 1:3 salt-to-polymer ratio, the contribution of grain-boundary conductivity in an inert-ceramic-based composite electrolyte is stronger than that of bulk conductivity via active ceramic matrix. One of the possible reasons for the reduced relative contribution of the active ceramics to the total conductivity of polymer-in-ceramic electrolyte is that the ceramic powder was not densified.
To conclude, despite strenuous efforts involving the preparation of highly amorphous polymers that are blended with modified ceramic fillers, the maximum conductivity of composite polymer electrolytes reported thus far remains at about 10−5 S.cm−1 with some unique studies where it approaches 10−4 S.cm−1 at room temperature. It is evident from the literature that researchers have identified the need to lower the polymer/ceramic interfacial resistance to improve the conductivity of the composites. Yet, what modification should the ceramic surface undergo remains an open question.
Our survey of the literature did not find any publications on composite electrolytes with use of PolyILs as a polymer component. One apparent reason could be that most of the researchers working with composite electrolytes have limited experience with polymer synthesis, and all the studies are based on commercially available polymers. As we discussed above, PolyILs are essentially single-ion conductors that provide significant advantages for use in batteries. 41,48 It is possible that using PolyILs will lead to significant improvements in the conductivity of composite electrolytes. Polymers with salt traditionally used in composite electrolytes have dual ionic conductivity. Combining these dual-ion conductors with active ceramics that are single-ion conductors might face a significant problem of gradients in ion concentration and polarizations. This might be one of the major contributions to the interfacial energy barrier for ions crossing from active ceramic to polymer and back. We suggest that the use of PolyIL with single Li+ (or Na+) ion conductivity in composite electrolytes might significantly reduce the interfacial-energy barrier and result in much higher conductivity.
Characterization techniques for the interfacial properties of composite electrolytes
The migration of ions in composite electrolytes is a multiscale process consisting of mechanisms that are manifested at different length scales, from the atomic scale up to the entire thickness of the film. Importantly, the final impedance of the composite electrolyte is a function of all these different mechanisms. The techniques that can be used to probe ion conduction at various scales are diverse and are often limited in their space or time resolution, making multi-technique approaches imperative for successful interpretation. 45 In this section, we lay out a span of techniques generally applied to interfacial phenomena 107 and Li-ion transport mechanisms in studies of composite electrolytes and their latest achievements. No characterization technique is omnipotent. Therefore, we encourage the incorporation of several surface-analytical techniques, such as time-of-flight secondary-ion mass spectroscopy (TOF-SIMS), scanning electron microscope (SEM), X-ray powder diffraction (XRD), X-ray photoelectron spectroscopy (XPS), as well as vibrational spectroscopies because of the complex interphase. 108
Relevant to composite electrolytes, Zagórski et al. 83 used solid-state NMR (ssNMR) to understand the mobility of Li+ ion in LLZO−PEO(LiTFSI) and the interaction between the composite components. The authors observed that the TFSI− anion interacts mainly with the polymeric chains. The authors implemented a 7Li 2D exchange spectroscopy (EXSY) NMR experiment, together with electrochemical methods to prove the existence of lithium exchange between the garnet and the polymer-electrolyte phases.
Zheng et al. 109 studied the LGPS−PEO(LiTFSI) composite-electrolyte system by high-resolution solid-state magic-angle-spinning (MAS) 6Li NMR Here, the authors tracked an isotope exchange, 6Li → 7Li, in a symmetric cell made of 6Li/7LGPS-PEO (7LiTFSI)/6Li. The authors observed an increase in the amount of 6Li on both LGPS-PEO interfaces and in the LiTFSI. They suggested that this may indicate that the main Li+ ion conduction is through the interface. To corroborate the conclusion that the lithium-ion transport is mainly through the LGPS-PEO interfaces, the authors performed conductivity measurements, which indeed correlated with the amount of LGPS in the composite electrolyte. Furthermore, the authors studied the formation of interfaces as a function of the concentration of LiTFSI and the mixing methods (ball-milling or stirring) of LGPS and PEO(LiTFSI). They found that both parameters affect the formation of the high-conductivity interface.
Chen et al. 19 used quasi-elastic neutron scattering (QENS) to study the influence of Li ion conducting ceramic (Ohara) on the segmental movement of a polymer electrolyte (PEO-LiTFSI) in a composite solid electrolyte. The authors noticed that a sample containing only polymer and a sample containing both polymer and ceramic, showed very similar relaxation times, thus suggesting that in the salt-free system, the segmental mobility of PEO is almost unaffected by the addition of Ohara ceramic. By contrast, adding ceramic particles to a system containing LiTFSI, resulted in slower segmental mobility of PEO. The authors also carried out conductivity measurements and detected a decrease in the conductivity of the samples following the addition of the Ohara ceramic. They concluded that the interfacial interactions between the PEO and Ohara surface determine the lithium-ion transport properties. Temperature-dependent 7Li and 19F NMR line shape analysis and spin−lattice relaxation (T1) measurements were used to examine the chemical environment and dynamics of Li+ and Tf− in PEO−LiTf polymer electrolyte and LICGC−PEO−LiTf composite electrolyte. 110 Because of the presence of LICGC (OHARA) ceramic, DSC results reveal a slightly decreased Tg and Tm of CPE compared to those of PE. The line-shape analysis and spin−lattice relaxation characterization of 7Li and 19F suggest that both the cation and the anion in PE are composed of two components: a mobile component and an immobile component. The ions corresponding to the broad component of NMR spectra with smaller T1 are largely immobile, while ions showing a narrow line width and much larger T1 are associated with the more mobile ion components. At temperatures below the melting point of PEO and much above Tg, a large ratio (>70%) of immobile Li+ and Tf− was observed in PE, suggesting that in the semicrystalline state, the ion mobility is controlled by the presence of the crystalline regions rather than by the Tg. Above the melting temperature of PEO, the amounts of mobile cations and anions increased significantly. In the presence of LICGC ceramic, an increased ratio of immobile Li+ and Tf− and reduced mobility of the mobile components were observed, indicating that the ceramic had a negative impact on both the cations and the anions. The NMR characterization was corroborated by conductivity results, which revealed that the intrinsic ionic conductivity of the polymer phase in CPE was only between 0.15 and 0.52 compared to that of PE. This issue appears to be endemic to polymer/ceramic composites and is attributed to interfacial resistance between the polymer and ceramic surface.
Relevant to our topic, Park et al. 111 used TOF-SIMS to study the LiCoO2/LLZO cathode/electrolyte interface. With TOF-SIMS, the authors were able to obtain elemental maps showing the ionic distribution, therefore observing the diffusion of several elements between the two layers. Blanga et al. 70 used TOF-SIMS to determine the lateral distribution of the polymer in composite films. They concluded that the polymer coating is homogeneous throughout the components of the composite.
Wang et al. 112 used FTIR spectroscopy to investigate chemical complexation between the two phases of ceramic LATP and polymer PEO-LiClO4 nanocomposite electrolyte films. They found indications of complexation between PEO-LiClO4 but not for LATP-(PEO-LiClO4) complexes.
Xu et al. 113 wanted to improve the solid-electrolyte/electrode interface and characterized the system by Raman spectroscopy. The authors studied a Li7La3Zr1.5Ta0.5O12 (LLZT) liquid-electrolyte hybrid system with addition of n-BuLi, a superbase, to suppress the possibility of Li+/H+ exchange. Jurado-Meneses et al. 114 studied the intra- and inter-polymer chain interactions in the (PEO)10CF3COONa+Al2O3 composite system. They monitored these interactions by Raman spectroscopy with varied Al2O3 concentration. The increase in amorphous-phase fraction of composites (PEO)10CF3COONa + x wt.% Al2O3 evidenced by changes in the intensity and broadness of IR and Raman bands, and by loss of the intensity in XRD diffractogram peaks was found to improve the conductivity by migration of Na+ ions through the pathways of amorphous phase surrounding the filler. However, Al2O3 concentrations greater than 3% resulted in a loss of ionic conductivity due to the blocking effect of filler particles, which hinders the motions of mobile ions.
Dam et al. 97 studied the DC conductivity and relaxation phenomena in PEO20-LiCF3SO3-ZrO2 nano-composite electrolyte. The authors suggested that the DC conductivity is affected by the polymer's segmental relaxation since it followed the Vogel-Tamman-Fulcher (VFT) behavior. The authors observed a frequency-independent plateau over a frequency range attributed to dc conductivity. In addition, through BDS, the authors could separate segmental relaxation from conductivity relaxation. This indicates that ion and segmental dynamics have different time scales. Detailed studies of segmental relaxation (by rheology) and conductivity relaxation (by BDS) in several PolyILs revealed that both exhibit VFT behavior although their characteristic time-scales may differ by more than six orders of magnitude. 67,97 The latter emphasize strong decoupling of ion dynamics from motions of polymer segments. Thus, VFT behavior of conductivity or conductivity-relaxation time does not necessarily imply coupling of ion and polymer dynamics, as many authors assumed. It just indicates that the energy barrier for conductivity varies with temperature. 48
For lithium aluminate-based polymer-in-ceramic electrolytes we have recently observed the complex non-Debye dielectric response (Fig. 5a), which can be described in terms of several distributed relaxation processes, separated by different frequency and temperature ranges and marked as 1–4. The low-temperature relaxation process (#1, below 280 °K) can be described by the Cole-Cole function. It reflects the interaction of Li+ ions with the matrix of the composite. Process #2 shows the structural changes in the sample including the observation of the PE melting point at temperatures close to 345 K. Process #3, which is associated with the percolation path, taking place at ambient temperatures, proves low-activation-energy, high-rate interfacial mobility of lithium cations. This percolation process has been observed in two samples. The first one is neat lithium aluminate (LiAlO2) pressed powder and the second contains 80% LiAlO2, 20% polyethylene oxide and LiI salt with a LiI:PEO ratio of 1:2. The amplitude of this process essentially decreases with increase in frequency and the position of maximum dielectric permittivity has almost no temperature dependence (See Fig. 5b). Finally, the low-frequency ac-conductivity increases with increasing temperature with S-shape dependency (Fig. 6) that is typical of percolation processes. 115,116

Figure 5. (a) 3D plot of dielectric losses of polymer-in-ceramic electrolyte; (b) Temperature dependence of dielectric permittivity of the polymer-in-ceramic electrolyte at different frequencies. Percolation temperature is marked by red color.
Download figure:
Standard image High-resolution image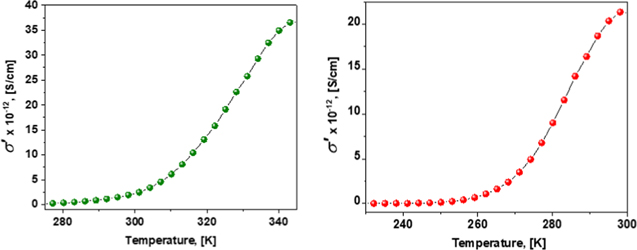
Figure 6. Temperature dependence of ac-conductivity at 0.01 Hz for the LiAlO2 (left) and composite electrolyte (right).
Download figure:
Standard image High-resolution imageThese features suggest that this process can be related to percolation phenomena. 115,117 Such a process was previously associated with the percolation of an apparent dipole moment excitation, within a developed fractal structure of connected pores in different porous systems, such as regular porous borosilicate glasses, 118 zeolites 119 and clays. 120 The discussed samples can be considered as porous materials and/or as confined-in-ceramics polymer electrolytes with non-homogeneous distribution of salt in polyethylene oxide, as presented by us in the past in the FIB-SEM tomography-segmented images Fig. 7. It can be seen that the confined LiI:P(EO)3−x particle-size distribution in LiAlO2 matrix is not uniform (Fig. 7c). The modal radial size of the confined PE entity is about 40 nm (Fig. 6c). For modelling of the polymer-in-ceramic electrolyte, the meshed 3D microstructure was then transferred into COMSOL with four different phases, acquired as: pore (electric insulator), ceramic bulk (the matrix), LiI:P(EO)3 electrolyte and randomly oriented grain boundaries. It was found that the presence of the grain-boundaries between the excess of lithium iodide salt and lithium aluminate, changes the initial potential gradient within the structure. The increase in temperature does not dramatically affect the potential distribution, since the major contribution to the conductivity, the GB conductivity, is not influenced by temperature. Therefore, we associate the percolation relaxation process in the current structures (Fig. 4) with the percolation of a charge excitation within the developed fractal structure of connected pores due to the interfacial migration of the charge carriers. Note that the percolation threshold for LiAlO2,:PEO:LiI electrolyte shifts to lower temperatures. By this is meant that a lower barrier is essential to reach the size of the percolation cluster and the grain-boundaries conduction path dominates.

Figure 7. 3D images of LiI:P(EO)3−x -in-ceramic electrolyte of a 4.5 μm3 volume (a), (b); LiI-PEO Particle Equivalent Spherical Radius Size (c); Electric potential gradient at room temperature (d), (e) and 70 °C (f), (g) of the 3D composite system free-of-GB (d), (f) and containing-2% GB(e), (g). Reproduced with permission of Blanga et al. 121 Copyright (2016) Elsevier.
Download figure:
Standard image High-resolution imageElectrochemical impedance spectroscopy (EIS) is the "bread-and-butter" of any advanced study on composite electrolytes. 122 It is a powerful method of characterizing the electrical properties (ion transport, diffusion, etc.) of both bulk and interface. It is very similar to dielectric spectroscopy and is employed to investigate the dynamics of bound or mobile charge in the bulk or interfacial regions of solid and liquid materials: ionics, semiconductors, mixed electronic–ionic, and even insulators (dielectrics). Impedance is defined as the response of an electrochemical system to an applied potential/current. 123 The general approach is to apply an electrical stimulus (a known voltage or current) to the electrodes and observe the response (current or voltage) that results from the fact that the sample is situated between the electrodes. EIS data are analyzed by fitting the curve to an equivalent-electrical-circuit model consisting of resistors, capacitors and certain electronic-like components. The data can be graphically represented in two convenient ways: the Nyquist plot, in which the negative imaginary part of the impedance, -Im(Z(ω)), is plotted against its real part, Re(Z(w)). And the Bode plot, in which absolute values of log (Z*(ω)) or φ (phase angle) are plotted against the frequency. From the Nyquist plot, the specific electrical resistance can be extracted and the bulk and grain-boundary conductivity values are calculated. Using the Arrhenius equation enables calculation of activation energy for ionic conductivity.
Liu et al. 124 applied EIS to study the interface between an inorganic solid electrolyte, LLZO, and a liquid electrolyte, LP30. The authors used a four-electrode system, which enabled the elimination of the resistance contribution from electrodes, thus were able to measure the Li+ ion transport within the solid/liquid interface. Keller et al. 125 studied the ionic conductivity of LLZO over a range of temperatures. They monitored two systems: a hybrid system of LLZO-P(EO)15LiTFSI (HSE), and P(EO)15LiTFSI solid polymer electrolyte (SPE). From EIS measurements, the authors reported that while at temperatures around 50 °C–80 °C, both compositions exhibit activation-energy values of about 40 kJ.mol−1, at 20 °C, SPE presents the highest conductivity values, thus indicating that the Li+-ion transport is mainly through the amorphous polymer phase. In addition, from their Arrhenius-plot analysis, the authors noted that although the ionic conductivity of LLZO is higher, the SPE presents a higher conductivity value than the HSE. The authors suggested that the incorporation of LLZO results in low conductivity values since they impose higher tortuosity in the polymer matrix, consequently, longer pathways for the Li+ cations to travel. See Fig. 8.
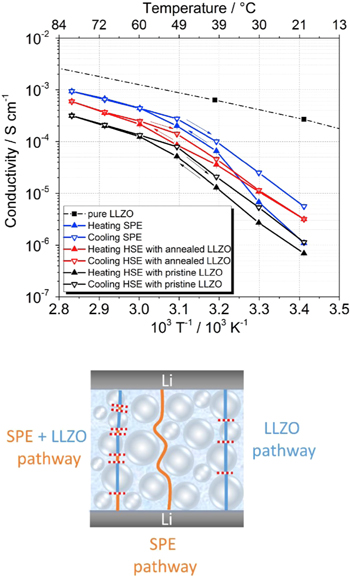
Figure 8. Top: The Arrhenius conductivity vs temperature plot of the SPE and HSE (containing pristine and annealed LLZO) samples. Bottom: schematic model of possible lithium-conducting pathways through the HSE samples. Reproduced with permission from Keller et al. 109 Elsevier (2017).
Download figure:
Standard image High-resolution imageTable II summarizes the major findings gained from studies described in the above section.
Table II. Characterization techniques and ion transport discoveries in exemplary composite electrolytes.
| Technique | Tests | Findings | References |
|---|---|---|---|
| High-resolution solid-state NMR | Examination of the local structural environments of Li+ in different phases | PEO-LiTFSI/LLZO (10–70 vol% ceramics) | Zagórski et al. 76 |
| MAS NMR Study of Li+ exchange between different phases by 7Li | Li ions can locally exchange between the garnet surfaces and the surrounding polymer chains | ||
| Study of conduction pathways by tracking an isotope exchange, 6Li → 7Li | Li-ion transport of the composites is governed by the polymer matrix, as a consequence of the high interfacial resistance between the garnet particles and the PEO-LiTFSI matrix | ||
| PEO-LiTFSI/LGPS (20–90 vol% ceramics) | Zheng et al. 91 | ||
| Li ion conduction is mainly through the LGPS−PEO interface. | |||
| The conductivity enhancement correlates with the amount of LGPS-PEO interface. Maximal conductivity is achieved at 70 vol% LGPS. | |||
| QENS | Study of the effect of interfacial interactions between different phases in the composite solid electrolyte on Li ion transport properties. | PEO-LiTFSI/Ohara (30 vol% ceramics) | Chen et al. 19 |
| Decrease in conductivity of PEO- LiTFSI electrolyte with addition of Ohara ceramic particles | |||
| Slowing down the segmental motion of PEO chains in the vicinity of the ceramics due to coordination with surface-bound lithium ions | |||
| Increase of the barrier for ion transport across the polymer–ceramic interface | |||
| FTIR spectroscopy | Study of the chemical complexation between the different phases in the composite solid-polymer electrolyte. | PEO-LiClO 4 /ceramic filler (5–20 wt.%): LATP, TiO2 and SiO2 | Wang et al. 94 |
| LiClO4 forms complexes with PEO, but LATP does not. One to two orders of magnitude enhancement in ionic conductivity by LATP is attributed to cation transport in the interphase region surrounding the particles. | |||
| Raman spectroscopy | Identification of ionic species. Study of the intra-chain and inter-chain interactions in the composite solid-polymer electrolyte. | PEO-LiCF 3 SO 3 /OIC (40–70 wt%) | Joo et al. 126 |
| OIC- organic–inorganic composite based on 3-glycidyloxypropyl trimethoxysilane and aluminum tri-secbutoxide. | |||
| Inclusion of more than 40% of OIC suppresses formation of ion aggregates and the crystallinity of PEO. Li salts mediate the interaction between the polymer-rich phase and the OIC | |||
| PEO-NaCF 3 SO 3 /Al 2 O 3 (3–30 wt%) | Jurado-Meneses et al. 96 | ||
| Interaction of sodium cation with PEO results in the formation of complex. Addition of alumina reduces crystallinity of Na-PEO complex. Improved ionic conduction is due to the migration of Na+ through the pathways of amorphous phase surrounding the filler. | |||
| TOFSIMS | Lateral and bulk distribution of polymer and ceramic phases. Interactions between salt, polymer and ceramics | PEO-LiI/LiAlO 2 (75–85 wt% ceramics) | Menkin et al. 68 |
| The complex species LiOCH2 and LiOC2H5 evidence the formation of the PEO-LiI complex. One-nm-thick interfacial layer on the top of LiAlO2 composed of the interaction products of ceramics with lithium salt is indicated by the appearance of AlIO and LiIAlO clusters | |||
| EIS and BDS | Study of ion transport, in the bulk and via the interface. The dynamics of bound and mobile charges. the separation between segmental relaxation and conductivity relaxation. | P(EO)-LiTFSI/LLZO (70 wt% ceramics) | Keller et al. 109 |
| Li+ transport is mainly through the amorphous polymer phase. | |||
| Addition of LLZO results in lower conductivity values since ceramic particles impose higher tortuosity in the polymer matrix. | |||
| The ion transfer at LLZO-polymer grain boundary represents the key issue to be addressed | |||
| PEO-LiCF 3 SO 3 /ZrO 2 (3–20 wt% ceramics) | Dam et al. 97 | ||
| The ion transport occurs via polymer phase. Temperature dependent dc conductivity, conductivity relaxation time and segmental relaxation time follow VTF behavior, suggesting strong correlation between ion conduction process and polymer segmental relaxation process. |
Computational models
When addressing a complex system, the combination of computational modelling and experimental methods, in most cases deliver a deeper understanding of the elusive ion-transport mechanism in solid-electrolyte composites. Unfortunately, combined studies of transport phenomena in composite electrolytes are scarce. Still, we refer the reader to a few of these.
Such a rare case is the work of Brooks et al. 127 that dealt with the ionic-diffusion mechanism in PEO-LiTFSI polymer electrolytes. The authors studied the diffusion coefficient as a function of temperature, molecular weight and salt concentration. On the basis of their MD simulations, Brooks et al. hypothesized that the diffusion coefficient changes in assent with the above-mentioned parameters. They corroborated their hypothesis by experimental measurements. The authors concluded that at 360 K intrachain diffusion is the most probable diffusion path for Li+ ion. At higher temperatures, interchain diffusion, responsible for higher diffusion rates, is more favorable. The authors also calculated by mean-square displacement (MSD), the diffusion of Li+ ions through the PEO matrix. They suggested that rigid polymers could increase the lithium diffusion.
Nevertheless, pure modelling also leads to profound understanding. Mogurampelly et al. studied the influence of aluminum-oxide (Al2O3) nanoparticles on the Li+ ion conduction mechanism in the PEO-based polymer electrolytes (solvated with LiBF4 salt) with the use of atomistic molecular dynamics (MD) simulations combined with trajectory-extending kinetic Monte Carlo simulations (TEKMC). 128 The authors observed that the addition of Al2O3 nanoparticles causes a decrease in ionic conductivity. They concluded that the addition of nanoparticles alters the polymer segmental dynamics, thus influencing the ionic mobilities. They also suggested that the size and shape of nanoparticles also influence the ionic conductivity.
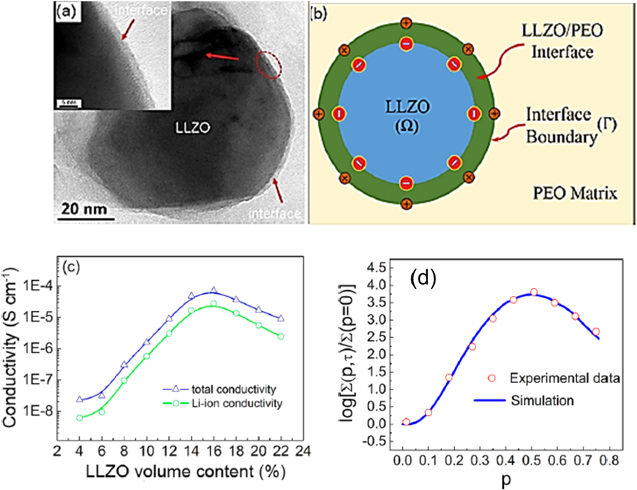
Another example is the recent paper by Li et al., 129 that modelled and measured the effect of nanoparticles on the ionic conductivity in a composite electrolyte. The authors varied the volume fraction of Li6.25Ga0.25La3Zr2O12 (Ga-LLZO) nanoparticles embedded in PEO. Li et al., noticed that the ionic-conductivity profile exhibits a percolation-type behavior suitable for two-phase mixture systems with high interfacial conduction (Fig. 9). On the basis of Dudney's work, 130 they proposed that at the Ga-LLZO/PEO interface, a space-charge region is formed. The defect concentration in the region provides a new kinetic pathway for ionic conduction that results in increased ionic conductivity. The authors developed a conduction model integrating a random resistor model for the two-phase mixture and a Monte-Carlo simulation to demonstrate that the enhanced ionic conductivity can be attributed to the space-charge region. Their conclusion, on the basis of their model, was that when nanoparticles are in contact or close proximity their own space-charge regions overlap to form a continuous space-charge pathway that significantly enhances the ionic conduction. The authors also compared their Monte-Carlo simulations of PEO:Ga-LLZO ionic conductivities to experimental data (Fig. 9).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 9. TEM images of the Ga-LLZO/PEO interface (a). Schematic illustration of a Ga-LLZO particle in the PEO:Ga-LLZO composite (b). Total conductivity and Li-ion conductivity as a function of Ga-LLZO nanoparticles volume fraction (c). The ionic-conductivity plot of PEO:Ga-LLZO composite: experimental data vs Monte-Carlo simulation data (d). Adapted with permission from Li et al. 129 Copyright (2019) American Chemical Society.
Download figure:
Standard image High-resolution image{kind=link}
Conclusions
This review is exclusively focused on composite electrolytes and does not address the origins of the high electrode/electrolyte interfacial resistance and sluggish charge transfer, the detrimental shuttle effects, and decomposition reactions between the electrolyte and the electrodes. Currently, composite electrolytes are considered to be the most promising solution for solid-state batteries. They should combine high ionic conductivity of ceramics with good mechanical properties, flexibility and adhesion of polymers. However, low ionic conductivity remains the major obstacle in employing composite electrolytes in batteries. Our analysis of the literature demonstrates the great importance of interfaces in ionic transport of composite electrolytes. By treating grain boundaries in ceramic media, one can achieve high ionic conductivity through percolating interfaces. In addition, surface modification of superionic conductors and even poor conducting ceramic materials can significantly reduce the energy barrier for cations to move from ceramic to polymer and back. This should increase the total ionic conductivity of composite electrolytes. Another important direction is the proper choice of a polymer matrix. Our analysis revealed no studies of composite electrolytes based on polymerized ionic liquids. PolyILs are essentially single-ion conductors, the same as the superionic ceramics. Moreover, this compatibility of ionic transport might help to resolve many interfacial problems in composite electrolytes. We assume that surface modification of ceramic particles, and proper choice of chemical structure and volume fraction of PolyIL can significantly improve ion transport in composite electrolytes.
High-ceramic-content composite electrolytes, modified by PolyIL will be relatively flexible and will conformally follow the complex surface geometry of electrodes. These electrolytes have a dual-purpose use—as protective cathode layers and as ion-conducting medium in batteries. Because dry polymers have conductivity that is significantly lower than that of superionic ceramics, decrease of the volume fraction of the polymer should improve the overall ionic conductivity in the composite without losing its flexibility and adhesive properties. However, the major problem of interfacial barriers to ion transport remains critical. Therefore, development of fundamental understanding of the parameters controlling interfacial barriers to ion transport between ceramic particles and polymers in composite electrolytes should include the following approaches: (i) unravelling the effect of "active" and "inert" ceramics and polymer-to-ceramic ratio on ion-transport phenomena; (ii) reduction of the interfacial barriers for Li+ and Na+ transport by modification of the polymer chemistry and surface functionalization of ceramic nanoparticles and (iii) synthesis of novel polymer electrolytes, based on polymerized ionic liquids and functionalized ceramic particles with a large number of defects and low-activation-barrier interface.