
Abstract
Stable colloidal tetrapropylammonium manganese oxide (TPA-MO) was formed by the reduction of tetrapropylammonium permanganate with 2-butanol at room temperature. Thin films of TPA-MO were prepared using the sol-gel process by dip-coating directly onto clean nickel foils followed by heat-treatment under controlled conditions. The microstructure evolution of TPA-MO films at various calcination temperatures was characterized using Brunauer-Emmett-Teller method, X-ray diffraction, and scanning electron microscopy. The performance of these films as supercapacitors was evaluated using cyclic voltammetry in various aqueous electrolytes. These thin films exhibited excellent capacitive behavior with a specific capacitance of 720 F/g. These films also showed good reversibility and cycling stability, losing little more than 20% of their charge capacity after 1500 cycles. © 2002 The Electrochemical Society. All rights reserved.
Export citation and abstract BibTeX RIS
Manganese oxides in their various forms have been widely studied and developed for applications in electrochemical energy conversion and storage systems.1
2
3
4
5 The superior electrochemical performance of these materials have made them attractive candidates for cathode materials in  alkaline cells,1 as an intercalation host for lithium batteries,2
3 and more recently as electrodes in ultracapacitors.4
5 As compared to other transition metal oxides such as
alkaline cells,1 as an intercalation host for lithium batteries,2
3 and more recently as electrodes in ultracapacitors.4
5 As compared to other transition metal oxides such as  CoO,
CoO,  and
and  these materials have received much attention and interest due to the low cost of raw materials, low toxicity, and environmentally friendly character.
these materials have received much attention and interest due to the low cost of raw materials, low toxicity, and environmentally friendly character.
Studies of Pang et al.4
5 have shown that sol-gel-derived  thin films are a promising electrode material for supercapacitors due to their high reversibility, good cycling stability, and their high specific capacitance of 700 F/g. However, in spite of their favorable characteristics, the potential use of such sol-gel-derived
thin films are a promising electrode material for supercapacitors due to their high reversibility, good cycling stability, and their high specific capacitance of 700 F/g. However, in spite of their favorable characteristics, the potential use of such sol-gel-derived  films for fabricating practical devices is limited by the very dilute concentration
films for fabricating practical devices is limited by the very dilute concentration  of the
of the  colloidal suspension employed in coatings. Stable nanoparticles of manganese oxides having higher concentrations are difficult to prepare because of the strong tendency of manganese oxides to precipitate or coagulate during synthesis. Any increase in the
colloidal suspension employed in coatings. Stable nanoparticles of manganese oxides having higher concentrations are difficult to prepare because of the strong tendency of manganese oxides to precipitate or coagulate during synthesis. Any increase in the  concentration invariably leads to the destabilization of the colloidal suspension. Considerable efforts have therefore been directed toward exploring alternative precursors and approaches in order to prepare stable colloidal suspensions with higher concentrations of manganese dioxide particles.
concentration invariably leads to the destabilization of the colloidal suspension. Considerable efforts have therefore been directed toward exploring alternative precursors and approaches in order to prepare stable colloidal suspensions with higher concentrations of manganese dioxide particles.
Recently, stable colloidal manganese oxides with concentrations as high as 0.57 M have been successfully prepared by incorporating tetralkylammonium cations ( propyl, and butyl) to prevent agglomerization of the negatively charged manganese oxide particles.6 Materials derived from such colloidal manganese oxides are reported to have lamellar structure with tetraalkylammonium cations intercalated between the manganese oxide layers. The manganese oxides have an average oxidation state of 3.70-3.79.6 The pore size distribution of materials derived from such organic cation template-based manganese oxides can be well controlled by the size of the particles that constitute the templates, which can be subsequently removed by heat-treatment.
propyl, and butyl) to prevent agglomerization of the negatively charged manganese oxide particles.6 Materials derived from such colloidal manganese oxides are reported to have lamellar structure with tetraalkylammonium cations intercalated between the manganese oxide layers. The manganese oxides have an average oxidation state of 3.70-3.79.6 The pore size distribution of materials derived from such organic cation template-based manganese oxides can be well controlled by the size of the particles that constitute the templates, which can be subsequently removed by heat-treatment.
In this paper, we report on the microstructural characterization of sol-gel-derived tetraproplyammonium manganese oxide (TPA-MO) thin films on nickel substrates and the electrochemical characterization of these films when employed as electrochemical capacitors.
Experimental
Stable colloidal suspension of tetrapropylammonium manganese oxide (TPA-MO) was prepared by the reduction of tetrapropylammonium permanganate with 2-butanol in aqueous solution at room temperature following the method reported by Brock et al.6 with some modifications in the synthesis procedure.  salt is added to a mixture of ultrapure (Milli-Q) water and 2-butanol in a volume ratio of 1:1 and stirred continuously for 2 h. The 2-butanol layer was then removed using a separation funnel. The manganese concentration in the resulting TPA-MO suspension was measured by ICP/AES (inductively coupled plasma with atomic emission spectrometry). The size of the TPA-MO particle was determined by laser light-scattering technique using a Zetasizer 3000HS advanced mobility analyzer (Malvern Instruments, Inc). Mobility measurements were performed using the same instrument. The pH value was adjusted by adding nitric acid,
salt is added to a mixture of ultrapure (Milli-Q) water and 2-butanol in a volume ratio of 1:1 and stirred continuously for 2 h. The 2-butanol layer was then removed using a separation funnel. The manganese concentration in the resulting TPA-MO suspension was measured by ICP/AES (inductively coupled plasma with atomic emission spectrometry). The size of the TPA-MO particle was determined by laser light-scattering technique using a Zetasizer 3000HS advanced mobility analyzer (Malvern Instruments, Inc). Mobility measurements were performed using the same instrument. The pH value was adjusted by adding nitric acid, 
Xerogels were prepared by evaporating a suspension of TPA-MO at ambient temperature. The Brunauer-Emmett-Teller (BET) surface area and pore volume were determined by the nitrogen gas adsorption-desorption method at 77 K using Micrometrics ASAP 2010. The pore size distribution was calculated by the Barrett-Joyner-Halenda (BJH) method using the desorption branch of the isotherm. A Netszch STA 409 analyzer was used to perform the simultaneous thermogravimetric and differential thermal analyses (TG/DTA). Samples were heated from 25 to 800°C at a heating rate of 2°C/min under a flowing air atmosphere. The flow rate was 20 mL/min and  was used as the reference material. A Scintag diffractometer using Cu Kα radiation was used to obtain the diffraction patterns of the sample calcined at various temperatures. A LEO scanning electron microscope (SEM) was used to characterize the microstructure of the TPA-MO thin films.
was used as the reference material. A Scintag diffractometer using Cu Kα radiation was used to obtain the diffraction patterns of the sample calcined at various temperatures. A LEO scanning electron microscope (SEM) was used to characterize the microstructure of the TPA-MO thin films.
Thin films of TPA-MO were formed directly on clean nickel foils (0.125 mm in thickness) by dip-coating under a controlled withdrawal velocity. All nickel foils were thoroughly cleaned prior to deposition according to the method reported by Pang et al.4 A measured amount of sol was mixed with 0.8% (v/v) of surfactant (0.05% sodium dodecyl sulfate) in order to improve the adherence of sol unto the nickel substrate. The electrochemical properties of these thin films were studied with an EG&G model 6310 potentiostat using a standard three-electrode cell configuration. Cyclic voltammetry was scanned over the potential range of 0.0-0.9 V, with a scan rate of 50 mV/s in various mild aqueous electrolytes. The reference electrode was a saturated calomel electrode (SCE) fitted with a bridge of Vycor frit, and the counter electrode was a platinum foil  The TPA-MO thin films were heat-treated at 100, 200, 300, 400, and 500°C in air for 1 h. The charge capacity, which is directly related to the differential capacitance of the thin films, was determined from the areas of the cyclic voltammograms. The mass of deposited
The TPA-MO thin films were heat-treated at 100, 200, 300, 400, and 500°C in air for 1 h. The charge capacity, which is directly related to the differential capacitance of the thin films, was determined from the areas of the cyclic voltammograms. The mass of deposited  films on the test electrodes was determined by dissolving the films in an
films on the test electrodes was determined by dissolving the films in an  mixture and using inductively coupled plasma/atomic emission spectrometry (ICP/AES). Based on the measured concentration of Mn per unit volume, the total mass of deposited
mixture and using inductively coupled plasma/atomic emission spectrometry (ICP/AES). Based on the measured concentration of Mn per unit volume, the total mass of deposited  per unit area of electrode was calculated using the formula weight of stoichiometric
per unit area of electrode was calculated using the formula weight of stoichiometric  of 86.94 g/mol.
of 86.94 g/mol.
The effect of electrolytes on the cycling performance of TPA-MO thin films was investigated using aqueous electrolytes containing cations of varying ionic radii, such as  (1.51 Å),
(1.51 Å),  (1.02 Å),
(1.02 Å),  (0.59 Å) and different anions
(0.59 Å) and different anions 
 and
and  The effects of ionic strength of the electrolytes on the cycling performance are studied by using aqueous
The effects of ionic strength of the electrolytes on the cycling performance are studied by using aqueous  solution of different concentrations. The ionic strength
solution of different concentrations. The ionic strength  was calculated based on the equation
was calculated based on the equation
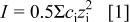
where  is the ionic concentration and
is the ionic concentration and  is the charge of the ion.
is the charge of the ion.
Results and Discussion
The mean diameter of the TPA-MO particles was determined by the light-scattering technique to be about 50 nm with diameters ranging from 36 to 72 nm. The larger particle size suggests clustering of primary particles to form larger agglomerates in the colloids. The manganese oxide concentration in the suspension was determined to be 0.2 M. The isoelectric point (IEP) of sol was determined by the mobility analyzer to be at around pH 2.8 (Fig. 1).
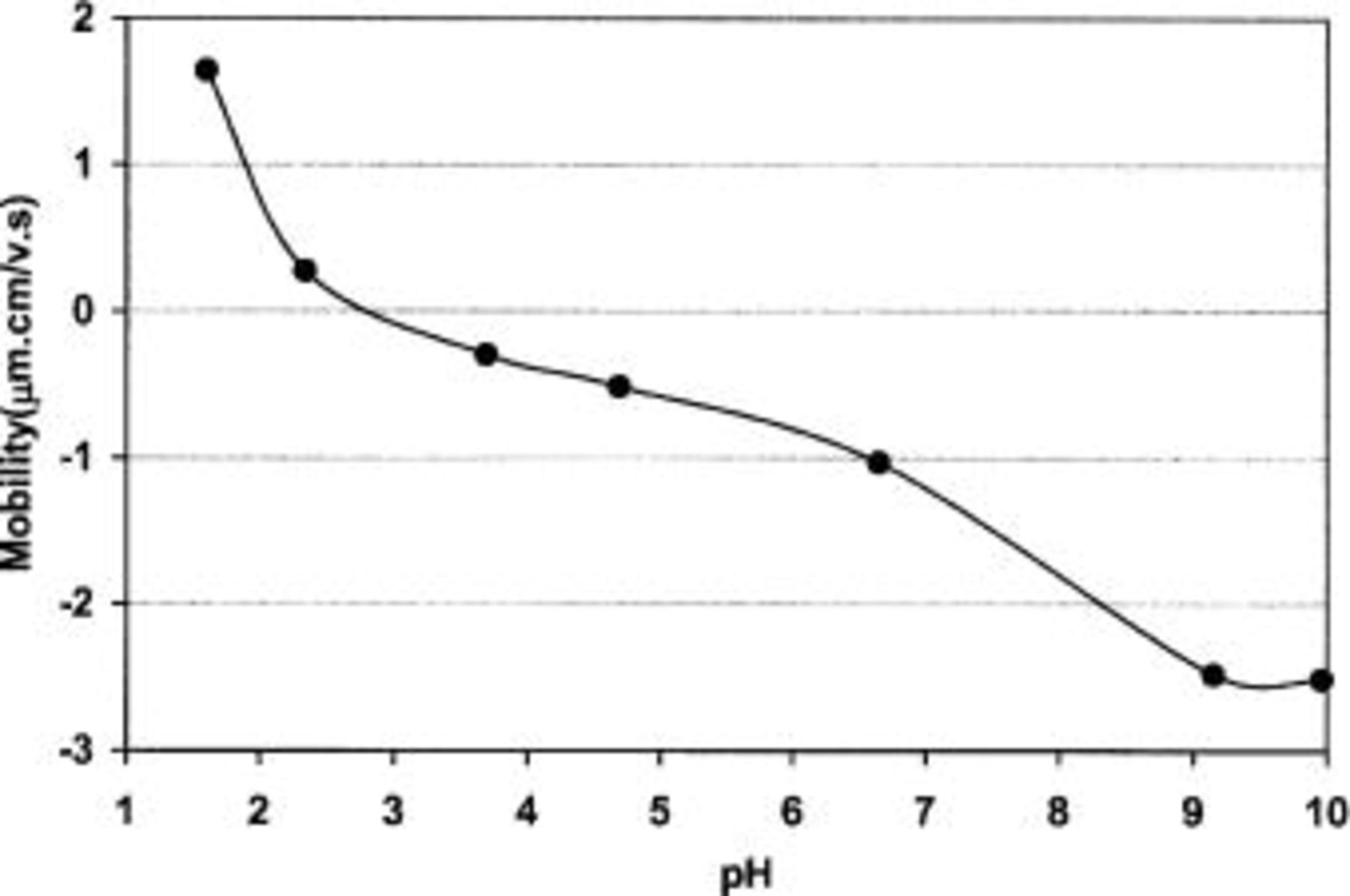
Figure 1. Mobility curve of TPA-MO sol.
Nitrogen absorption-desorption (BET) analysis
Figure 2a shows the typical nitrogen absorption-desorption isotherm of TPA-MO xerogel. According to IUPAC classification, this is a type IV isotherm, which is indicative of mesoporosity with a hysteresis loop of H1.7
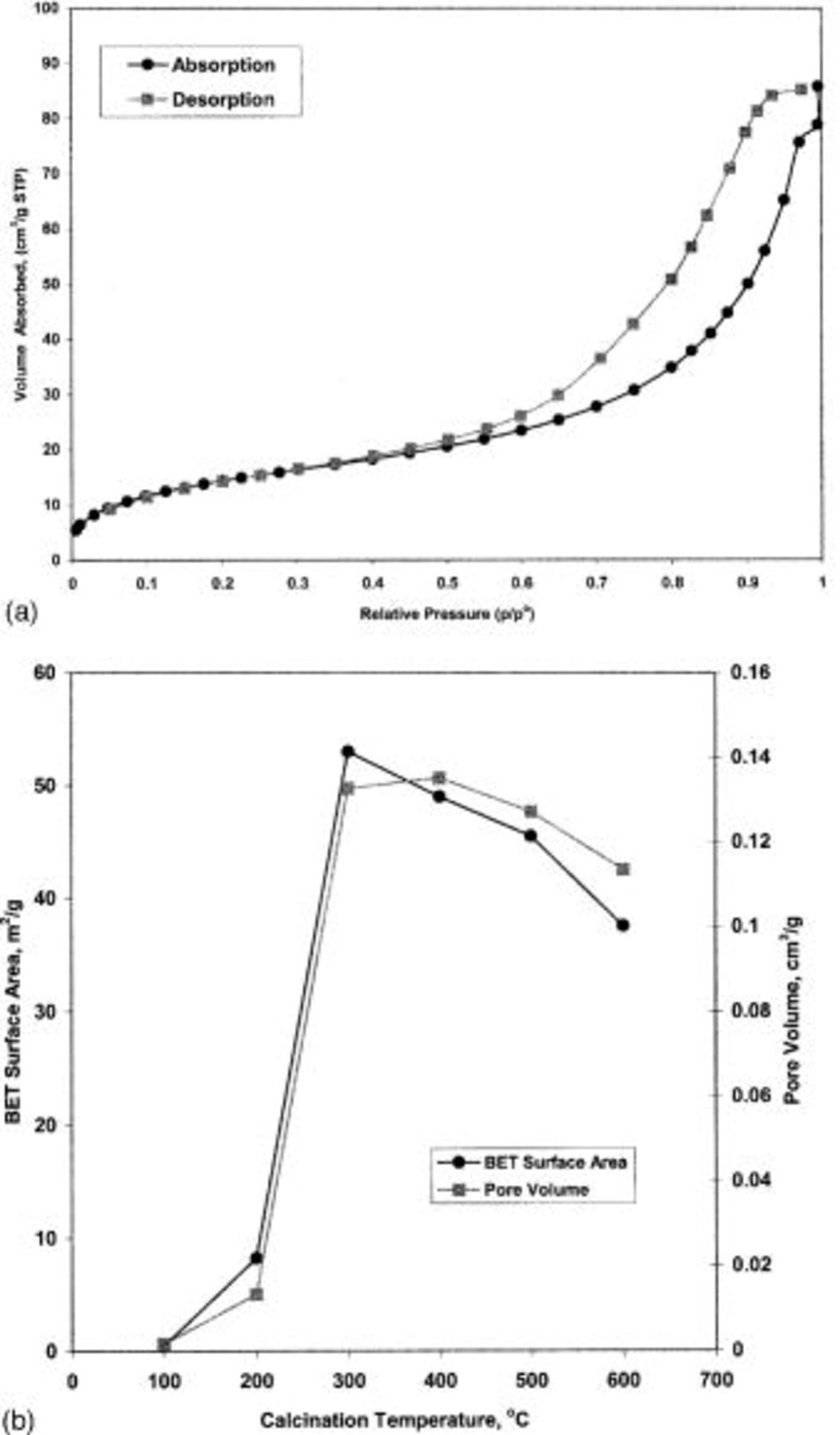
Figure 2. (a) Typical nitrogen absorption-desorption isotherm of TPA-MO xerogel. (b) Effect of calcination temperatures on the BET surface area and total pore volume of TPA-MO xerogel.
The BET surface area and total pore volume of TPA-MO xerogel calcined at various temperatures are shown in Fig. 2b. Both the BET surface area and pore volume increased drastically with increase in temperature from 100 to 300°C. The BET surface area reached a maximum of  at 300°C. Above 300°C, the BET surface area decreases from
at 300°C. Above 300°C, the BET surface area decreases from  at 400°C to
at 400°C to  at 600°C. The pore volume, however, increased slightly above 300°C, reaching its maximum at 400°C, and then showed a decreasing trend from 400 to 600°C. The substantial increase in BET surface area and pore volume (about 99% increase) from 100 to 300°C could be attributed to the removal of physically and chemically adsorbed water molecules, the solvent molecules (2-butanol), and the combustion of the organic template to form pores within the materials. The observed decrease in the BET surface area accompanied by a slight increase in pore volume at 300-400°C suggested the onset of densification through the removal of residual organic matter and the formation of larger pores. At temperatures beyond 400°C, the xerogels were further densified with the collapse of smaller pores and an observed decrease in the BET surface areas and pore volumes.
at 600°C. The pore volume, however, increased slightly above 300°C, reaching its maximum at 400°C, and then showed a decreasing trend from 400 to 600°C. The substantial increase in BET surface area and pore volume (about 99% increase) from 100 to 300°C could be attributed to the removal of physically and chemically adsorbed water molecules, the solvent molecules (2-butanol), and the combustion of the organic template to form pores within the materials. The observed decrease in the BET surface area accompanied by a slight increase in pore volume at 300-400°C suggested the onset of densification through the removal of residual organic matter and the formation of larger pores. At temperatures beyond 400°C, the xerogels were further densified with the collapse of smaller pores and an observed decrease in the BET surface areas and pore volumes.
The changes in pore size distributions of TPA-MO xerogels with heat-treatments are shown in Fig. 3. In general, the pore volume was observed to increase significantly from 100 to 300°C, with much more defined pore size distributions at higher temperatures. While three distinct modes of pore size were observed in xerogels calcined at 300°C, only one distinct mode of pore size was observed in xerogels calcined at 600°C. These pore size distributions showed that the dominant peak shifted gradually toward larger pore diameters at higher temperatures, indicating the formation of larger pores as a result of a higher degree of densification.
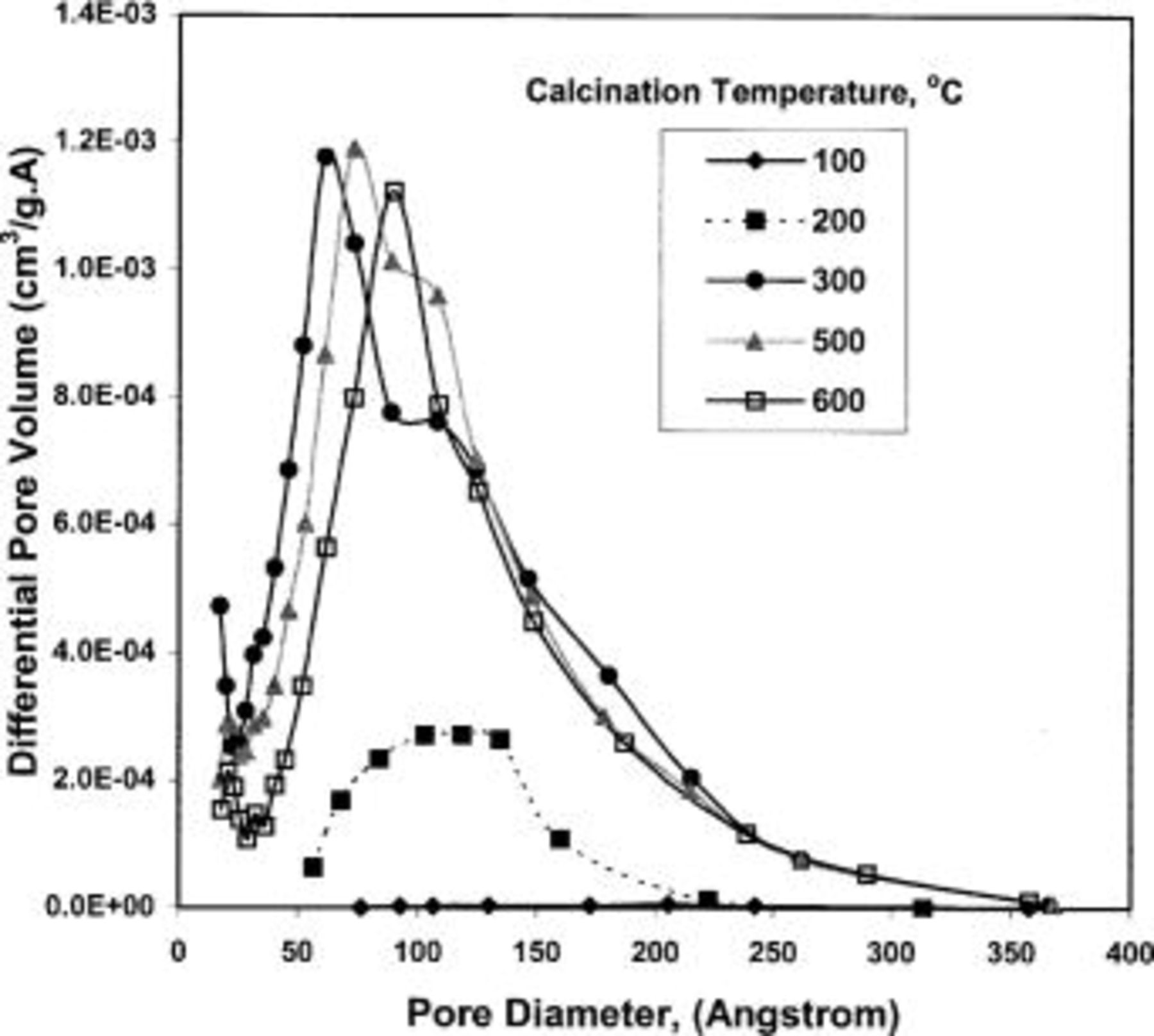
Figure 3. Changes in pore size distributions of TPA-MO xerogels with heat-treatments.
Scanning electron microscope (SEM)
Figure 4 represents the SEM micrographs of TPA-MO thin films on the supporting nickel substrate that were fired at various temperatures in air for 1 h. These SEM micrographs showed that TPA-MO films prepared at room temperature consist of densely packed nanoparticle clusters due to the presence of adsorbed water, solvent, and organic template molecules, which are believed to occupy the interlayer spaces and pores. This is evidenced by the negligibly low pore volume and undefined pore size distribution. Void spaces among loosely packed nanoparticle clusters were observed in films calcined at 200°C as a consequence of the partial removal of adsorbed water, solvent, and organic template molecules. Films calcined at 300°C showed distinct and densely packed particles of uniform sizes, following the complete removal of both adsorbed water molecules and organic residues. The observable grain size of about 40-50 nm in the micrograph is consistent with the mean particle size measured by the laser light-scattering method. Significant densification was observed in samples calcined at 500°C, with smaller particles coalesced to form larger particles.

Figure 4. SEM micrographs of TPA-MO thin films on nickel substrate at various firing temperatures.
Thermal analysis
The TG/DTA curves of TPA-MO xerogel is shown in Fig. 5. Two major weight losses were observed to occur between room temperature and 800°C. The initial weight loss of about 10% associated with endothermic reactions occurred at around 100°C. This could be attributed to the removal of residual 2-butanol solvent and physically absorbed water molecules. A sharp exothermic peak accompanied by substantial weight loss was observed at about 180°C, which could be attributed to the decomposition of the low molecular organics, particularly 2-butanol retained within the porous matrix. The sample was observed to have lost up to 70% of its initial weight on heating up to 200°C. A very gradual subsequent weight loss of approximately 5% was observed when heated up to 220°C. This temperature coincides with the onset of the second exothermic peak, probably associated with the decomposition of any residual organic matter. The broad exothermic peak, as observed over the temperature range 300-800°C, contains two smaller exothermic peaks at around 500 and 600°C. This could be attributed to a phase transformation of the xerogels from an amorphous to a crystalline phase.
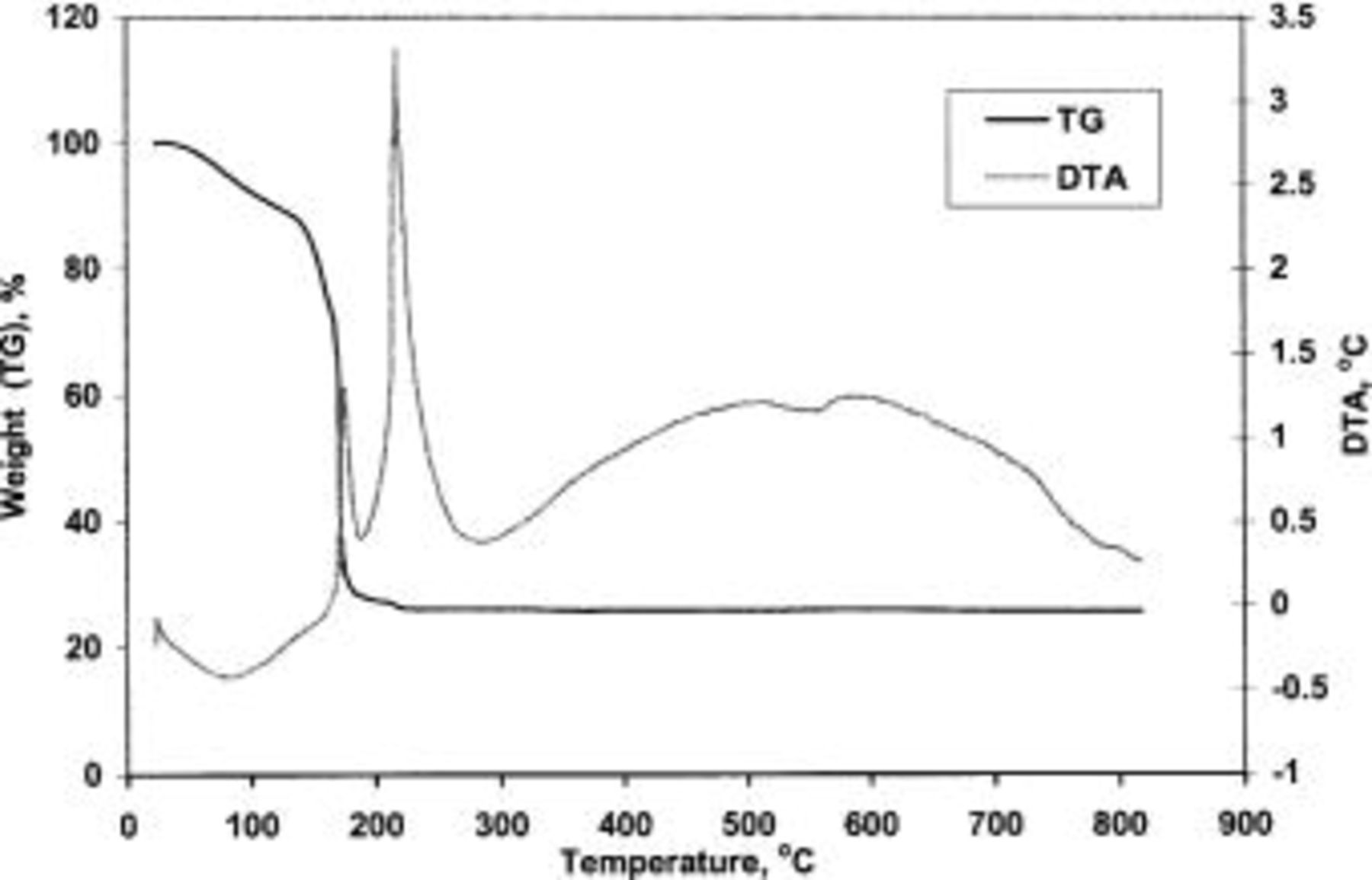
Figure 5. TG/DTA curve of TPA-MO xerogels.
X-ray diffraction
Figure 6 shows the combined X-ray diffraction (XRD) patterns of TPA-MO samples calcined at various temperatures in air for 1 h. The absence of any distinct diffraction peaks showed that TPA-MO samples remained largely amorphous in nature, even after being calcined at temperatures as high as 400°C. Distinctive diffraction peaks are only observable at calcination temperatures of 500°C. This result concurred with the TG/DTA data that the phase transformation of xerogels from an amorphous to a crystalline phase only occurs at about 500°C. According to the charge-storage mechanism of  proposed by Pang et al. ,4 such an amorphous phase allows unhindered dilation of manganese oxide lattice during proton intercalation or deinteracalation associated with the charge and discharge processes, thereby enhancing the cycling stability and reversibility of these films.
proposed by Pang et al. ,4 such an amorphous phase allows unhindered dilation of manganese oxide lattice during proton intercalation or deinteracalation associated with the charge and discharge processes, thereby enhancing the cycling stability and reversibility of these films.
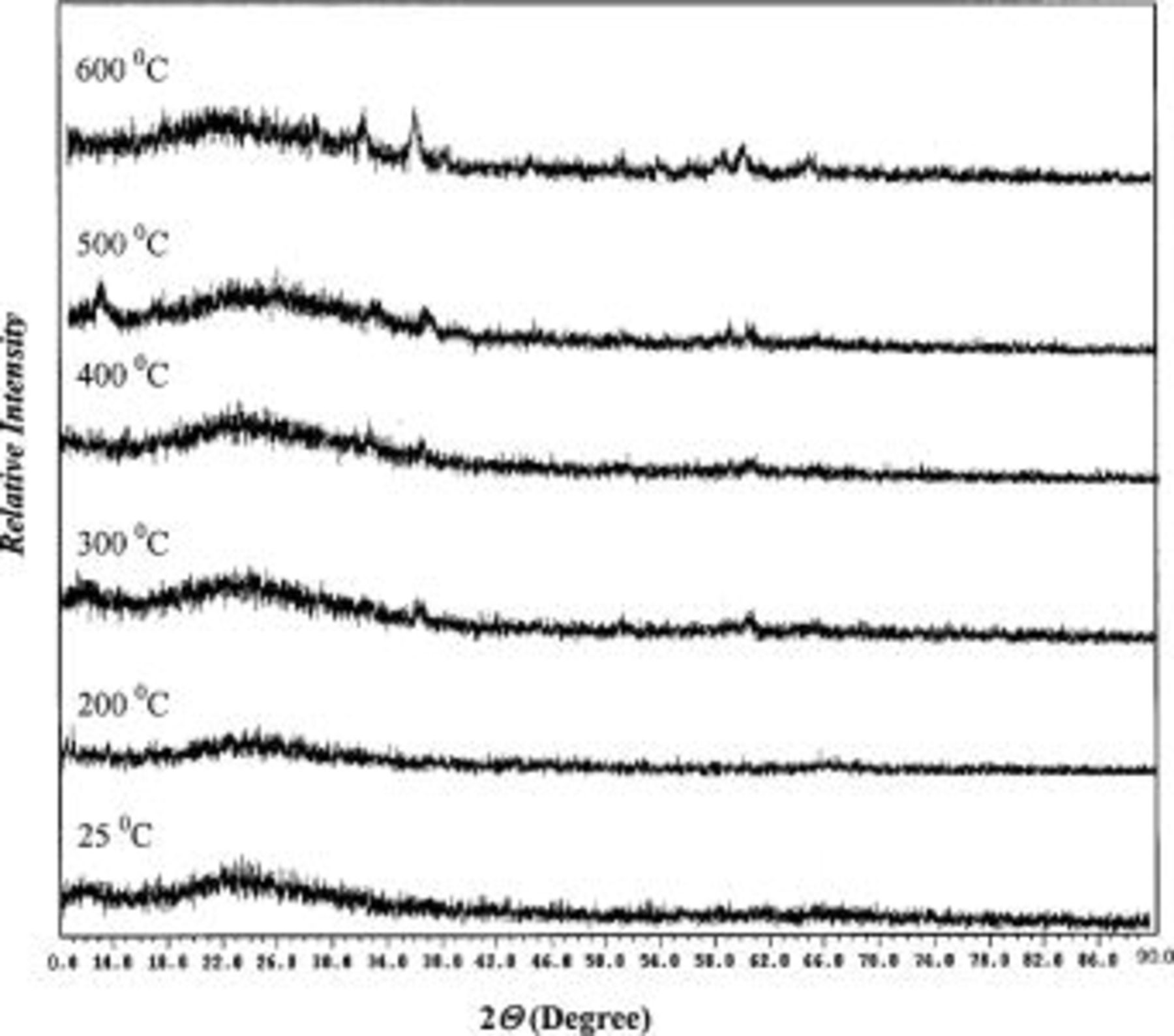
Figure 6. XRD spectra of TPA-MO xerogel.
Cyclic voltammetry
Figure 7 shows the effect of calcination temperature on the charge capacity of TPA-MO thin films. A maximum charge capacity was obtained for films calcined at 300°C; this temperature coincides with the highest BET surface area. There appears to be a positive relation between the charge capacity and BET surface area (Fig. 2b and 7). The relatively higher BET surface area and pore volume at 300°C allows easy accessibility of electrolyte within the oxide matrices during voltammetric cycling.
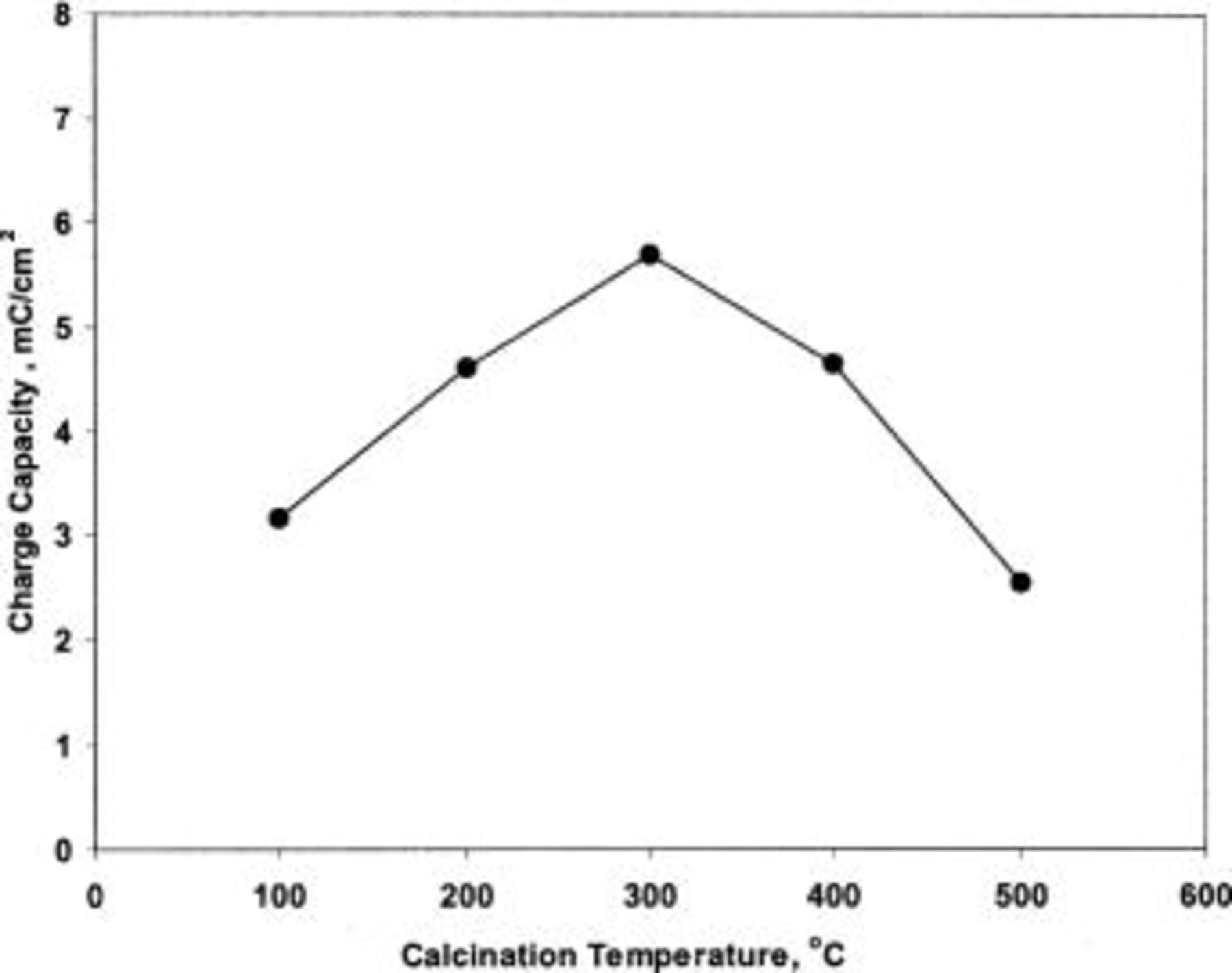
Figure 7. Effect of calcination temperature on the charge capacity of TPA-MO thin films.
It has been reported that the presence of structural water promotes proton diffusivity in manganese oxide, but its resistivity also increases with increasing water content.8 Heat-treatment at 300°C in air for 1 h has the effect of lowering the water content and consequently increases the electronic conductivity of the TPA-MO films. These factors are believed to have contributed collectively to the substantial higher charge capacity of films calcined at 300°C.
Cycling stability and reversibility
The results of the long-term cycling of TPA-MO thin films in aqueous 1.0 M  solution are shown in Fig. 8. The charge capacity of these films was observed to fade gradually, with a net loss of about 20% of their initial charge capacity after 1500 cycles. However, as shown in Fig. 9, the shape of these cyclic voltammograms (CVs) changed very little over 1500 cycles, indicating high cycling stability and reversibility. As reported by Pang et al. ,4 the observed charge capacity fading could be attributed to the partial dissolution of the TPA-MO film into the electrolytes during cycling rather than any structural or chemical degradation. However, it is speculated that other factors may also be involved, such as changes in the electronic conductivity of the film during cycling associated with oxidation states or structural water content. It is noteworthy that these films showed good reversibility, as indicated by the almost constant anodic and cathodic charge capacity ratios over the span of 1500 cycles (Fig. 8).
solution are shown in Fig. 8. The charge capacity of these films was observed to fade gradually, with a net loss of about 20% of their initial charge capacity after 1500 cycles. However, as shown in Fig. 9, the shape of these cyclic voltammograms (CVs) changed very little over 1500 cycles, indicating high cycling stability and reversibility. As reported by Pang et al. ,4 the observed charge capacity fading could be attributed to the partial dissolution of the TPA-MO film into the electrolytes during cycling rather than any structural or chemical degradation. However, it is speculated that other factors may also be involved, such as changes in the electronic conductivity of the film during cycling associated with oxidation states or structural water content. It is noteworthy that these films showed good reversibility, as indicated by the almost constant anodic and cathodic charge capacity ratios over the span of 1500 cycles (Fig. 8).
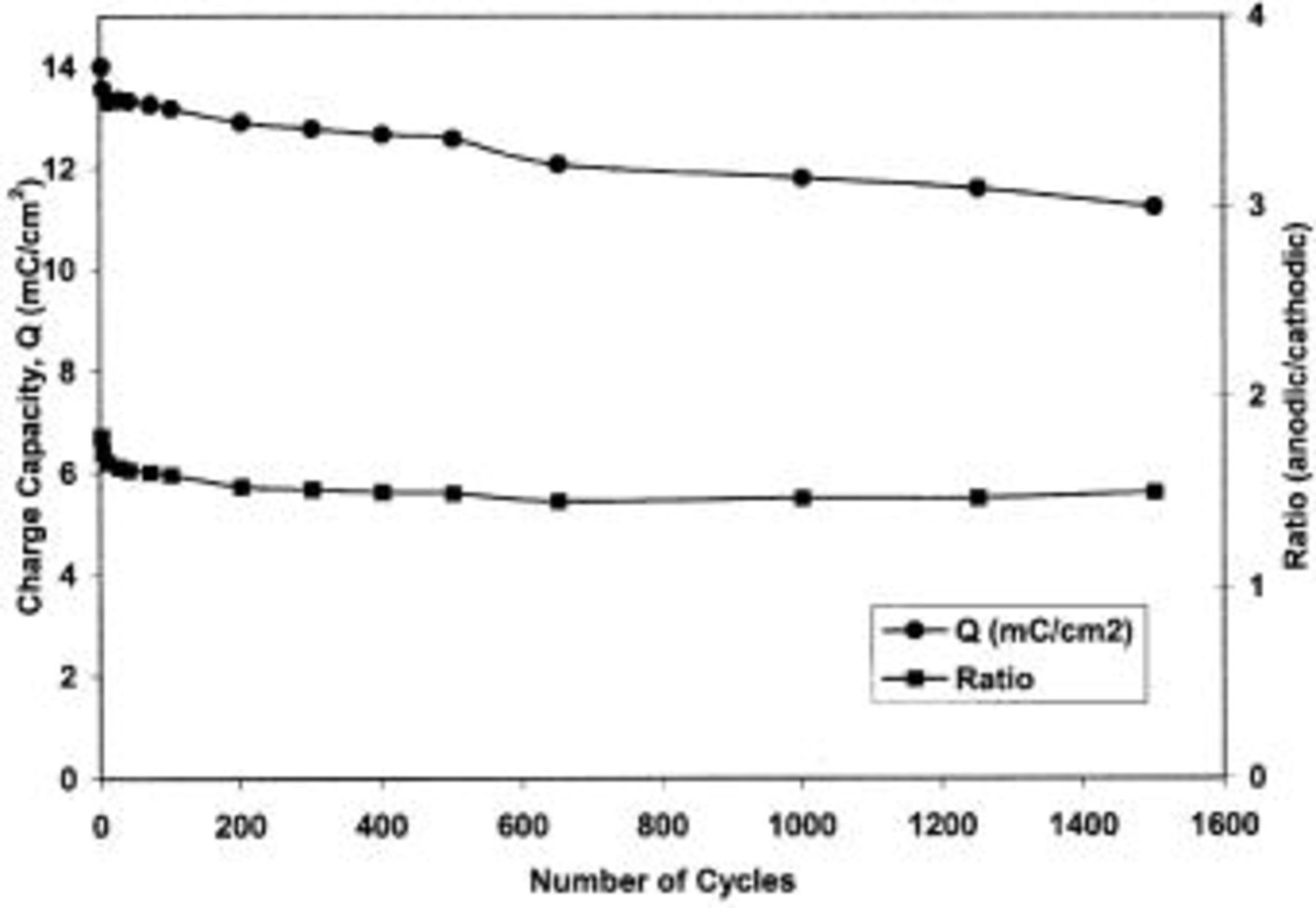
Figure 8. Charge capacity variation as a function of cycle number for TPA-MO thin films.
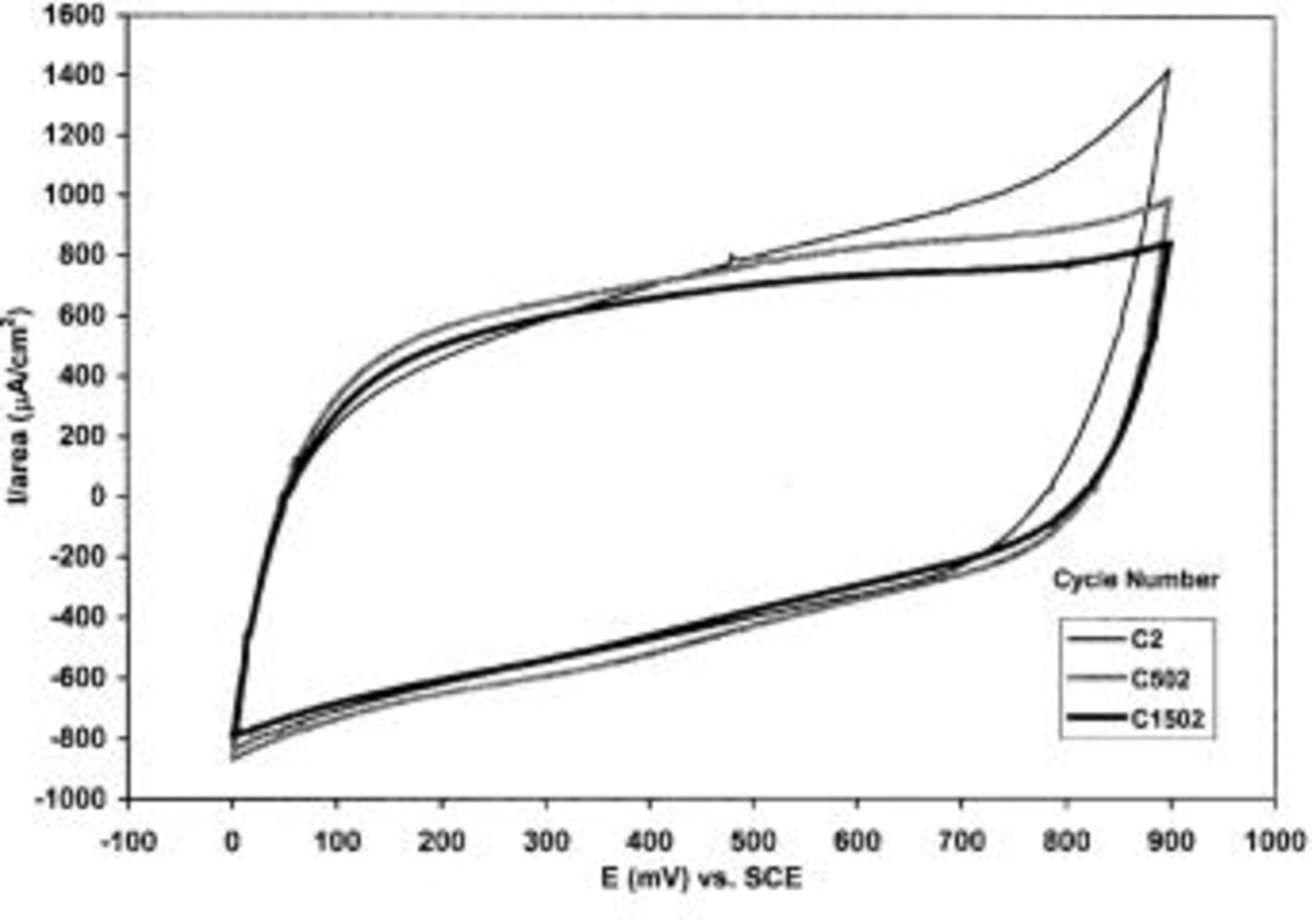
Figure 9. CVs of TPA-MO at cycle number 2, 502, and 1502.
Effect of electrolytes composition
CVs of TPA-MO thin films in various aqueous electrolytes over the potential range 0.0 to  vs. SCE are shown in Fig. 10. All the CVs exhibited roughly rectangular mirror images, which are indicative of capacitive behaviors.
vs. SCE are shown in Fig. 10. All the CVs exhibited roughly rectangular mirror images, which are indicative of capacitive behaviors.
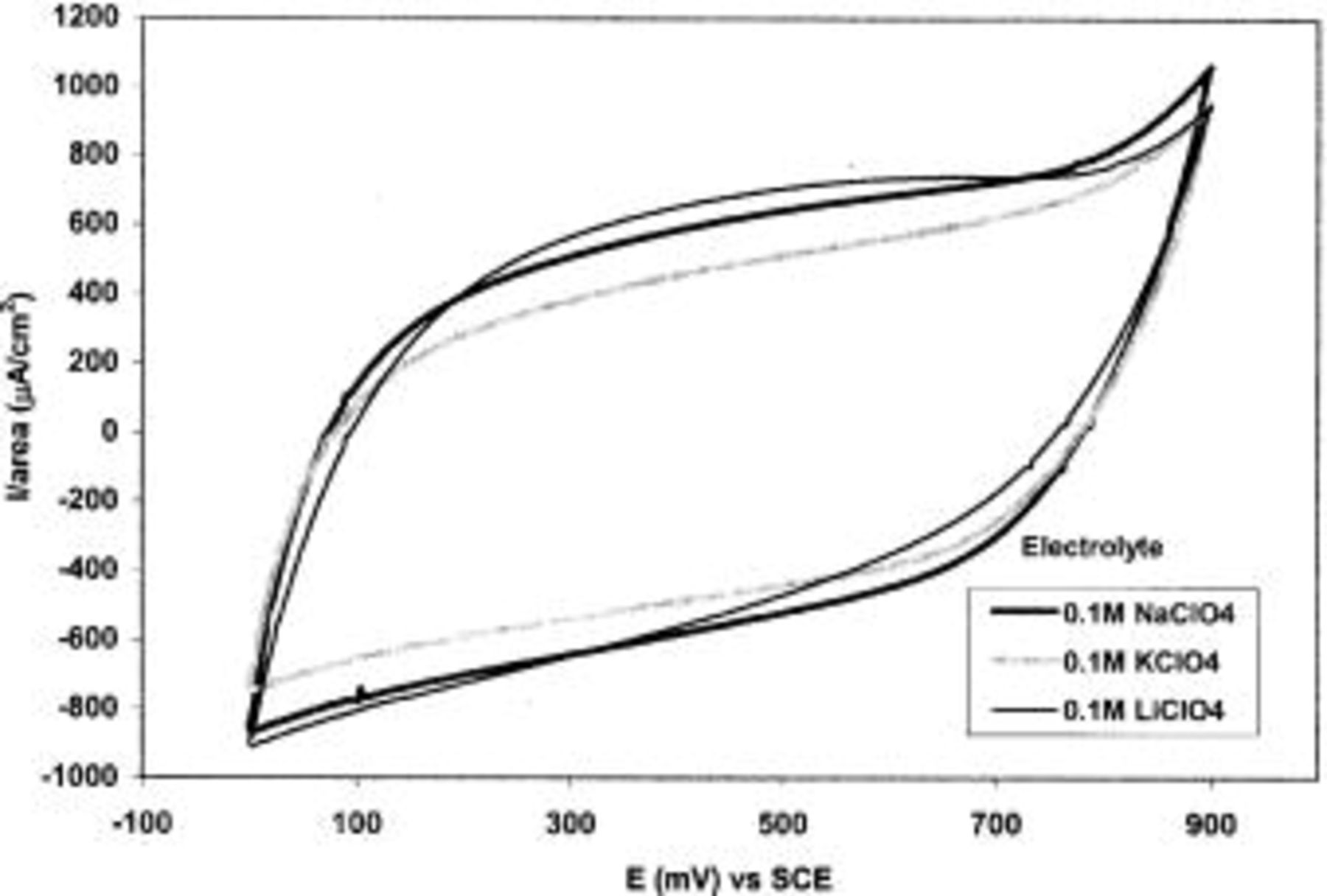
Figure 10. CVs of TPA-MO in various mild aqueous electrolytes of the same concentration.
As shown in Table I, the cation type in the electrolytes significantly influences the charge capacity of TPA-MO films. Higher charge capacity was obtained with electrolyte containing cationic species of smaller ionic radii, as evidenced by the higher charge capacity obtained with aqueous  solution. The charge capacity with
solution. The charge capacity with  is about 34 and 4% higher than that for
is about 34 and 4% higher than that for  and
and  solutions, respectively. The higher charge capacity could be attributed to enhanced ionic mobility of the smaller
solutions, respectively. The higher charge capacity could be attributed to enhanced ionic mobility of the smaller  ions within the oxide matrices during intercalation or deintercalation. Moreover, the smaller size of
ions within the oxide matrices during intercalation or deintercalation. Moreover, the smaller size of  ions allows the permeability of the ions even into the micropores, which might not be reached readily by larger cations. The nature of anions (
ions allows the permeability of the ions even into the micropores, which might not be reached readily by larger cations. The nature of anions (
 and
and  ) appears to have negligible effect on the charge capacity.
) appears to have negligible effect on the charge capacity.
Table I.
| The effect of electrolyte composition and ionic strength on the charge capacity of TPA-MO. | |
|---|---|
| Electrolyte composition | Charge capacity 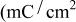 ) ) |
0.1 M 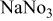 | 10.46 |
0.1 M 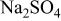 | 11.35 |
0.1 M 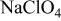 | 10.89 |
0.1 M 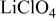 | 11.32 |
0.1 M 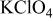 | 8.44 |
Ionic strength of 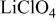 (M) (M) | Charge capacity 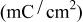 |
| 0.1 | 11.32 |
| 0.3 | 16.00 |
| 0.5 | 19.34 |
| 1.0 | 21.79 |
Effect of ionic strength
Figure 11 shows the CVs of TPA-MO films in  solutions of various concentrations, ranging from 0.1 to 1.0 M. The charge capacity of TPA-MO films was observed to increase with the concentration or ionic strength of the electrolyte. The charge capacity increased by up to 11.23% in 1.0 M
solutions of various concentrations, ranging from 0.1 to 1.0 M. The charge capacity of TPA-MO films was observed to increase with the concentration or ionic strength of the electrolyte. The charge capacity increased by up to 11.23% in 1.0 M  as compared to that of 0.1 M
as compared to that of 0.1 M  (Table I).
(Table I).
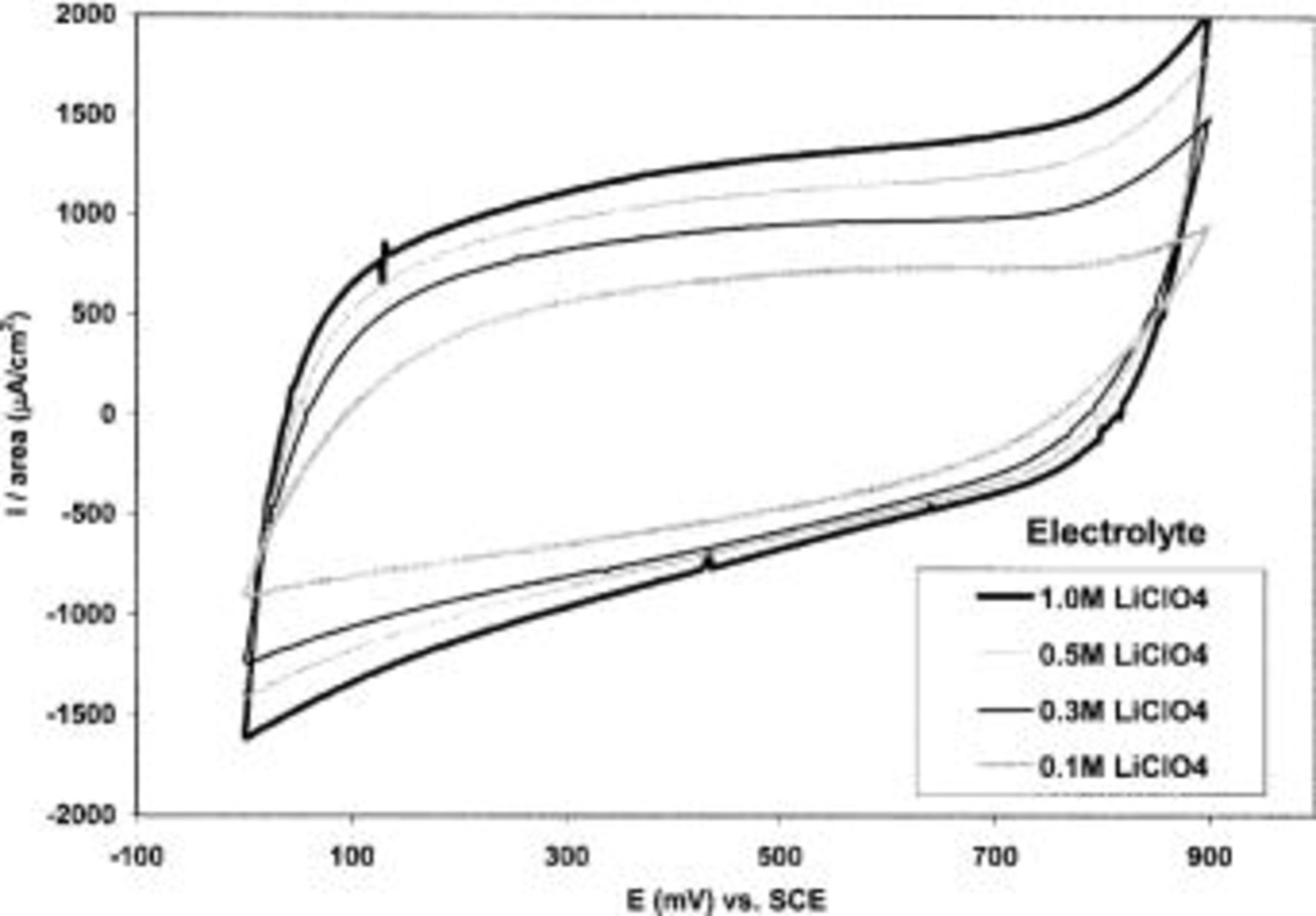
Figure 11. CVs of TPA-MO in aqueous  solution of various concentrations.
solution of various concentrations.
The thickness of the electric double layers formed at the electrode/electrolyte interface is related to the Debye length  of the double layer, which is inversely proportional to the square root of the ionic strength of the electrolyte solution.9 Increasing the ionic strength of the electrolyte has the effect of reducing the thickness of the electric double layer, thereby increasing the double-layer capacitance. An increase in the ionic strength also resulted in higher ionic conductivity and mobility, which consequently contributed to enhanced capacitance of TPA-MO films.
of the double layer, which is inversely proportional to the square root of the ionic strength of the electrolyte solution.9 Increasing the ionic strength of the electrolyte has the effect of reducing the thickness of the electric double layer, thereby increasing the double-layer capacitance. An increase in the ionic strength also resulted in higher ionic conductivity and mobility, which consequently contributed to enhanced capacitance of TPA-MO films.
Conclusion
We demonstrated that TPA-MO thin films are promising novel electrode materials for the fabrication of electrochemical capacitors. These films exhibit high charge-storage capacity, good cycling stability, and reversibility in appropriate electrolytes and operating conditions. Further optimization in the electrochemical performance and cycling behavior of these thin films is envisaged by optimizing the synthesis parameters such as volume ratio of 2-butanol and water, using different chain lengths of tetraalkylammonium (methyl, butyl, or ethyl) as organic templates, as well as the microstructure of these films.
Acknowledgments
This work was jointly funded by the Environmental Chemistry and Technology Program, University of Wisconsin-Madison, and a research grant under the Defense Army Research and Procurement Agency-Mesoscopic Integrated Conformal Electronics (DARPA-MICE) program.
The University of Wisconsin-Madison assisted in meeting the publication costs of this article.