
Abstract
Some new unsymmetrical diaryl sulfones were synthesized by anodic oxidation of fast violet B in aqueous/ethanol solutions in the presence of arylsulfinic acids at a carbon electrode. The data show that the electrogenerated N-acetyl-p-benzoquinone diimine was converted to the unsymmetrical diaryl sulfones by Michael type addition reaction with aryl sulfinic acids. This work has led to the development of a high yield, green, reagentless and facile electrochemical method for the synthesis of the described sulfones. In addition, four possible products were optimized at the BP86/def2-SVP level of theory by the parameters, natural charge, LUMO coefficient of Michael acceptor, thermodynamic and kinetic stability.
Export citation and abstract BibTeX RIS
Aryl and diaryl sulfones has attracted much attention in agrochemicals,1 medicinal chemistry, biological activities and provided (synthesis) numerous potent sulfone derivatives for different therapeutic (remedial) targets, for example, anti-inflammatory,2,3 antimicrobial,4,5 antiproliferative,6,7 anticancer,8 antimalarial,9 cysteine protease inhibitors,10,11 thyroid receptor ntagonist,12 EPAC2 antagonist,13 PI3K/Akt/mTOR signaling pathways inhibitors,14 and γ-secretase inhibitors.15 Also, aryl sulfones are found in various drugs such as 4,4'-diamino-diphenyl sulfone (dapsone)16 and aryl pyrrolyl (and aryl indolyl) sulfones.17 In addition numerous procedures for the chemical synthesis of sulfones have been reported, including oxidation of thioethers,18–20 sulfonylation of arenes,21,22 transition-metal-free,23–25 transition-metal-catalyzed.26–28 Furthermore, the majority of the available protocols still suffer from high temperature, toxic transition metal, use of oxidative condition and undesirable solvent. In continuation of our experience in electrochemical synthesis of organic compounds,29–31 we decided to synthesize new diaryl sulfone compounds via electrochemical oxidation of fast violet B (FVB) in the presence of arylsulfinic acids (1) at a carbon electrode. On the other hand, fast violet B and its derivatives are biologically active compounds and used in medicinal chemistry32–35 and histochemistry.36–40 Also, a literature survey reveals that, no paper has reported on the electrochemical behavior of FVB. Therefore, this paper pursues two main objectives. (1) Further insights into the electrochemical oxidation of FVB in aqueous solutions and (2) the synthesis of new diaryl sulfone derivatives via an environmentally friendly method. To achieve these aims, in the first step, the electrochemical oxidation of FVB in the presence and the absence of arylsulfinic acids as nucleophiles was studied, and in the next step, the obtained electrochemical data was used in the electrochemical synthesis of diaryl sulfone derivatives. In addition, the possible products were optimized at the BP86/def2-SVP level of theory by the parameters, natural charge, LUMO coefficient of Michael acceptor, thermodynamic and kinetic stability. This method represents a green and one-pot electrochemical method for the synthesis of new diphenylsulfone derivatives in good yields and purities.
Experimental
Apparatus and reagents
Cyclic voltammetry, controlled-potential coulometry and preparative electrolysis were performed using an Autolab model PGSTAT 20 potentiostat/galvanostat. The working electrode used in the voltammetry experiments was a glassy carbon disc (1.8 mm diameter) and a platinum wire was used as the counter electrode. The working electrode used in controlled-potential coulometry and macroscale electrolysis was an assembly of four carbon rods (31 cm2), while a large platinum gauze constituted the counter electrode. The working electrode potentials were measured vs. Ag/AgCl (all electrodes from AZAR Electrodes). Fast Violet B (FVB), 4-toluenesulfinic acid (pKa = 1.99),41 benzensulfinic acid (pKa = 1.84)41 and 4-chlorosulfinic acid (pKa = 1.81),41 perchloric acid and ethanol were obtained from commercial sources.
Computational study
The geometries of all species in the gas phase were fully optimized at BP86/def2-SVP level of theory using Gaussian 09.42 The frequencies were computed at mentioned level of theory. All minima and transition structures were confirmed to have none and one imaginary frequency, respectively. Transition structures were located using the Berny algorithm. Intrinsic reaction coordinates (IRC)43,44 calculations were used to confirm the connectivity between transition structures and minima. Also, NBO analyses45 were carried out at the mentioned levels of theory. A starting molecular-mechanics structure for the ab initio calculations was obtained using the HyperChem 5.02 program.
Electroorganic synthesis of 3a-3c
An ethanol/aqueous perchloric acid (c = 0.1 M) mixture (80 ml) (30/70, v/v) solution containing FVB (0.25 mmol) and aryl sulfinic acids sodium salt (1a–1c) (0.25 mmol) was electrolyzed in an undivided cell at 0.56 V vs. Ag/AgCl. The electrolysis was terminated when the current decreased by more than 95% (after consumption of about 50 coulombs). After neutralization by addition of sodium bicarbonate, the precipitated solid was washed with water. After washing and drying, the products were characterized by IR, 1HNMR, 13C NMR, and MS.
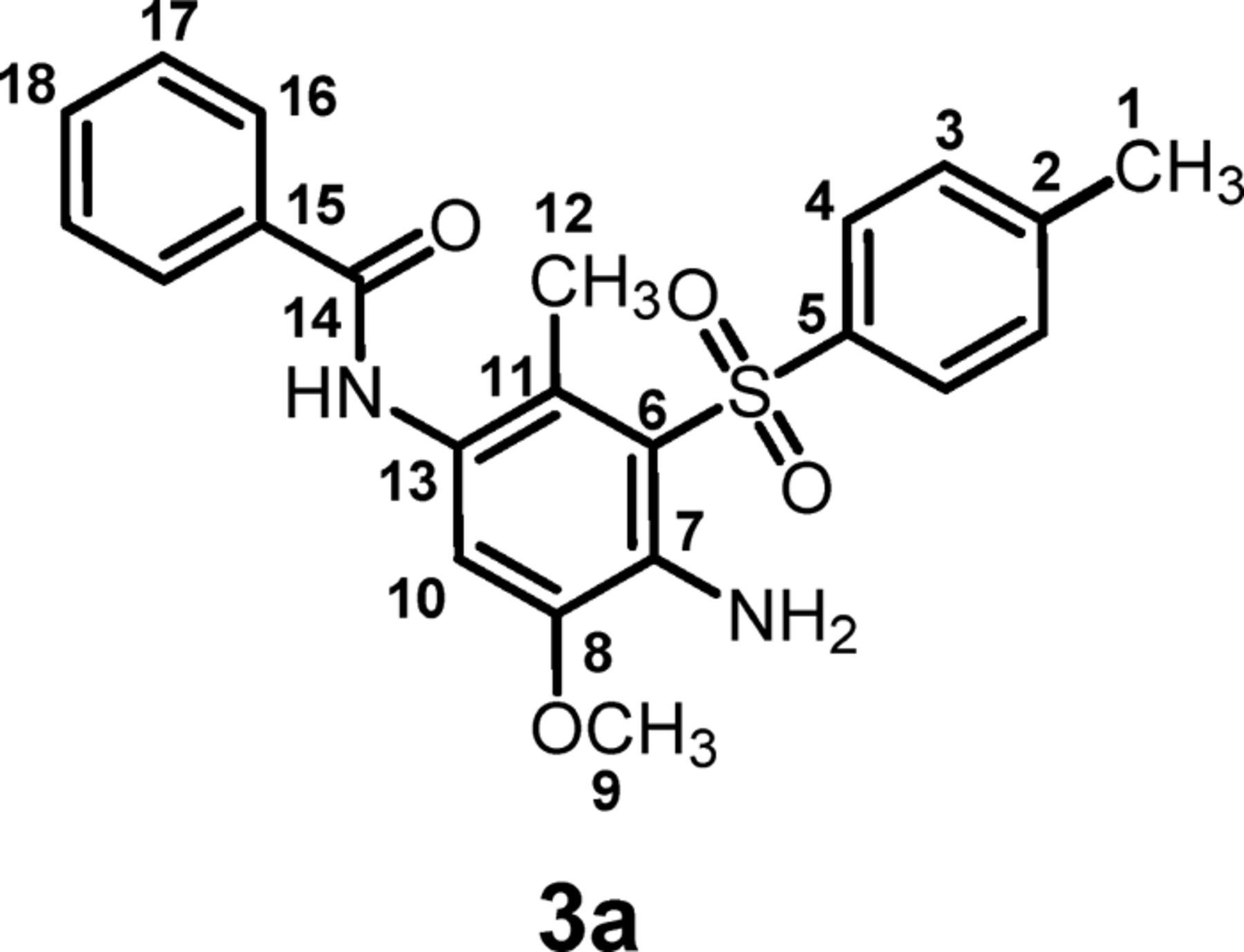
N-(4-amino-5-methoxy-2-methyl-3-tosylphenyl)benzamide (3a) C22H22N2O4S
Isolated yield: 88%. mp 142-144°C. 1H NMR (400 MHz, DMSO-d6) δ: 2.31 (s, 3H, methyl), 2.57 (s, 3H, methyl), 4.0 (s, 3H, methoxy), 6.5 (broad, ∼2H, NH2), 7.14 (s, 1H, aromatic), 7.61 (d, J = 8.4 Hz, 2H, aromatic), 7.68 (t, 2H, aromatic), 7.75 (t, 1H, aromatic), 7.94 (d, J = 8.0 Hz, 2H, aromatic), 8.12 (d, J = 7.2 Hz, 2H, aromatic), 9.95 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d6) δ: 15.1 (C-12), 21.0 (C-1), 56.1 (C-9), 114.3 (C-10), 117.1 (C-11), 124.6 (C-7), 125.4 (C-6), 125.6 (C-13), 127.5 (C-16), 128.3 (C-4), 129.8 (C-17), 131.5 (C-3), 134.1 (C-18), 138.7 (C-15), 140.1 (C-5), 144.0 (C-2), 144.9 (C-8), 165.4 (C-14); IR (KBr) v: 3497, 3393, 3236, 2935, 1640, 1579, 1515, 1476, 1302, 1241, 1139, 1121, 843, 814, 597, 555 cm−1; MS (EI, 70 eV) (m/z) (relative intensity %): 410 (M, 63), 382 (7), 210 (32), 181(11), 105 (100), 77(88), 43 (20).
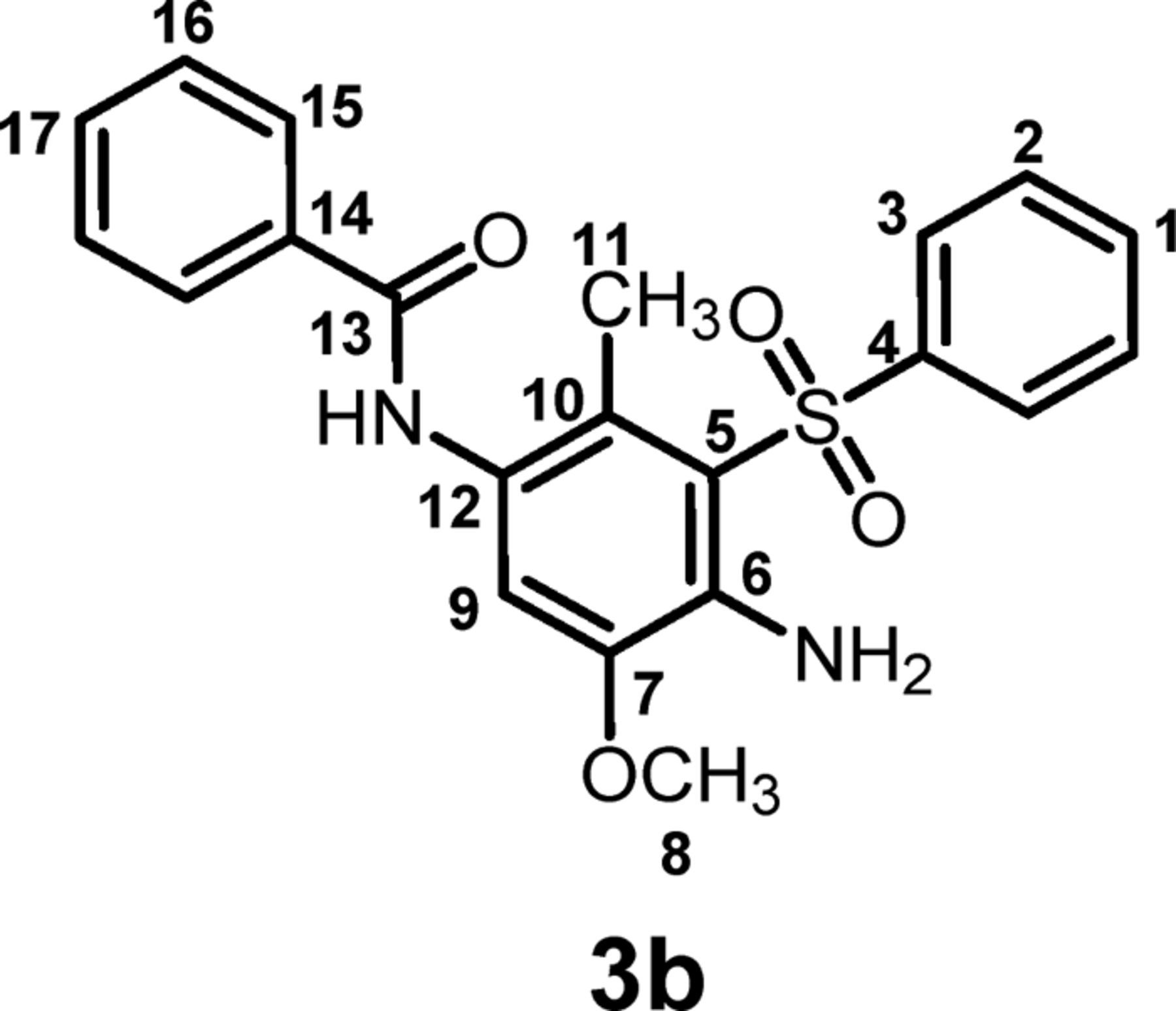
N-(4-amino-5-methoxy-2-methyl-3-(phenylsulfonyl)phenyl) benzamide (3b) C21H20N2O4S
Isolated yield: 85%. mp 160-163 oC. 1H NMR (400 MHz, DMSO-d6) δ: 2.32 (s, 3H, methyl), 4.02 (s, 3H, methoxy), 6.5 (broad, ∼2H, NH2), 7.17 (s, 1H, aromatic), 7.68 (t, 2H, aromatic), 7.76 (t, 1H, aromatic), 7.82 (t, 2H, aromatic), 7.89 (t, 1H, aromatic), 8.06 (d, J = 7.6 Hz, 2H, aromatic), 8.13 (d, J = 7.2Hz, 2H, aromatic), 9.97 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d6) δ: 15.1 (C-11), 56.1 (C-8), 114.5 (C-9), 116.7 (C-10), 124.7 (C-6), 125.5 (C-12), 127.5 (C-5), 128.3 (C-15), 129.5 (C-3), 131.5 (C-16), 133.5 (C-2), 134.1 (C-17, C-1), 138.9 (C-14), 142.9 (C-4), 144.9 (C-7), 165.4 (C-13); IR (KBr) v: 3477, 3380, 3242, 2940, 1635, 1579, 1521, 1475, 1306, 1245, 1143, 720, 694, 607, 555 cm−1; MS (EI, 70 eV) (m/z) (relative intensity %): 396 (M, 78), 331 (6), 196 (30), 149 (14), 105 (100), 77 (62), 43 (38).
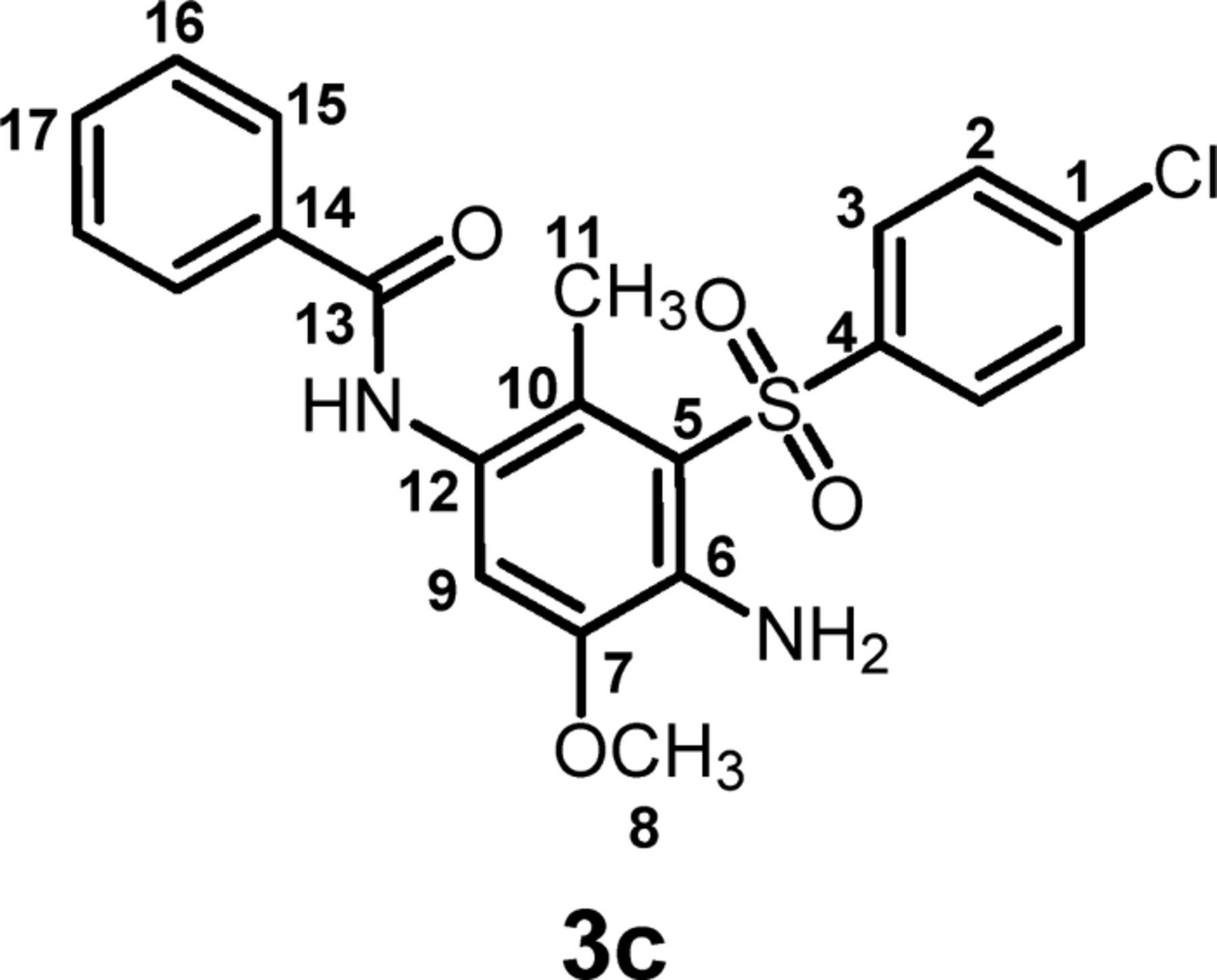
N-(4-amino-3-(4-chlorophenyl)sulfonyl)-5-methoxy-2-methylphenyl)benzamide (3c) C21H19ClN2O4S
Isolated yield: 96%. mp 122-124°C. 1H NMR (400 MHz, DMSO-d6) δ: 2.13 (s, 3H, methyl), 3.83 (s, 3H, methoxy), 6.36 (broad, 2H, NH2), 6.99 (s, 1H, aromatic), 7.50 (t, 2H, aromatic), 7.57 (t, 1H, aromatic), 7.71 (d, J = 8.8 Hz, 2H, aromatic), 7.88(d, J = 8.4 Hz, 2H, aromatic), 7.94 (d, J = 7.2 Hz, 2H, aromatic), 9.79 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d6) δ: 15.1 (C-11), 56.1 (C-8), 114.6 (C-9), 116.2 (C-10), 124.8 (C-6), 125.3 (C-12), 127.48 (C-5), 127.51 (C-1), 128.3 (C-15), 129.6 (C-16), 131.5 (C-3), 134.1 (C-2), 138.3 (C-17), 139.0 (C-14), 141.6 (C-4), 145.0 (C-1), 165.4 (C-13); IR (KBr) v: 3479, 3386, 3371, 3230, 3095, 1635, 1578, 1478, 1308, 1243, 1145, 1121, 755, 696 cm−1; MS (EI, 70 eV) (m/z) (relative intensity %): 430 (M, 16), 382 (79), 266 (53), 152 (16), 105 (60), 43 (100).
Results and Discussion
pH studies
The electrochemical oxidation of FVB has been studied at various pH values (Fig. 1, Part I). As can be seen, the cyclic voltammograms of FVB show one anodic peak (A1) in the positive going scan which can be ascribed to the oxidation of FVB to (Z)-N-(4-imino-5-methoxy-2-methylcyclohexa-2,5-dien-1-ylidene)benzamide N-acetyl-p-benzoquinonediimine (FVBox) and corresponding cathodic peak (C1) which is attributed to the reduction of FVBox to FVB (Scheme 1). Under these conditions, the peak current ratio (IpC1/IpA1) is close to one, which can be considered as a criterion for the stability of electrogenerated FVBox under the experimental conditions. In other words, any side reactions, such as hydrolysis, dimerization,46 or oxidative ring cleavage47 on electrochemically generated FVBox are too slow to be observed at the time scale of cyclic voltammetry.
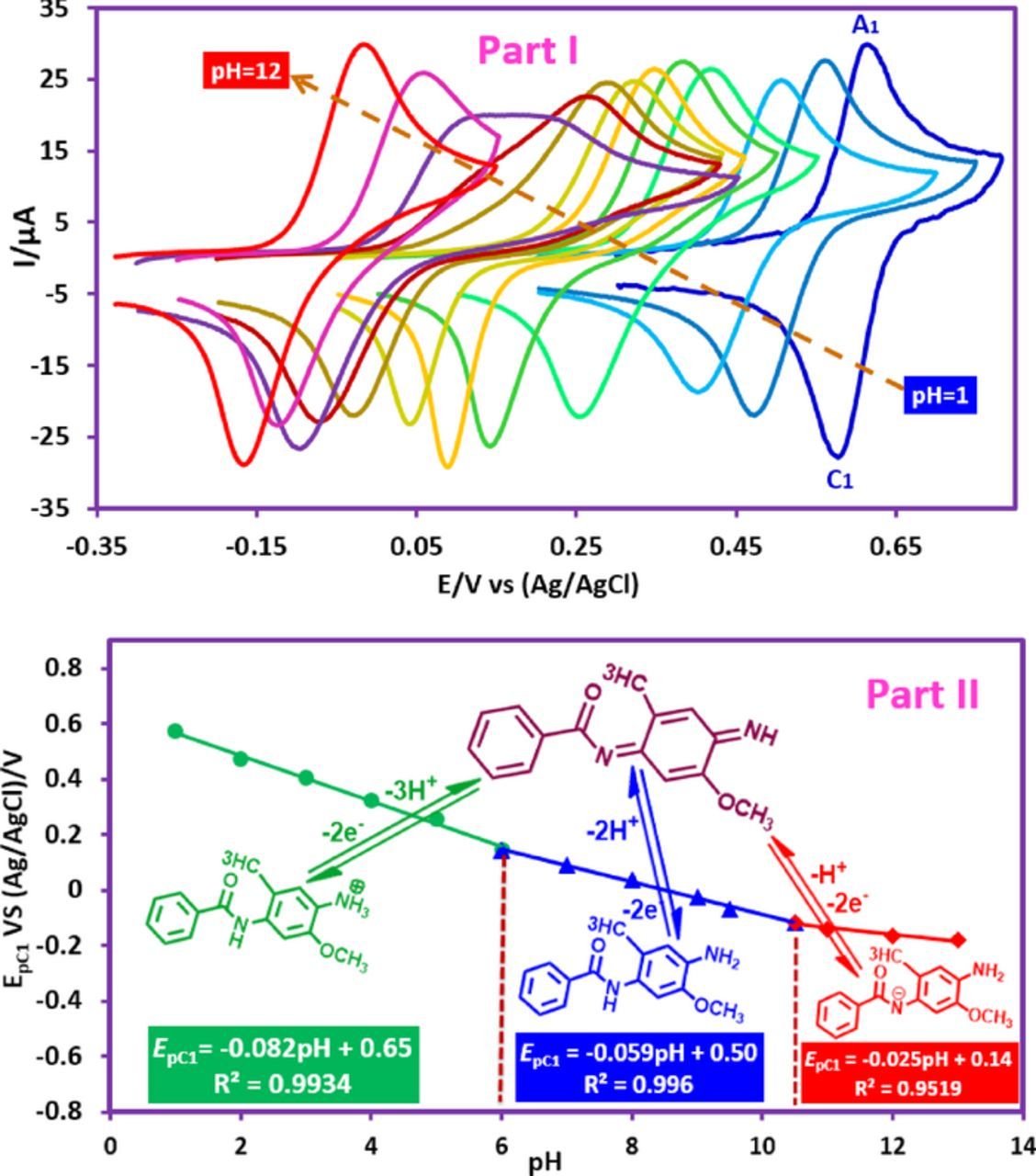
Figure 1. Part I: Cyclic voltammograms of FVB (1.0 mM) at glassy carbon electrode, in aqueous buffer solution/ethanol mixture (70/30, v/v) with different pH values. pHs are: 1.0, 2.0, 3.0, 5.0, 6.0, 7.0, 8.0, 9.0,9.5, 10.0, 11.0 and 12.0. Scan rate: 500 mV/s. Temperature = 25 ± 1 °C. Part II: The potential-pH diagram of FVB.
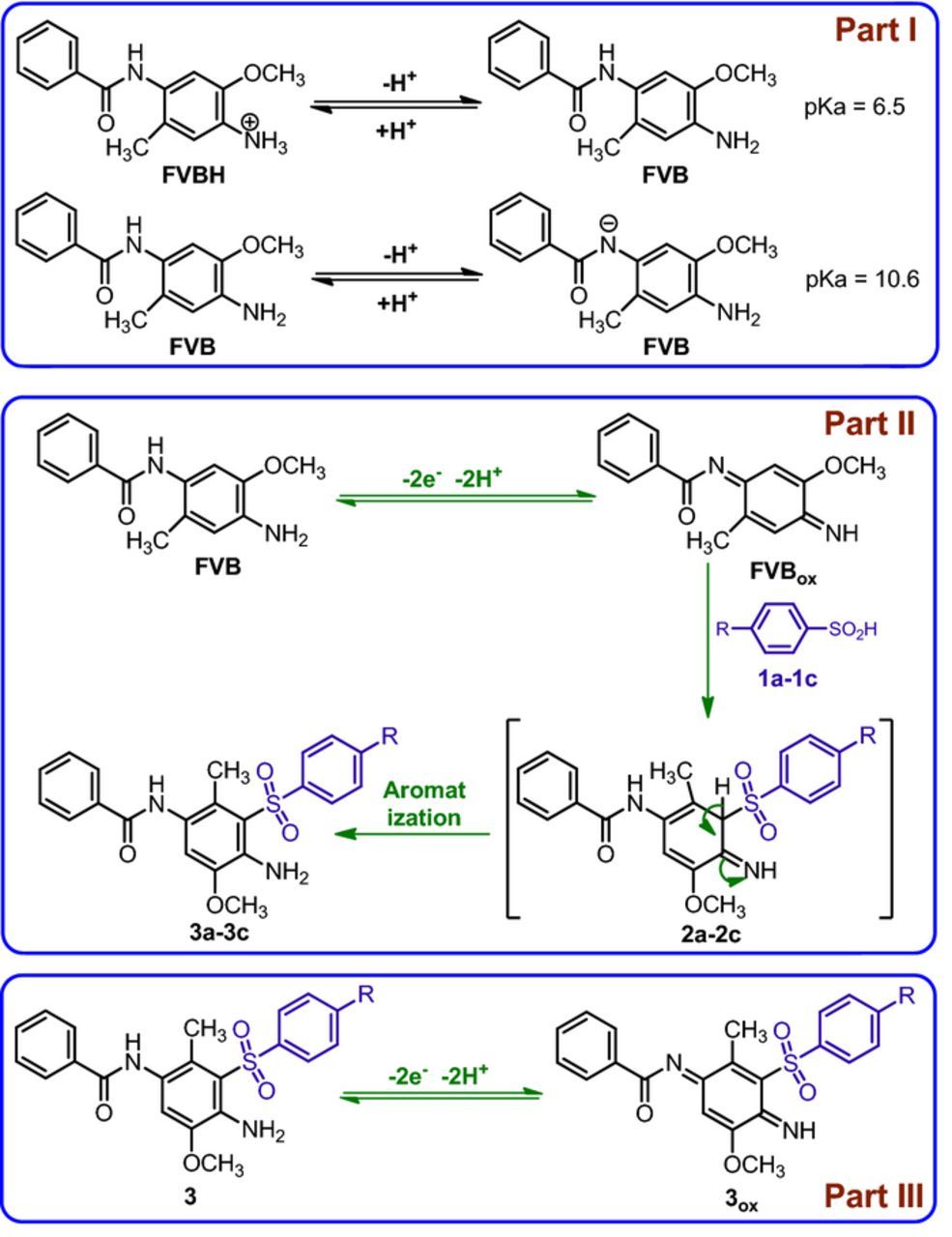
Scheme 1. Part I: Acid/base behaviors of FVBH/FVB and FVB/FVB−. Part II: Proposed mechanism for the electrochemical oxidation of FVB in the presence of arylsulfinic acids. Part III: Redox behavior of 3/3ox.
By increasing pH, EpA1 and EpC1 shifts to negative values. This confirms the participation of proton(s) in the redox behavior of FVB/FVBox. The variation of EpC1 with pH is described by:
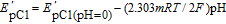
where m is the number of protons involved in the reaction and E'pC1(pH = 0) is the peak potentials at pH = 0.0. R, T and F have their usual meanings. The Pourbaix diagram for peak C1 includes three lines with different equations and slopes around pH values 6.5 and 10.6 (Fig. 1, Part II).
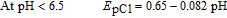
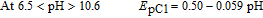
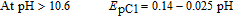
It can be concluded that the electrode reaction occurring at the pH lower than 6.5 is a two-electron/three-proton process involving the oxidation of the "protonated-FVB" (FVBH) to FVBox and vice versa. On the other hand, at 6.5 < pH > 10.6, the EpC1 value shifts by −59 mV per pH, indicating that the electrode reaction is a two-electron/two-proton process involving the oxidation of FVB to FVBox and vice-versa. Whereas, in the pH > 10.6, the EpC1 value shifts by −25 mV per pH, indicating that the electrode reaction is a two-electron/one-proton process involving the oxidation of FVB− to FVBox and vice versa. The pKa' for acid/base equilibria, FVBH/FVB and FVB/FVB− is 6.5 and 10.6 respectively (Scheme 1, Part I).
Another noteworthy behavior was observed in the basic solutions (pHs 9 and 10). In this pH region, CV of FVB in water (bicarbonate buffer, c = 0.2 M, pH 9.0)/ethanol (70/30, v/v) mixture show two anodic (A1 and A2) and two cathodic (C1 and C2) peaks (Fig. 2, Part Ia and inset). In addition, the cyclic voltammogram of FVB in the same conditions, however in water/ethanol (50/50, v/v) mixture is shown in Fig. 2, Part Ib. As shown, with increasing ethanol percentage in the solvent mixture, the voltammogram tends to become regular in shape.
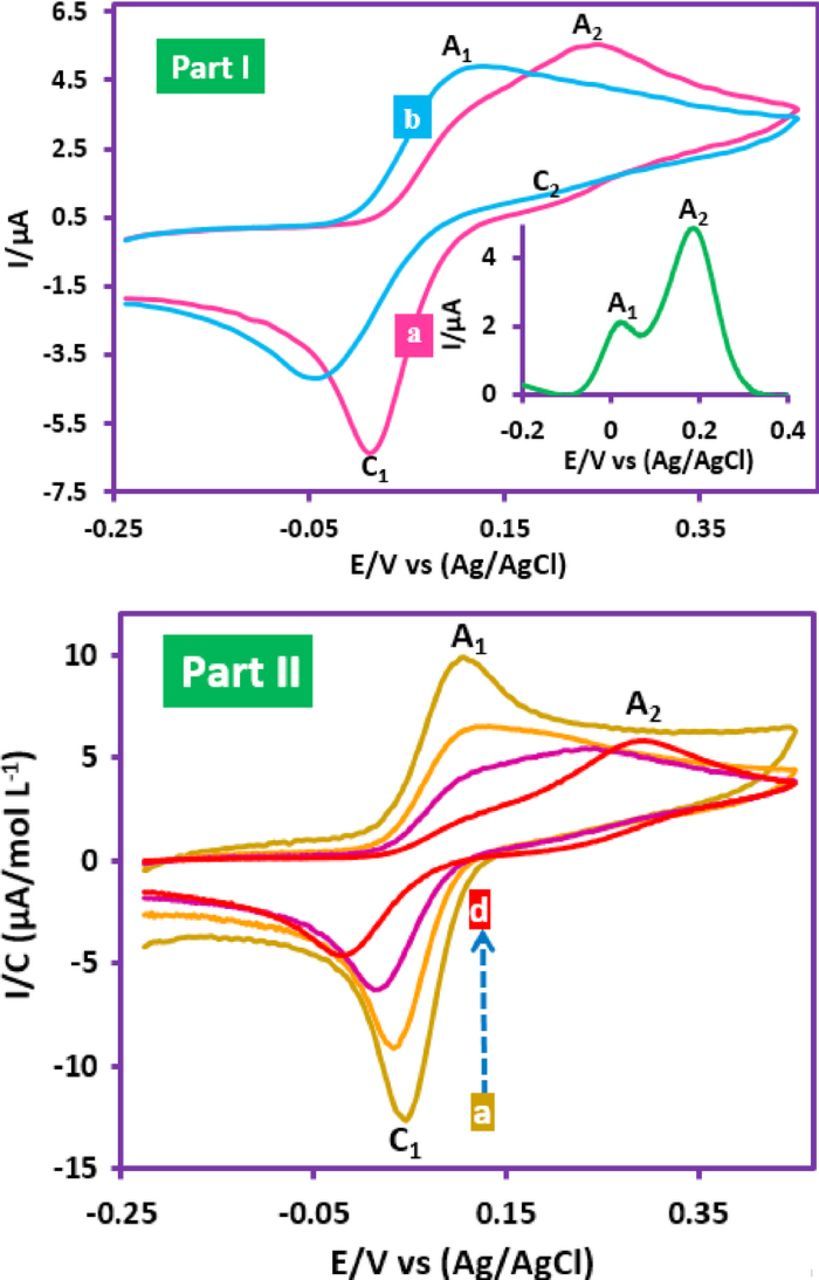
Figure 2. Part I: Cyclic voltammograms of FVB (1.0 mM) in water (bicarbonate buffer, c = 0.2 M, pH, 9.0)/ethanol mixtures. (a) water/ethanol 70/30, v/v. (b) water/ethanol 50/50, v/v. Inset: Differential pulse voltammogram of FVB at (a) conditions. Part II: Normalized CVs of 1.0 mM FVB in water (bicarbonate buffer, c = 0.2 M, pH 9.0)/ethanol (70/30, v/v) mixture in different concentration of FVB. Concentration from (a) to (d) are: 0.25, 0.5, 1.0 and 2.0 mM. Scan rate: 50 mV/s. Temperature = 25 ± 1 °C.
In addition, the effect of FVB concentration on the normalized cyclic voltammograms of FVB is shown in Fig. 2, Part II. Normalized cyclic voltammograms are obtained by dividing the current by the FVB concentration (I/c). It indicates that with increasing FVB concentration, the anodic peak current ratio (IpA1/IpA2) decreases. These data show the adsorption phenomena in electrochemical oxidation of FVB in the basic solutions.
Electrochemical oxidation of FVB in the presence of aryl sulfonic acids
The cyclic voltammogram of FVB in a water (perchloric acid, 0.1 M)/ethanol (70/30 v/v) mixture is shown in Fig. 3a. The cyclic voltammogram of FVB in the presence of p-toluenesulfinic acid (1a) as a nucleophile at the same conditions, was shown in Fig. 3b. Compared with Fig. 3a, the cathodic peak current (IpC1) significantly decreased and two new anodic peaks (A3 and A4) appears at more positive potentials. The decrease in IpC1 indicates that the electrochemically generated FVBox is consumed by a chemical reaction with 1a.48 In this Figure, curve c is the cyclic voltammogram of 1a in the absence of FVB. Comparison of curve b with curve c, shows that peak A4 is related to the oxidation of 1a.
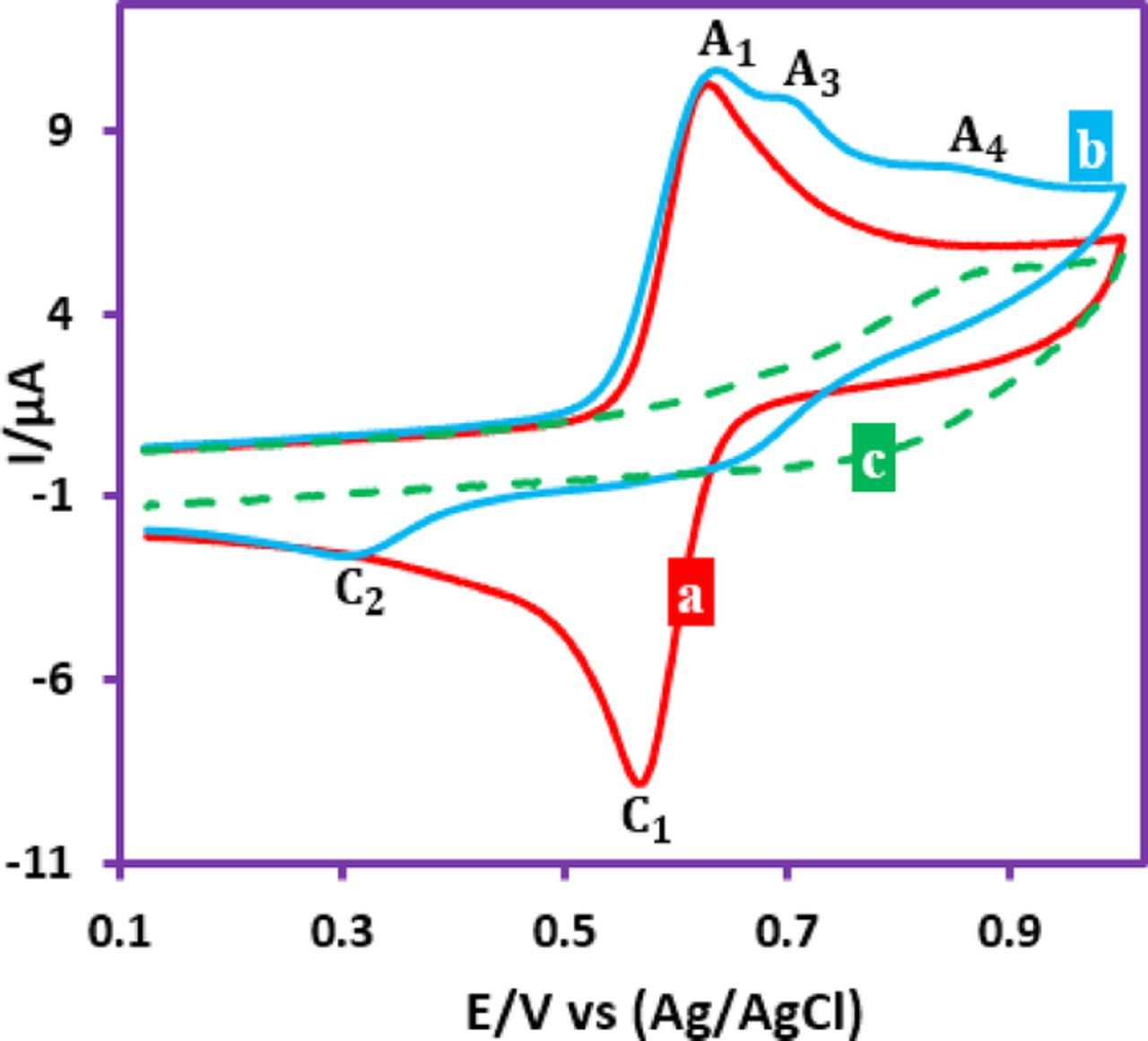
Figure 3. Cyclic voltammograms of FVB (1.0 mM): (a) in the absence, (b) in the presence of p-toluenesulfinic acid (1a) (1.0 mM) and (c) cyclic voltammogram of p-toluenesulfinic acid (1.0 mM) at glassy carbon electrode, in water (perchloric acid, 0.1 M)/ethanol mixture (70/30, v/v). Scan rate: 100 mV/s. Temperature = 25 ± 1°C.
More data were obtained by changing the potential scan rate in a solution of FVB in the presence of 1a (Fig. 4, Part I). The results indicate that the peak current ratio (IpC1/IpA1) is dependent on the potential scan rate, so that, it increases with increasing scan rate. The increasing IpC1/IpA1 at faster scan rates is related to the decrease of the experimental time scale. At this condition, a small amount of FVBox could be reacted with 1a. The same result was obtained by decreasing the concentration of 1a (Fig. 4, Part II). As can be seen, with increasing 1a concentration: (1) peak C1 has been removed. (2) Peaks A3 and A4 have been increased. The increasing of 1a concentration causes an increase in the reaction rate of FVBox with 1a and thus decreasing the IpC1. Since, peak A3 is related to the oxidation of the product formed after the reaction of 1a with FVBox, it increases with increasing 1a concentration (Part II) and decreasing potential scan rate (Part I).
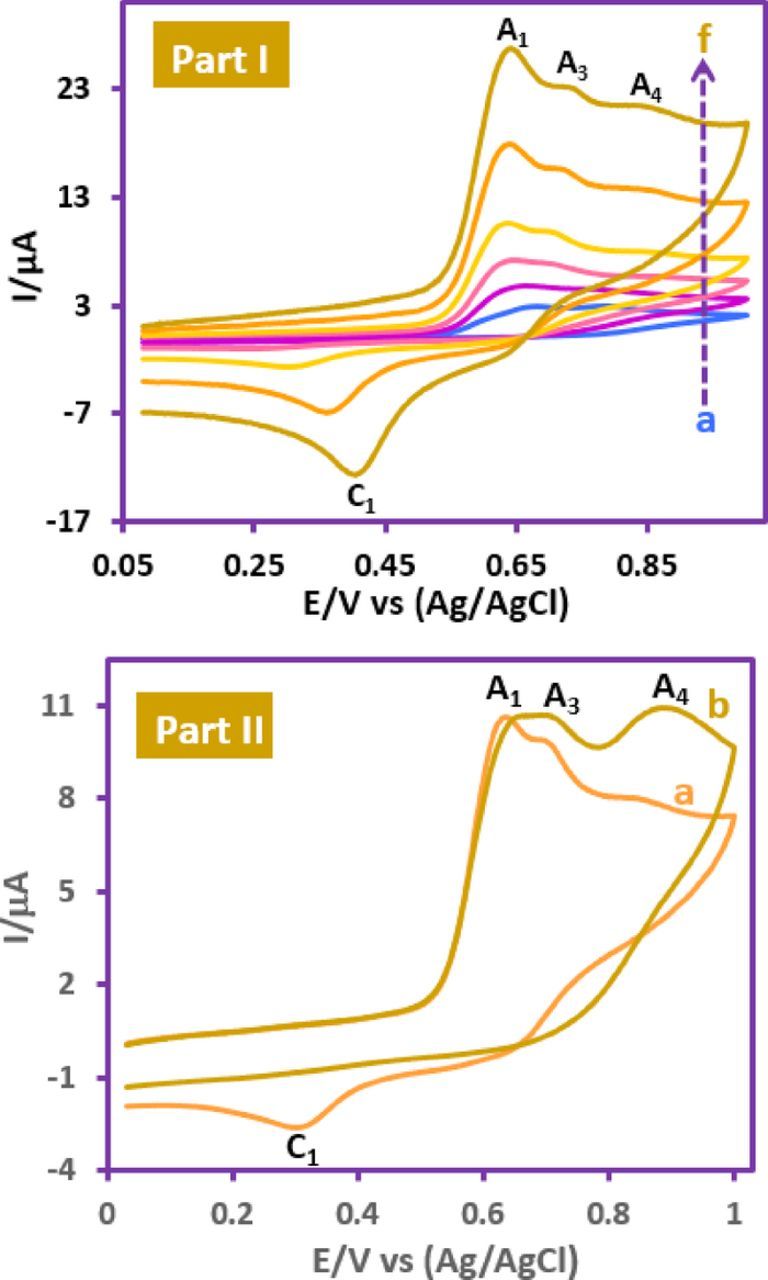
Figure 4. Part I: Cyclic voltammograms of FVB (1.0 mM) in the presence of p- toluenesulfinic acid (1a) (1.0 mM) at various scan rates. Scan rates from (a) to (f) are: 10, 25, 50, 100, 250 and 500 mV/s, respectively. Part II: Cyclic voltammograms of FVB (1.0 mM) in the presence of (a) 1.0 mM of p-toluenesulfinic acid (1a) and (b) 2.0 mM of 1a, at glassy carbon electrode in water (perchloric acid, 0.1 M)/ethanol mixture (70/30, v/v). Temperature = 25 ± 1°C.
Controlled-potential coulometry (CPC) was used to determine the number of electrons involved in the oxidation of FVB in the presence of 1a. CPC was performed in water (perchloric acid, 0.1 M)/ethanol (70/30 v/v) mixture containing FVB (0.25 mmol) and 1a (0.25 mmol) at 0.56 V vs. Ag/AgCl. The electrolysis progress was monitored using cyclic voltammetry (Fig. 5). It was found that, proportional to the progress of coulometry, both anodic peaks (A1 and A3) decreased. These peaks disappeared when the charge consumption was 2.3 e− per molecule of FVB.
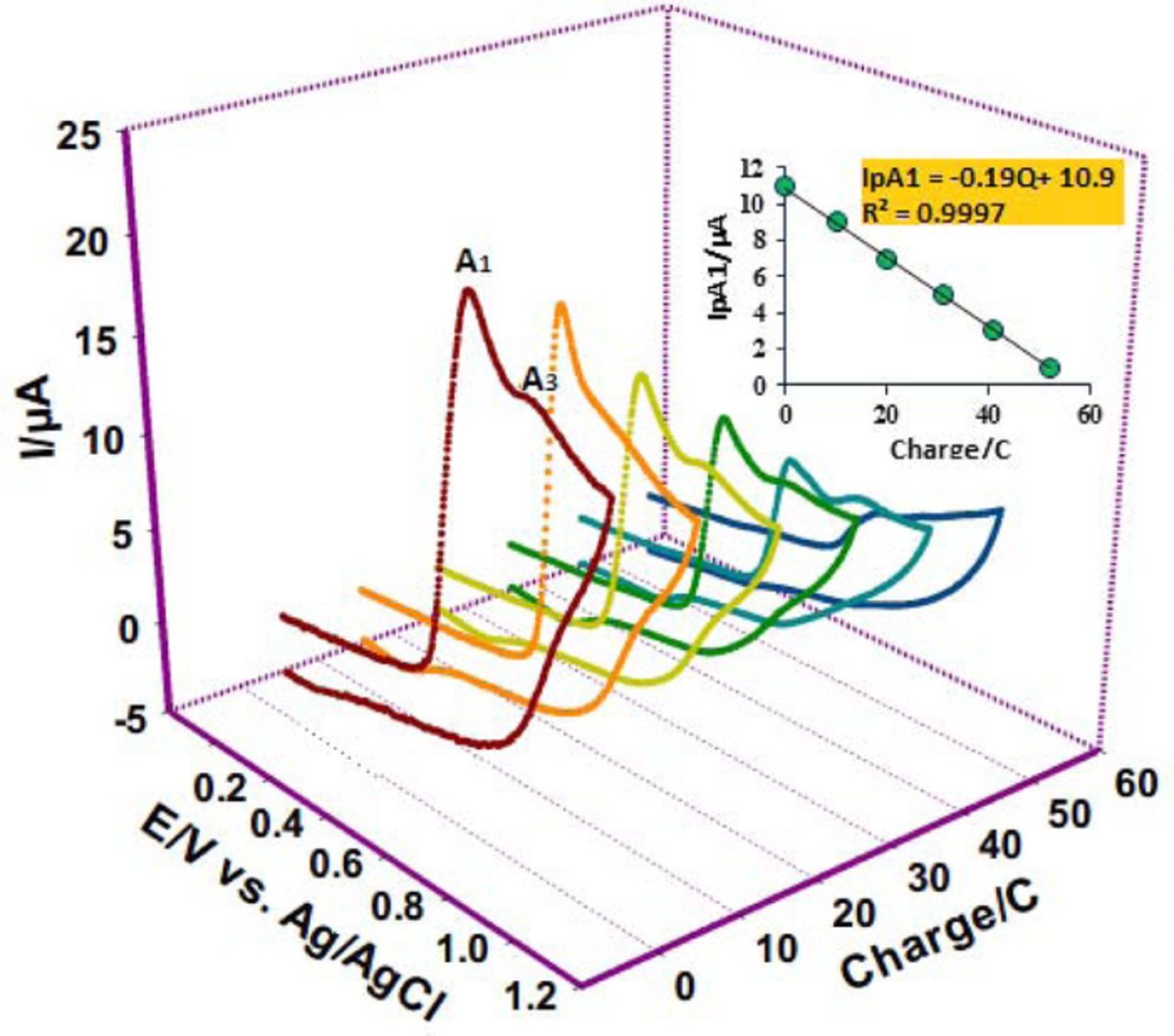
Figure 5. Cyclic voltammograms of FVB (0.25 mmol) in the presence of 1a (0.25 mmol) during controlled-potential coulometry at 0.56 V vs Ag/AgCl after consumption of: (a) 0 (b) 10, (c) 20, (d) 30, (e) 40 and (f) 50 coulomb in water (perchloric acid, 0.1 M)/ethanol mixture (70/30 v/v). Scan rate: 100 mV/s .Temperature = 25 ±1°C.
Based on our experience in the electrochemical generation of quinone diimine49–51 along with the electrochemical and spectroscopic data of the final product, we propose the following mechanism for the electrochemical oxidation of FVB in the presence of 1 (Scheme 1, Part II). In the proposed mechanism, for simplification, the protonation of species is omitted. According to this Scheme, the generation of the FVBox is followed by a Michael type addition reaction of 1 producing the 2 which after aromatization to afford the 3 as the final product.
The oxidation of 3 is more difficult than the oxidation of FVB due to the presence of the electron-withdrawing phenylsulfonyl group as well as by the insolubility of the final products (3) in water (perchloricacid, 0.1 M)/ethanol (70/30 v/v) mixture. According to Scheme 1, Part 3, the anodic peak A3 pertains to the oxidation of 3a to its oxidized form 3aox.
Computational study
The suggested mechanism for the reaction of FVBox with 1a has been studied using density functional theory (DFT). In order to study the Michael acceptor (FVBox) reactivity toward the reaction with 1a, we have investigated four parameters, natural charge and LUMO coefficient of FVBox and thermodynamic and kinetic stability of possible products. FVBox can be attacked by 1a from sites A, B,C or D to yield four types of compounds (3a, 4a, 5a and 6a) (Scheme 2). However, 1H NMR results derived in this work (presence of a singlet aromatic peak at 7.14 ppm) are in contrast with the nucleophilic attacked of 1a to FVBox from N situation. Thus, further theoretical studies will be done according to the paths A and B. The geometry of the compounds 3a and 4a investigated here were optimized at BP86/def2-SVP level and are given in Fig. 6.
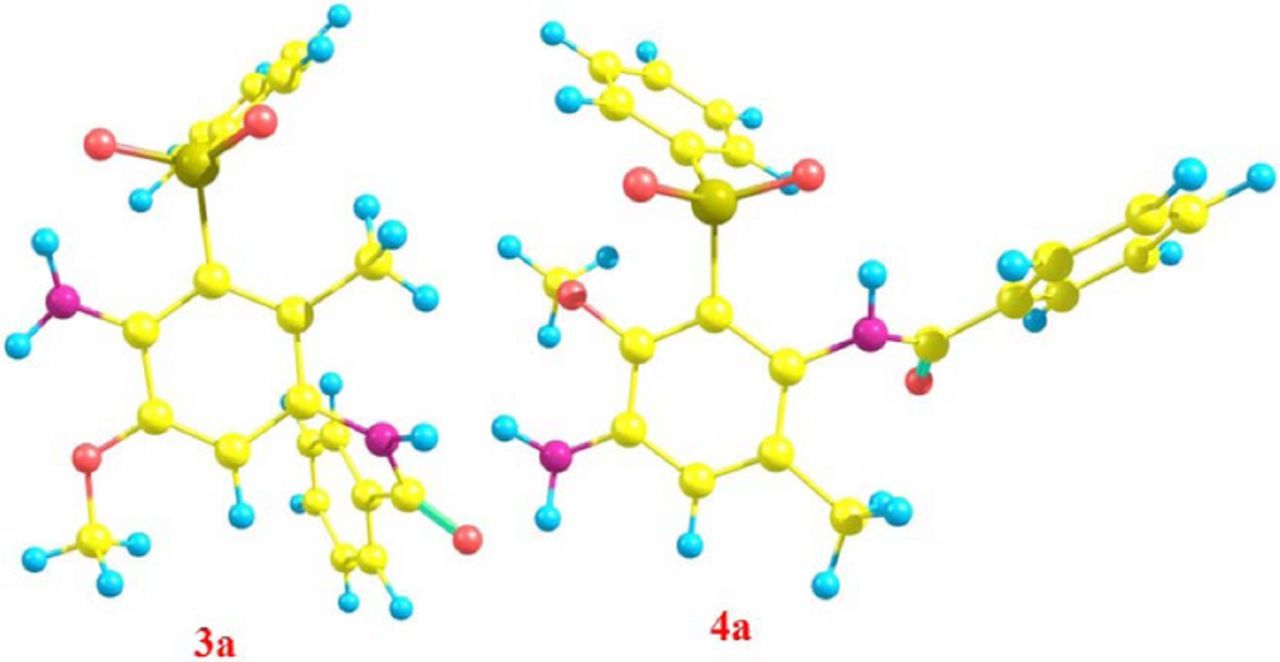
Figure 6. The optimized structures of 3a and 4a at BP86/def2-SVP level of theory.
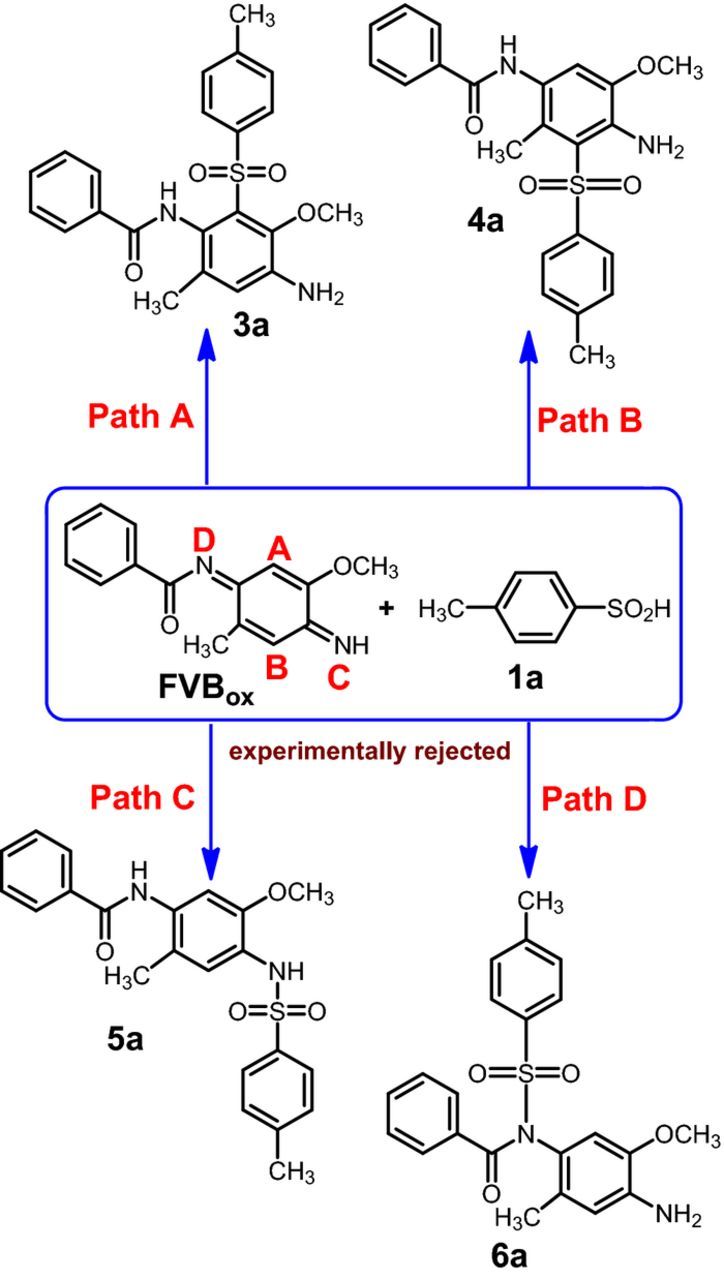
Scheme 2. The structures of possible compounds from the oxidation of NAP with 1a.
The NBO analysis was used for calculation of the partial charge as well as LUMO coefficient of each atom in FVBox investigated here at BP86/Def2-SVP level of theory. As shown in Fig. 7, the natural charges of C1 and C4, in FVBox are about −0.31 and −0.26e. This result shows that C4 is slightly positive than C1. On the other hand, the calculated LUMO coefficient for latter carbon in excellent agreement with NBO analysis shows that the C4 with LUMO coefficient about 0.25 is more suitable site than C1 with LUMO coefficient about −0.30. Thus it can be concluded that the NBO analysis results as well as LUMO coefficient in conjugation with experimental data confirms that the C4 position, is more favored for latter electrochemical reaction.
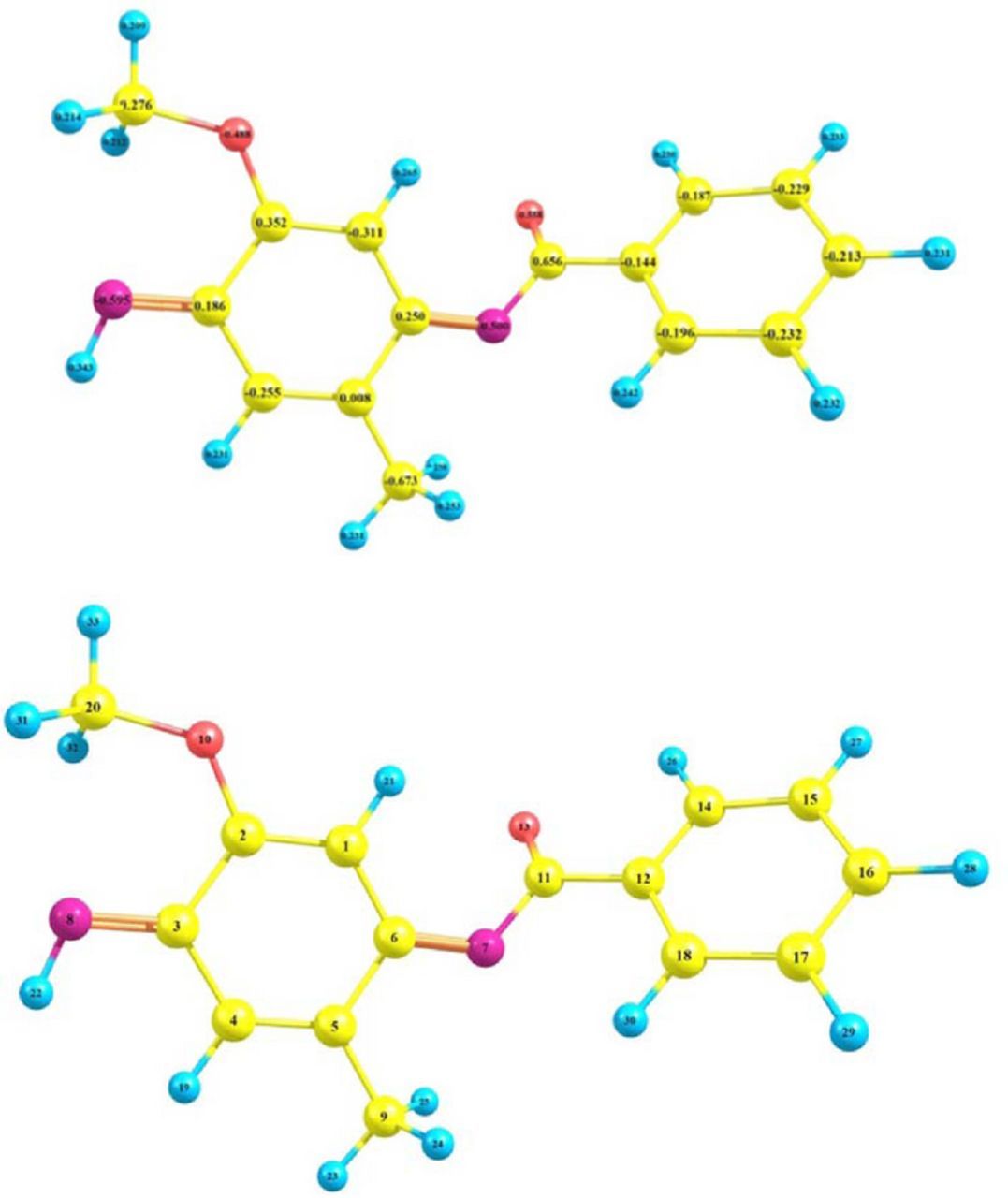
Figure 7. The natural charge of NAP calculated at BP86/def2-SVP levels of theory.
In continuation the thermodynamic stability of two possible products (3a and 4a) of latter electrochemical reaction were investigated. Fig. 6 shows the structure of two possible products according to proposed mechanism for latter electrochemical reaction. A comparison between the relative Gibbs free energies of two possible products 3a and 4a, are 0.0 and 4.6 kcal/mol, respectively. As can be seen, in good agreement with NBO analysis and calculated LUMO coefficient the relative Gibbs free energies show that the compound 3a is more stable than compound 4a.
The mechanism for the formation compounds 3a and 4a from the electrochemical reaction of FVBox with 1a has also been studied kinetically (Scheme 3). On the basis of proposed mechanism here, we could consider one intermediate as well as two transition states for the latter electrochemical reaction. The minimum energy path of the initial reaction starts with the nucleophilic attack of 1a to C4, C1 (Scheme 3, paths i and ii) of FVBox which led to the formation of Int-1 and Int-1-2 in paths I and II.
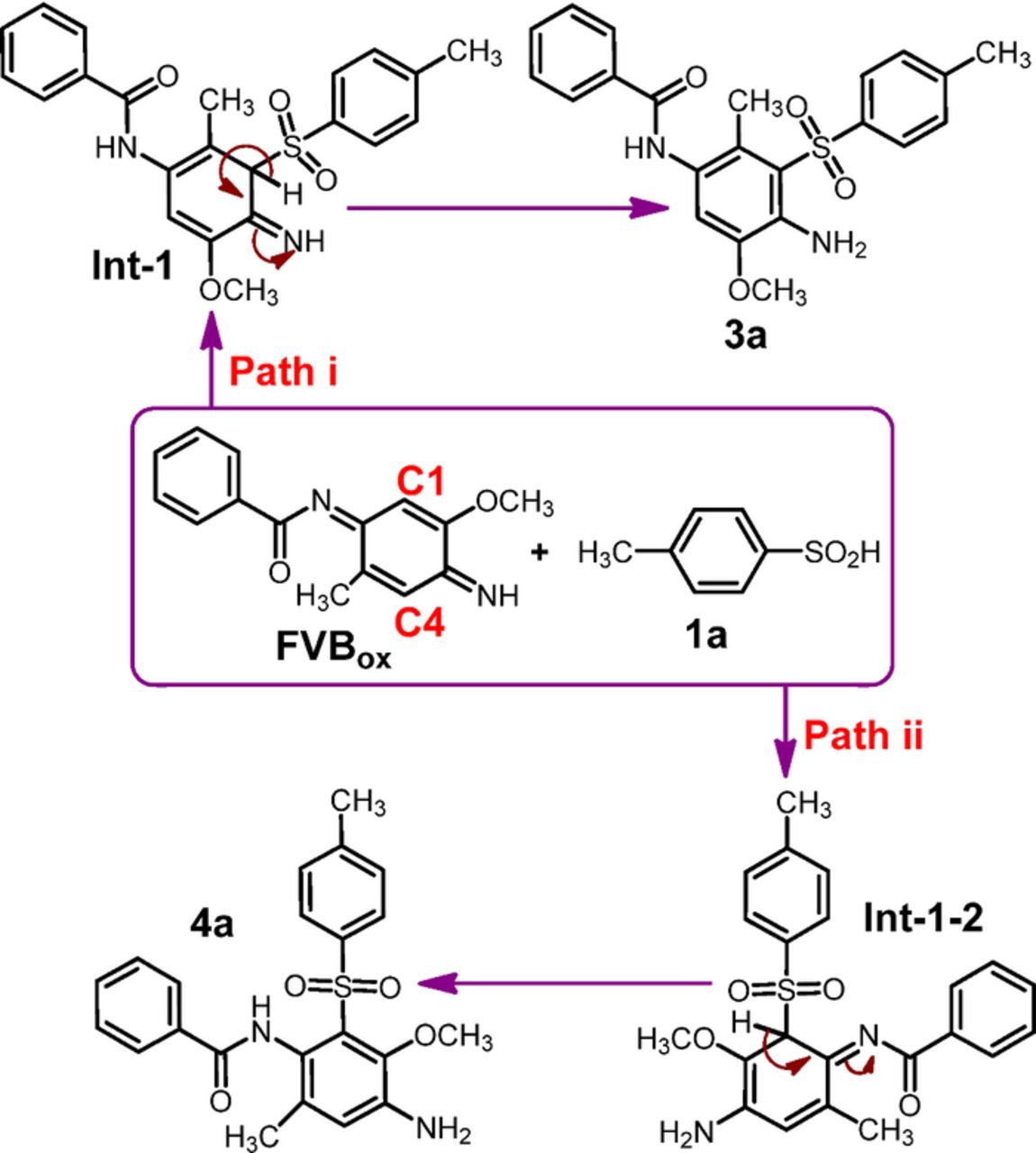
Scheme 3. The proposed pattern for the formation of compound 3a and 4a.
The calculated barrier energies for the formation of Int-1 and Int-1-2 are −7.7 and −3.5 kcal/mol and those for corresponding TS-1 and TS-1-2 are about 46.5 and 91.6, respectively (Scheme 3 and Fig. 8). The calculated barrier energies for the formation of compounds 3a and 4a are −31.8 and −27.2 kcal/mol, respectively and those for corresponding TS-2 and TS-2-2 are about 31.2 and 33.5 kcal/mol (Scheme 3 and Fig. 8). The result confirms that the formation of 3a not only thermodynamically but also kinetically more favorable than 4a, and transition states in the path I is less endothermic than path II.
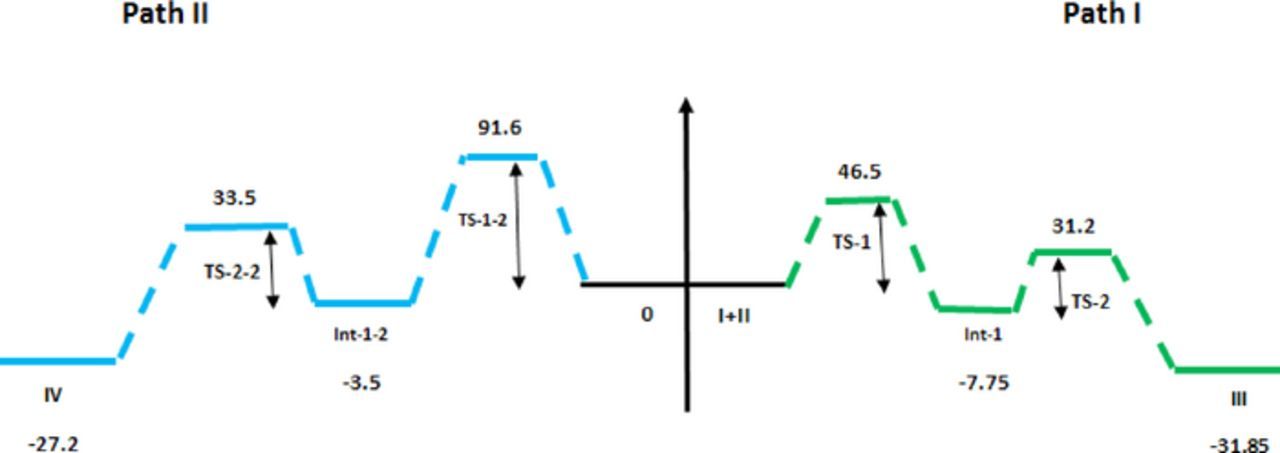
Figure 8. Calculated energy profile for the electrochemical reaction of NAP with 1a.The relative Gibbs free energy obtained at BP86/def2-SVP.
According to these data, it can be declared that final products influenced by four parameters, natural charge and LUMO coefficient of Michael acceptor and thermodynamic and kinetic stability of possible products. Also according to above data we can conclude that the 3a is the most favorable product obtained from the electrochemical reaction.
Constant current electrolysis
Among the electrochemical parameters for the synthesis of organic compounds, the current density is one of the most important factors influencing the yield and purity. The synthesis of 3a was performed via the constant current electrolysis under the same conditions as described for controlled-potential electrolysis when the theoretical amount of electricity (50 C) was passed. In this study, the current density varied from 0.01 to 0.75 mA/cm, while the other parameters (temperature = 298 K, FVB amount: 0.25 mmol, and 1a amount: 0.25 mmol) are kept constant. Our results show that the highest yield (88%) was obtained at a current density of 0.05 mA/cm2. Fig. 9, Part I, shows the effect of current density on the yield of 3a. In this current density, the electrode potential remained nearly constant at +0.56 V (Fig. 9, part II), which is necessary for the oxidation of FVB. Higher current densities result in an increase in side reactions such as the oxidation of 3a, solvent and ...., resulting in a decrease in product yield.
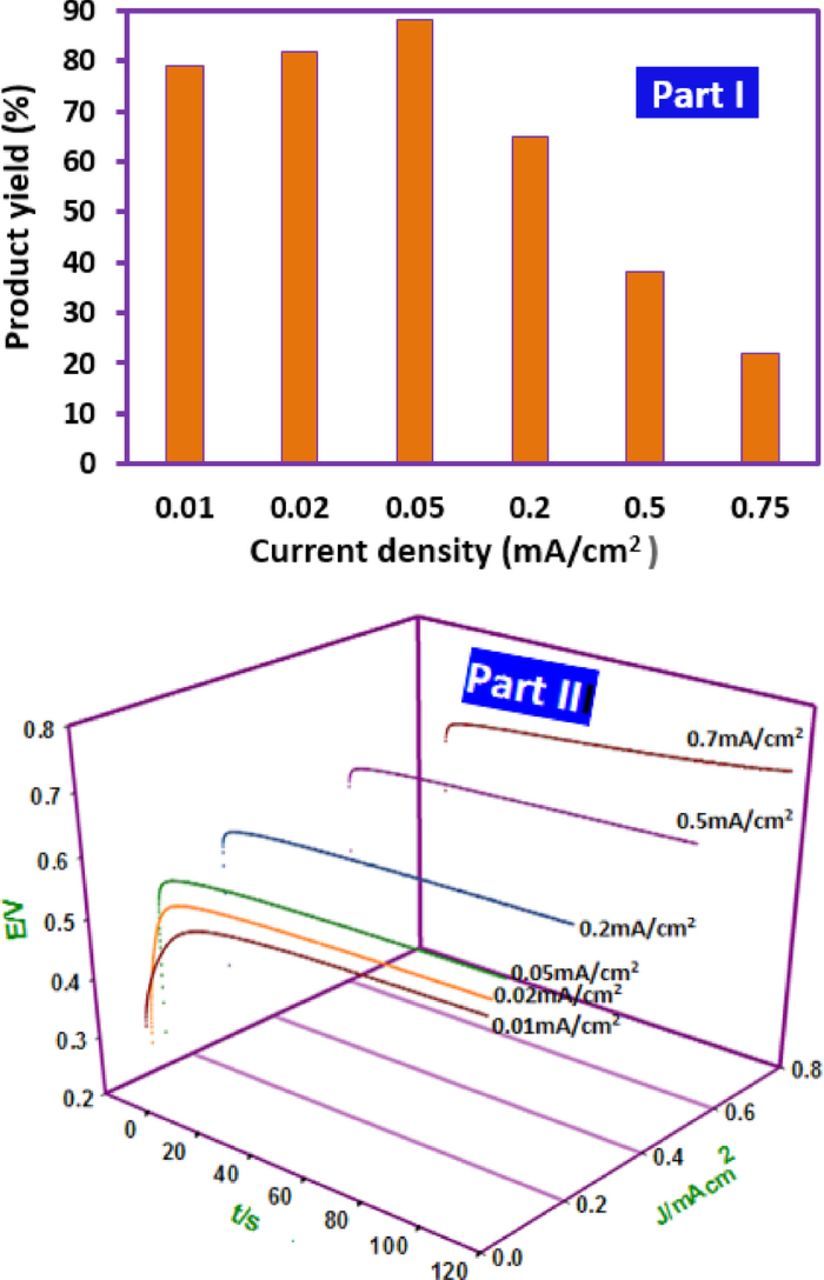
Figure 9. Part I: Effect of current density on the yield of 3a. Charge consumed, 50 C. Part II: The potential-time diagram during constant current electrolysis FVB (0.25 mmol) in the presence of 1a in ethanol/aqueous perchloric acid (c = 0.1 M) mixture (80 ml) (30/70, v/v) solution. Electrode rotation rate: 1000 rpm. Other conditions are as same as Fig. 5.
Conclusions
This paper follows two main objectives. First, further insights into the electrochemical oxidation of FVB in aqueous solutions and second, the synthesis of new diaryl sulfone derivatives via a green method. To achieve the first objective, electrochemical oxidation of FVB has been studied in different pH values and its redox behavior was investigated in detail and assigned as shown in Figure 1, Part II. To attain the second objective, electrochemical oxidation of FVB has been studied in the presence of arylsulfinic acids (1a-1c) and the obtained electrochemical data was used in the electrochemical synthesis of diaryl sulfone derivatives 3a-3c. In this work, some new unsymmetrical diaryl sulfones were synthesized via anodic oxidation of FVB in the presence of arylsulfinic acids (1a-1c) as nucleophiles in a water/ethanol mixture via a facile and environmentally friendly method under ambient conditions in good yields and purities. In addition, the structure of products were optimized at the BP86/def2-SVP level of theory by the four parameters, natural charge, LUMO coefficient of Michael acceptor, thermodynamic and kinetic stability.
Acknowledgments
We acknowledge the Bu-Ali Sina University Research Council and the Center of Excellence in Development of Environmentally Friendly Methods for Chemical Synthesis (CEDEFMCS) for their support of this work.