Abstract
Electrolyte instability in batteries at the electrolyte/Li interface is the bottleneck for transition from graphite to Li metal anodes. Hence, search for battery electrolytes with good chemical and electrochemical stability, for use in Li-ion and beyond Li-ion battery chemistries, is one of the important research areas in electrochemical energy storage. Room temperature ionic liquids (RTIL) feature wide electrochemical windows, low vapor pressures and good thermal stability, and due to their intrinsic properties, RTILs are researched as potential alternatives to currently used liquid electrolytes. In this paper, we report systematic investigation of chemical and electrochemical stability of barely researched cyclic phosphonium cation based ionic liquids (CylP+nA−; n = 5 or 6). We systematically screen a combinatorial library of linear alkane −, cyclic alkene −, ethers − and amines − functionalized CylP+nA− ionic liquids based on their electrochemical stability at Li interface and future possibilities to improve desired properties, and identify the candidate materials among the variants in the library. The electrochemical stability of linear alkane functionalized CylP+5 ions at the Li interface, and the possibilities to engineer thermophysical properties by facile chemical modifications, make them potential candidate materials for use as electrolytes in advanced Li batteries.
Export citation and abstract BibTeX RIS
Realizing high capacity Li-ion batteries and advanced Li batteries requires deploying electrodes with higher energy densities. Moving from graphite to Li metal anode multiplies the energy density of anodes by a factor of approximately 10, however, transition from graphite to Li metal anode is limited by electrode corrosion, Li plating and dendritic growth resulting from electrochemical instability of traditionally used organic electrolytes. Though a lot of research has been carried out over the past two decades in the direction of design and development of stable electrolytes and protective coatings for Li metal anodes, to limit electrode corrosion and structural degradation caused by electrolyte breakdown at the electrode/electrolyte interface, it still remains as one of the important and emerging research areas in electrochemical energy storage.1,2
The important features to look out for while selecting an electrolyte for batteries are the chemical and electrochemical stability (at electrode/electrolyte interfaces), thermal stability and good ionic conductivity. Room Temperature Ionic Liquids (RTILs) are organic salts composed of weakly bound cation/anionic species3 which have been reported as potential alternatives to currently preferred liquid electrolytes.4 RTILs feature wide electrochemical windows, low vapor pressures and good thermal stability and due to their intrinsic properties, RTILs have been used in both chemical and electrochemical applications such as catalysis,5,6 organic synthesis,7,8 batteries, fuel cells and organic solar cells.2,9 Utilizing wider electrochemical windows and thermal stability of RTILs, it is considered possible to design stable electrolytes and realize batteries with high energy density and enhanced safety.4,10 Hence ILs with the cations based on imidazolium, piperidinium, pyrrolidinium and tetraalkyl − phosphonium, and a few functionalized derivatives of the above cations, along with commonly used anions, were researched as potential alternatives to currently used organic electrolytes.4,11–22 However, compared to the enormous chemical space spanned by the combinations of cations plus their functionalized derivatives and anions that can form ILs, the amount of exploration done so far is minimal.23,24 There remains a lot of opportunity available in exploring the rich chemical landscape of ILs, and the potential to discover a better electrolyte for the next generation electrochemical energy storage devices exists.
In this paper, we present a systematic investigation of electrochemical stability of barely researched cyclic phosphonium (or phospholanium) cation CylP+n(n = 5 or 6) based ionic liquids. CylP+n (n = 5 and 6) are phosphorous analogs of pyrrolidinium and piperidinium cations (shown in Fig. 1). Despite being conceptualized and patented few years ago,25 and studied using first principles calculations in few publications,26,27 CylP+nA− ILs were not routinely synthesized and characterized,28,29 and detailed electrochemical experiments to qualify CylP+5 based ILs as battery electrolytes are yet to be performed. In this paper, we study the electrochemical stability of functionalized CylP+n (n = 5) ions to explore the chemical space spanned by functional modifications and to identify the relavant chemical structures that are potential alternatives to currently used organic electrolytes.
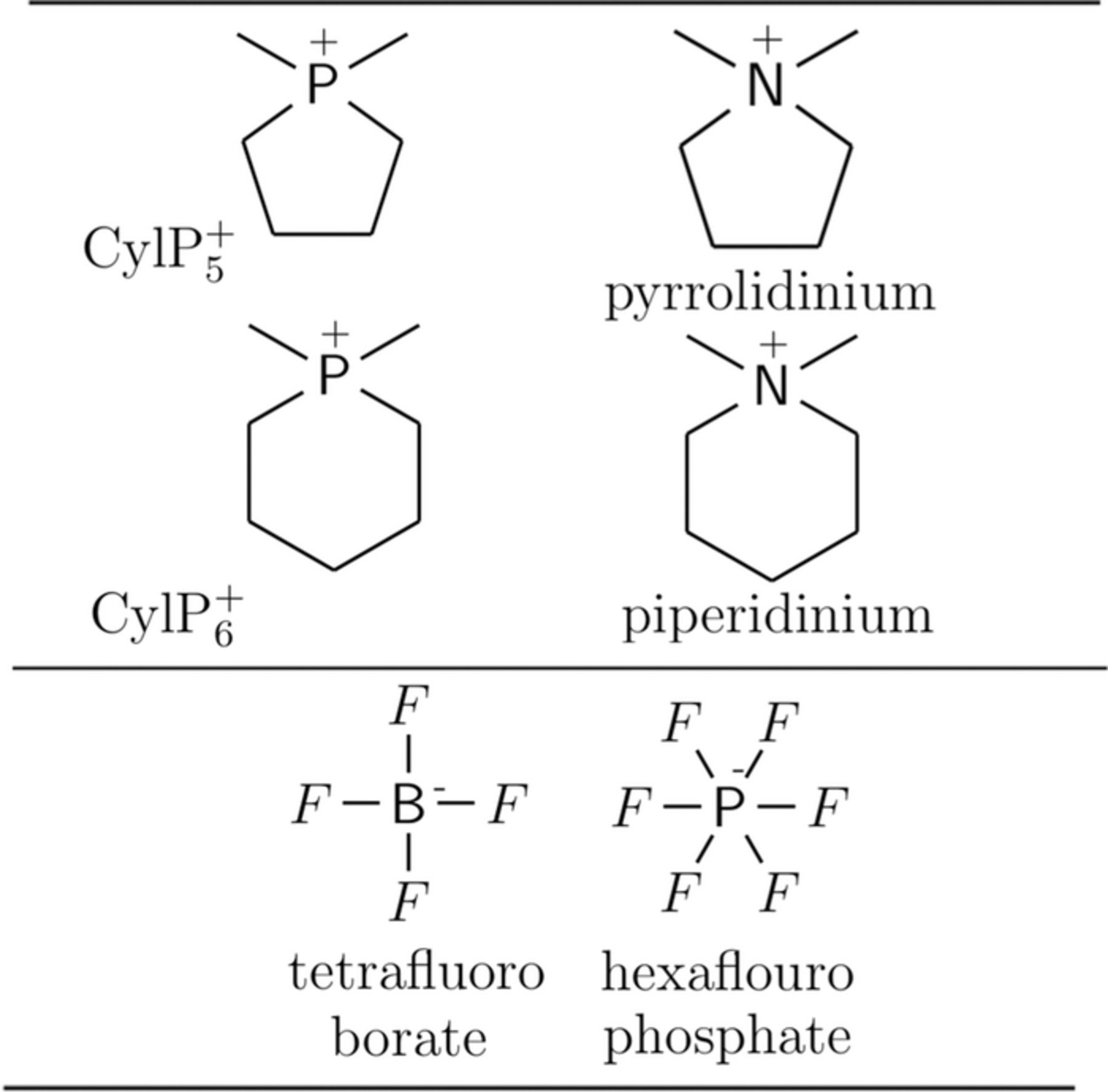
Figure 1. Phosphorous analogs of Pyrrolidinium, piperidinium ILs and commonly used anions.
In phosphonium cation IL family, functionalized acyclic tetraalkyl phosphonium cations (analogs of tetraalkylammonium ILs) were studied extensively and concluded in the literature as better candidates for electrolytes with higher reduction stability and lower viscosity compared to the nitrogen analogs.30–38 The general consensus from the first principles calculations on acyclic phosphonium cations is that the phosphonium ILs have wider electrochemical stability windows (EW) and hence higher cathodic limits relative to nitrogen analogs.26,27,39 The better reduction stability of phosphonium cations is ascribed to the empty d-orbitals in phosphorous that result in stronger binding of functionalities, and hence increased resistance to reduction.27
In previous studies, the reduction stability of phospholanium cations was calculated from 1e− reduction calculations resulting in annihilation of cation and subsequent dissociation of functionalities. Though it seems to be the dominant pathway, more than one pathway is probable for 1e− reduction when certain functionalities (such as ethers, aminophenyl) are used to functionalize base phospholanium cations. In this paper, we explore more than one reaction pathway for 1e− reduction (and subsequent electron transfers) to get insights into the electrochemical stability of CylP+n cations and the functional substitutents. We show that the inherent reduction/ oxidation stability of the functional groups (and the substituents on the functional group) is also important in determining the overall electrochemical stability of cations.
Here we report the calculations of model R − CylP+5 − R' ions – variants with alkane −, amine −, ether − and cyclic alkene − functional group substitution at the two free terminals (R and R') of the CylP+5 ions (shown in Figure 2). The choice of functional groups include some moieties with electron-withdrawing character and the others with electron-donating character since the substituent constants have significant effect on the reduction stability of CylP+5 ions. Electron donating/electron withdrawing character of the functional group affects the reduction stability of the cations where the propensity to acquire an electron decreases when functionalized by electron-donating groups (EDGs) and increases when functionalized by electron-withdrawing groups (EWGs).39 It is intuitive and we also find in our calculations that electron-donating groups (EDGs) like alkyls, cyclic alkenes and amines increase the reduction stability compared to ether substituents (EWGs). However, substituent constants alone cannot explain the electrochemical stability of R − CylP+5 − R' ions; solvation energies and enthalpy of lithiation that determine the inherent redox stability of substituents also play a significant role. We discuss the contribution of the above factors in determining the electrochemical stability of R − CylP+5 − R' ions in this paper.
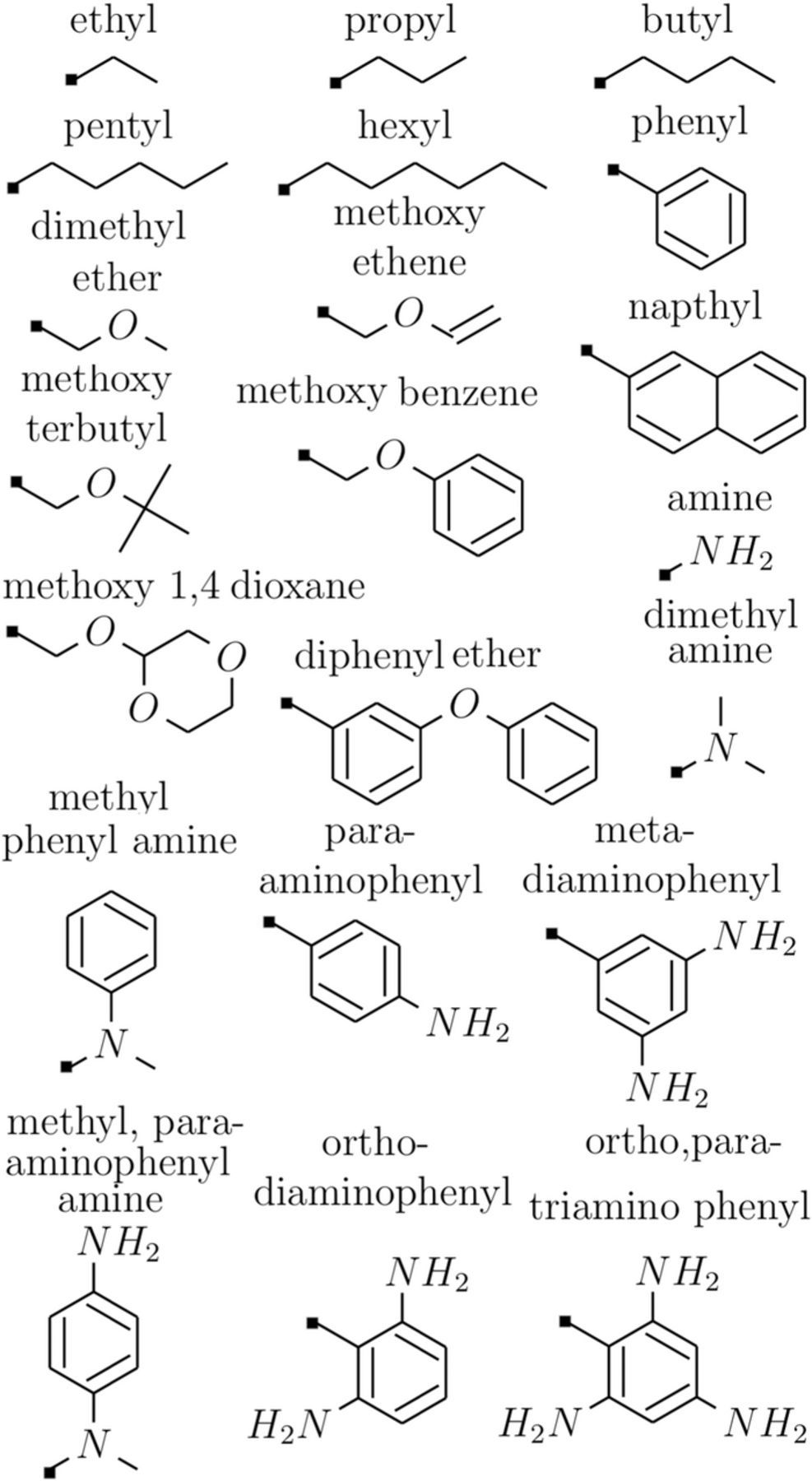
Figure 2. List of functionalities: linear alkyls, cyclic alkenes, ethers and amines.
Methodology
We have constructed a model of R − CylP+5 − R' ion with two methyl groups (as the starting point) and generated a combinatorial library with a new set of cations by symmetric (R = R') and asymmetric substitution (R = CH3; R ≠ R') of 23 functionalities categorzied into four sets: linear alkanes, cyclic alkenes, ethers and amines. We use DFT to calculate redox free energies and electrochemical stabilities of functionalized R − CylP+5 − R' ions. Theory and computational approach to calculate EWs and electrochemical stability against Li+/Li are presented here. The model reactions (schemes A–C) for 1e− reduction to determine the electrochemical stability are also presented.
Quantum chemistry based methods such as Density Functional theory (DFT) are very well suited to predict electrochemical stabilities of electrolytes and lithium salts and identify candidate materials.39–45 In the past, quantum chemistry calculations have been extensively used by multiple research groups to provide invaluable insights into electrolyte decomposition pathways that determine the inherent redox stability of electrolytes in the bulk and at the electrode/electrolyte interfaces.46–53 High-throughput computational methodology employing DFT was also reported to screen the combinatorial libraries of probable candidate materials for electrolytes based on the relative energy level alignment (HOMO/LUMO) at the electrode/electrolyte interfaces.54,55
Electrochemical windows
EW is a good descriptor to predict electrochemical stability of IL electrolytes. It is typically calculated as the difference between their oxidation and reduction potentials (anodic (VAL) and cathodic (VCL) limits).26,56 When ILs are deployed as electrolytes in Li-ion batteries, VAL and VCL are compared against the redox potential of Li electrode  ,57 and it can be concluded that electrolytes are electrochemically stable when its cathodic limit is lower compared to standard Li electrode potential and anodic limit is higher compared to a given cathode. The reduction stability of an ionic liquid electrolyte when interfaced with Li electrode is a function of electron affinity of the cation (or anion), stability of corresponding dissociated species and the solvation energy of the free radicals.26,27 Similarly, the oxidation stability is a function of ionization potential of the anion (or cation) and the solvation energies of oxidized species. The electrochemical stability window is then calculated as the difference between the minimum of oxidation and reduction potentials of cations and anions.
,57 and it can be concluded that electrolytes are electrochemically stable when its cathodic limit is lower compared to standard Li electrode potential and anodic limit is higher compared to a given cathode. The reduction stability of an ionic liquid electrolyte when interfaced with Li electrode is a function of electron affinity of the cation (or anion), stability of corresponding dissociated species and the solvation energy of the free radicals.26,27 Similarly, the oxidation stability is a function of ionization potential of the anion (or cation) and the solvation energies of oxidized species. The electrochemical stability window is then calculated as the difference between the minimum of oxidation and reduction potentials of cations and anions.
The cathodic/anodic limits are set by either cations or anions that are determined by the minimum of redox potentials of both species. The ΔSCF procedure56 reported in the literature to calculate reduction potential overestimates the EW as the dissociation since solvation of radicals post reduction are not taken into account.26,27 Solvation energy of the functionalities depend on their corresponding radical-solvent interactions that affect the electrochemical stability of the cations. Hence, in this paper, the cathodic limit is calculated from the reduction free energies of cation plus the solvation energies of the dissociated radicals as shown in Figure 3 and described in Reaction schematics section. The anodic limit of an anion is calculated from the oxidation potentials as the difference in free energies of the ion and the oxidized species (without dissociation). The dissociation of anions that can proceed through multiple channels is not considered here. Hence, the anodic limit of ILs calculated in this paper correspond to the upper limit of anodic stability of a given IL pair.
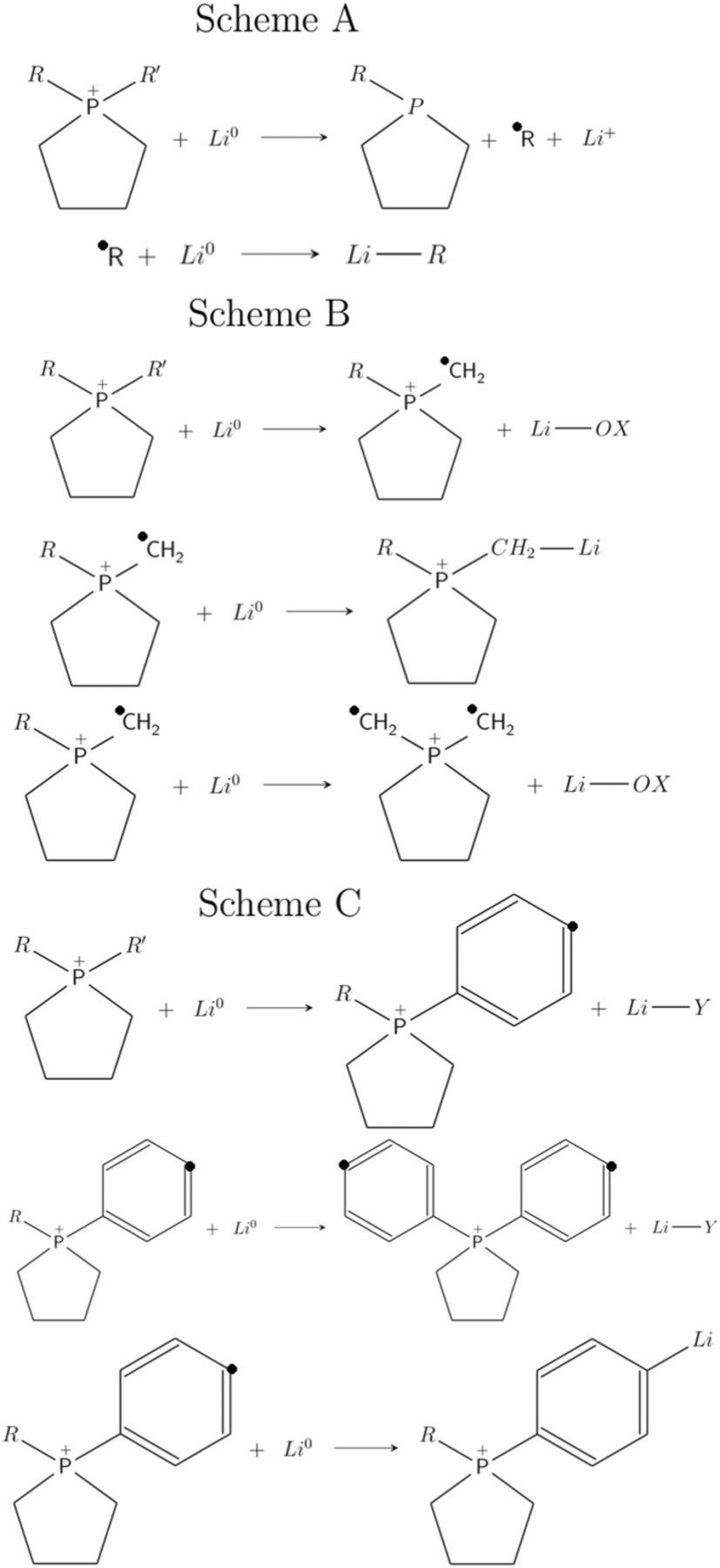
Figure 3. Schematics of 1e− and 2e− reduction reactions. Scheme A - Reduction-dissociation of cation followed by lithiation of solvated radicals; Scheme B and Scheme C – dissociation and lithiation of functional group followed by lithaition/reduction of phospholanium cation. R/R' in schemes B and C correspond to ethers and amino phenyl functionalities respectively. − OX in scheme B corresponds to organic groups, − oxymethyl, − oxyethene, − oxybenzene, − oxyterbutyl and − oxy1, 4 dioxane. Y in scheme C corresponds to organic groups NH2 and − OC6H5 (diphenyl ether).
Reaction schematics
The electrolyte decomposition at the Li interface can thus be summarized into four steps: (i) electron transfer from electrode to electrolyte, (ii) dissociation post reduction, (iii) solvation of radicals and (iv) lithiation of solvated radicals. The reaction free energies of the above four steps will determine the electrochemical stability of CylP+n ions at the Li electrode interface. Reduction potentials of the cations in the combinatorial library are thus calculated from the free energies of decomposition reactions (shown in Figure 3). The first and second electron reduction reactions were represented in Scheme A where the first electron transfer from Li electrode to cation results in (R − CylP+5) − R' bond dissociation and solvated neutral cyclic phosphonium and free radicals; subsequent electron transfer results in lithiation of the free radicals. For ether − and aminophenyl − functionalized cations, we explored other possibilities of decomposition, schemes B and C, where the lithiation of functional group precedes the reduction of phospholanium cation. The overall reduction stability is then determined from the minimum of reduction potentials from all the calculations. Oxidation potentials of cations (and anions) are calculated from the free energies of initial and corresponding oxidized species without dissociation.
Reaction free energies and redox potentials
For a given cation/anion, the 1e− reduction/oxidation reactions of cations are represented by R+ + e− → P and R+ → R2 + + e−, respectively, where R+ upon acquiring an electron is reduced to products, P, and upon losing an electron results in oxidized species R2 +. Oxidation of anions is represented by A− → A + e− where A− upon losing an electron results in neutral species A. The free energies of reduction and oxidation processes, ΔG°Red and ΔG°Ox, are calculated based on the thermodynamic cycles shown in Figure 4.
![Equation ([1])](https://content.cld.iop.org/journals/1945-7111/163/6/A1057/revision1/d0001.gif)
![Equation ([2])](https://content.cld.iop.org/journals/1945-7111/163/6/A1057/revision1/d0002.gif)
![Equation ([3])](https://content.cld.iop.org/journals/1945-7111/163/6/A1057/revision1/d0003.gif)

Figure 4. Thermodynamic chemical cycles for evaluating free energies of R − CylP+5 − R'A− redox reactions.
In the reduction cycle, R+ and P correspond to the reactants and products, ΔG°Rxn is the difference between free energies of products and reactants in gas phase, ΔG°s(P) and ΔG°s(R+) are the solvation energies of products and reactants. In oxidation cycles, R+/R2 + and A−/A correspond to initial and oxidized states respectively. EA(A) and IP are the electron affinity of the anion and ionization potential of the cation respectively. The cathodic and anodic limits with reference to Li electrode are then calculated as58
![Equation ([4])](https://content.cld.iop.org/journals/1945-7111/163/6/A1057/revision1/d0004.gif)
![Equation ([5])](https://content.cld.iop.org/journals/1945-7111/163/6/A1057/revision1/d0005.gif)
where 4.44 V and −3.05 V correspond to the SHE value in absolute potential scale at room temperature and the Li standard electrode potential Vs SHE for aqueous solutions, respectively.59 It is then concluded that the cation is electrochemically stable to reduction upto the cathodic limit against Li+/Li.
The free energies of lithiation resulting from subsequent electron transfers are calculated as
![Equation ([6])](https://content.cld.iop.org/journals/1945-7111/163/6/A1057/revision1/d0006.gif)
where GLi − R is the free energies of lithiated radical, and μLi is the chemical potential of lithium in Li electrode, respectively. μLi is calculated as the sum of the energy of Li atom in solution and the enthalpy of sublimation of lithium (ΔfH°gas(Li) = 1.65 eV).60 The free energies of lithiation for reactions shown in schemes B and C are also calculated using similar equation as above.
Computational methodology
Geometry optimization calculations in vacuo and in solution using a continuum solvation model were performed using a meta-GGA functional, M06-L, and triple-ζ basis, 6-311+G(2d,p) as implemented in Gaussian09 quantum chemistry package.61 Meta-GGA functionals in combination with double-ζ and triple-ζ basis sets were reported to result in lowest mean absolute deviation in interaction energies of 236 different ion-pair strcutures containing both protic and aprotic ionic liquids.62 In conjunction with M06-L, we have used SMD (universal solvation model)63 for implicit solvation calculations which is based on quantum mechanical charge density of a solute molecule interacting with a dielectric continuum. The solvation free energy comprises bulk electrostatic contribution calculated using self consistent reaction field approach in integral-equation-formalism polarizable continuum model (IEF-PCM) and cavity-dispersion-solvent-structure term arising from short-range interactions between the solute and solvent molecules. The dielectric constant of ε = 10.186 ( ∼ εRTIL) is used in all the calculations. Models of solvated ILs are important since stabilization of reduced/oxidized ions by the solvent molecules must be taken into account and typically performed in vacuo (gas phase) calculations lack proper description of molecule-solvent interactions. Since it is computationally expensive to use fully solvated IL models, solvent approximation in the form of a solute-molecule interacting with a dielectric continuum with a certain ε value (that matches with bulk ILs) is often employed to correct energies calculated in vacuo.45 All the energies reported in this paper correspond to energies including solvation and free energy contributions to the total energy were calculated from vibrational frequencies and statistical mechanics corrections.
Results
Calculated free energy profiles of one and two e− reduction reactions for representative models are shown in Figures 5a–5d. Calculated free energies of one and two e− reduction reactions for all symmetrically and asymmetrically substituted R − CylP+5 − R' ions are listed in Supp. Info, Tables S1 and S2. Li electrode reference is also shown in the figure, ΔG°ref = −1.4 eV. The reaction products with energies below the Li reference are unstable at the interface and the difference in the absolute values of ΔG°Red and ΔG°ref corresponds to the reduction stability of cations at the Li interface.
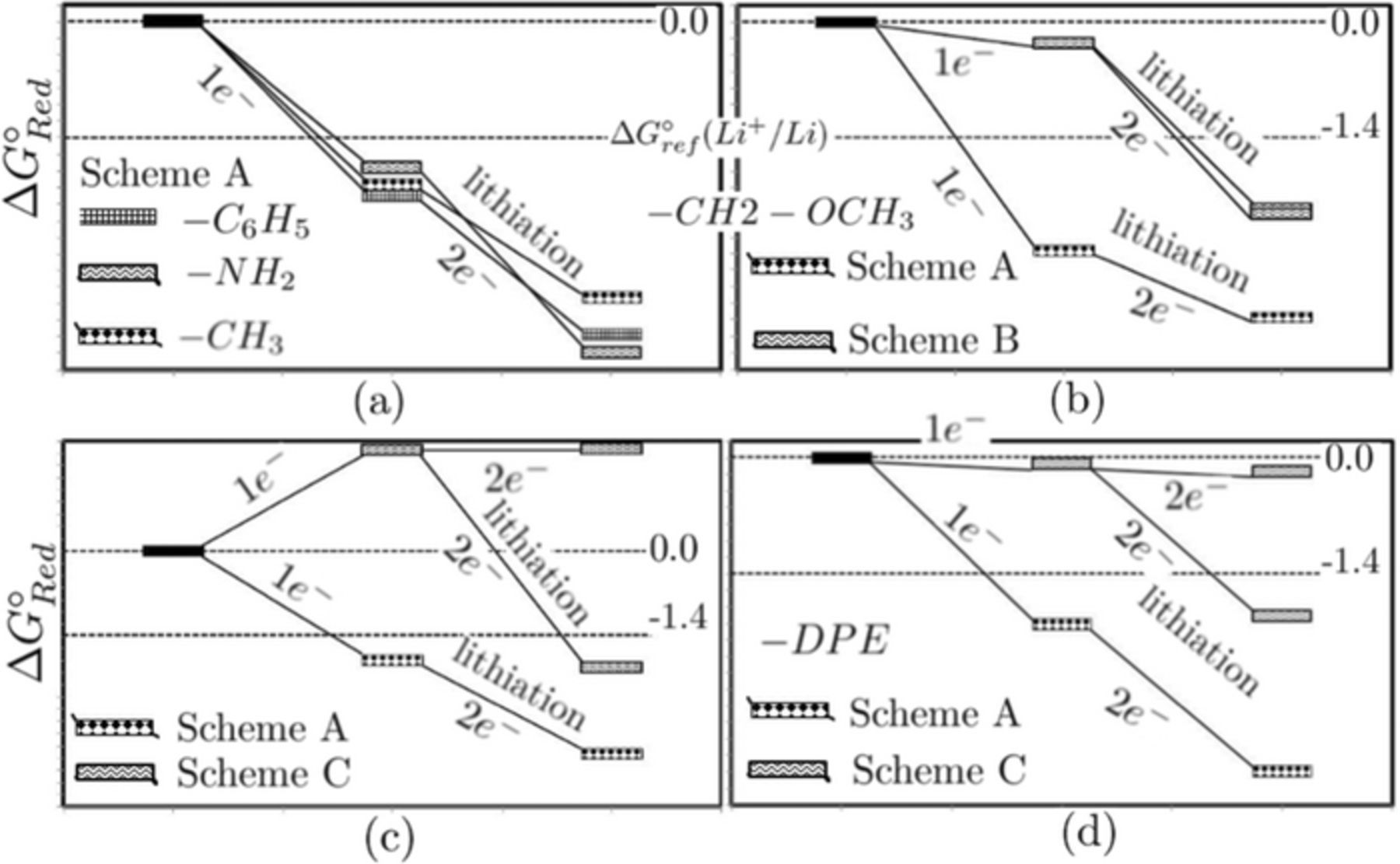
Figure 5. Free energy profiles of one and two e− reduction reactions of representative models. R/R' = CH3, NH2 and phenyl (a) dimethyl ether (b) para − aminophenyl (c) and diphenyl ether (d).
The general trends observed from the calculated data are (i) reductive decomposition of all the variants in the combinatorial library follow scheme A shown in Figure 3 and (ii) ΔG°Red for first electron transfer from Li electrode to cation is downhill in all the cases indicating instability at the Li interface and (iii) asymmetric substitution of functionalities (Vs symmetric substitution) did not improve the reductive stability of phospholanium cations. The first electron transfer is thus the rate determining step in all the cases. We further highlight the relative energetics of reductive decomposition in this section that reveal interesting trends in the reduction stabilities upon varying the substituent functional groups. In this study, energy barriers for decomposition of reduced phospholanium ions, and concomitant first and second electron transfers are not explicitly modeled. Hence the effect of stability of lithiated products (resulting from second electron transfer) on the thermodynamic barrier for first electron transfer is not evident from our calculations.
Reduction stability of R − CylP+5 − R' ions
- Linear alkane functionalized cations feature reduction stabilities on the order of 0.53–0.82 V vs Li+/Li; R/R' = −CH3 is the best candidate in the alkane series (Figure 5a). The first electron transfer from Li electrode is thermodynamically favorable with ΔG°Red = −1.96 eV, and the subsequent electron transfer resulting in the lithiation of free radical is further downhill with ΔG°Red = −3.33 eV.
- Reduction stabilities of longer alkyl chain substituted cations are lesser compared to − CH3 because of increased solvation energies of the free radicals of longer alkyl chains. In asymmetrically substituted models, dissociation of longer alkyl chains is favored compared to methyl and hence, the trend in reduction stabilities is similar to that of symmetrically substituted models.
- Previous theoretical studies39 reported an increase in the electron affinity of imidazolium cations as a function of increasing chain length of linear alkyl substituents and hence an increase in reduction stability, however, it is not observed in our calculations. We find that ethyl and longer alkyl chains have higher solvation energies compared to methyl, that add to the stability of the reduced products. Hence, we see an opposite trend in the reductive stabilities as a function of increasing alkyl chain length. Previous experimental studies64,65 also reported an increase in the reduction stability as a function of increasing alkyl chain length however, the increase in reductive stability was attributed to the increase in activation barrier for electron transfer due to shielding of cations by longer alklyl chains. The energy barriers for electron transfer are not calculated in this study, and hence the increase in kinetic stability is not evident from our calculations.
- Cyclic alkenes feature slightly lower reduction stabiltiies compared to linear alkyls with ΔG°Red on the order of −2.11 eV. The lower reduction stability of phenyl substiuted compared to methyl substituted cations is due to the difference in solvation energies of both groups. The reductive stability of cyclic alkene substituted CylP+5 cations is due to the localized electron density in the phenyl ring and the electron donating character of cyclic alkenes. Subsequent electron transfer is also strongly favored by 0.5 eV compared to methyl because of greater stabilization of lithiated phenyl radical. Similar to linear alkanes, asymmetric substitution did not improve the reduction stability of the cations.
- Moving from phenyl to napthyl further reduced the reduction stability; similar behavior is seen in both symmetric and asymmetric substitution. The solvation energies of phenyl and napthyl radicals are greater compared to C1 and hence reductive stability is lesser in asymmetric substitution compared to symmetric substitution.
- In ether substituted cations, ΔG°Red for dimethyl ether and diphenyl ether are shown in Figures 5b and 5d respectively. Diphenyl ether features better reduction stability in the ether series (0.62 V vs Li+/Li). The improvement in the reduction stability compared to other ether substitutions is due to the unstable − C6H5 − O − C6H5 radical, that is stabilized upon lithiation.
- Reductive decomposition of dimethyl ether functionalized cation proceeds via reaction scheme A with ΔG°Red(1e−) on the order of −2.76 eV. Similar to alkanes and alkenes, second electron transfer resulting in lithiation of methoxy radical is favorable by 1.8 eV. The difference in the reaction free energies of schemes A and B demonstrate the electrochemical stability of dimethyl ether compared to the bare phospholanium cation. Free energy of lithiation of − OCH3 and phospholanium cation (scheme B) is unfavorable by almost 2 eV compared to first electron transfer via scheme A. Hence, the subsequent electron transfer in scheme B results in lithiation of the charged phospholanium species that is highly favored by approximately 1.8 eV, compared to generation of another methoxy radical upon reduction. No significant difference in reductive stabilities is seen when dimethyl ether is replaced by other ether functionalities in the series.
- Primary amine features better reduction stability than − CH3 (0.36 V vs Li+/Li) as shown in Figure 5a however, subsequent electron transfer is favored by 0.7 eV compared to − CH3. When the hydrogens on the amine group are substituted by methyl or phenyl groups, the reduction stability decreases due to higher solvation energies of bulkier radicals compared to − NH2.
- The other variants where primary amines are attached to cyclic rings at ortho −, meta −, para − and ortho, para − positions, cations display different reductive stabilities that depend on the position of the amino group. For example, para − substitution, shown in Figure 5c, features better reductive stability (0.39 V vs Li+/Li) among the four variants and ortho − substitution is least stable.
Electrochemical windows
EW is typically calculated assuming that cathodic limit is set by reduction of cations and anodic limit is set by the oxidation of anions for a particular choice of anionic and cationic species. In some cases, the anodic and cathodic limits are set by the same ion (cation/anion), for example, the reduction potential of TFSI− is above the reduction potential of imidazolium cations,66 which leads to reduction and oxidation of the same species at the negative and positive electrodes, respectively. Hence, it is important to identify the ions that set cathodic and anodic limits to calculate the absolute EW for a given combination. Experimentally measured EWs of RTILs vary over a wide range and depend on the experimental conditions: purity of ILs, choice of reference electrode and cutoff current. Hence it is difficult to compare the calculated EWs against experiments. However, the trend in the relative stabilities can be reliably predicted using first principles calculations.26,39
The calculated oxidation potentials of two anions, BF−4 and PF−6, (shown in Figure 1) are on the order of 8.83 and 9.23 V, respectively and the order of oxidation stability of anions agree with the reported values.42,56,67 The oxidation potentials of R − CylP+5 − R' are also calculated here, and we find that oxidation potentials of all variants in the library are lower compared to both anions included this study. Hence, the calculated EWs in this paper correspond to the difference between cathodic and anodic limits of R − CylP+5 − R' ions. Calculated EWs predict that phosphorous based ILs feature wider EWs (shown in Table I and Supp. Info, Table S3) compared to nitrogen based ILs, consistent with previously reported trend in the electrochemical stability of two ILs.26,34
- Linear alkyl functionalied cations (R − CylP+5 − R') with R = CH3 features a wider EW on the order of 6.73 V. The EWs decrease with an increase in alkyl chain length. The reduction potential did not change significantly upon increasing the chain length from C1 to C6 ( ∼ 0.25 V) however, the oxidation potentials drop by approximately 1 V, resulting in decreased electrochemical stability on the order of 1.3 V. The aysmmetric substitution also shows similar trend in difference in the EWs as seen in symmetric substitution; EWs are in the range of 5.81 – 6.74 V with lesser values when the free radical is a longer alkyl chain compared to methyl group. The trends in electrochemical stability is due to increase the solvation energies of free radicals and oxidized cations; solvation energies of neutral phospholanium species with increasing varuing alkyl chain length are not significantly different.
- Cyclic alkenes substituted cations have similar reductive stability but lower oxidative stability compared to alkanes. Though both the functionalities feature similar reductive stability, cyclic alkene show a difference in the EWs (4.1 and 5.1 V) due to reduced oxidative stability of napthyl functionalized cations; unlike reductive stability, the oxidation potential is decreased by 1 V by increasing the ring count from one to two.
- Ether substitutions show a broader spread in EWs in the range of 3.5 – 4.6 V with dimethyl ether substituted cation being the best among the series. Asymmetric substitution with dimethyl ether and methyl further increases the value to 5 V. The increase in EW from asymmetric substitution is due to difference in the stability of dissociated cations with attached methyl group or ether group. Despite featuring wider EWs, all ether substitutions have poor reductive stability, and hence are not preferred candidate ILs compared to linear alkyl substituted ILs.
- Amine substitutions feature narrower EWs (3.4 – 3.9 V) compared ot linear alkyl substiuted cations because of reduced oxidation stability of amino-substituted cyclic rings.
Table I. Calculated Electrochemical Windows, Cathodic limits, VCL(Vs Li+/Li), and one and two e− reduction free energies of symmetrically substiuted R − CylP+5 − R' ions; R/R' = alkyls, ethers, cyclic alkenes and amines.
| VCL | VAL | ||
|---|---|---|---|
| R − CylP+5 − R' | EW | Vs Li+/Li | |
| R/R' = alkanes | |||
| methyl | 6.73 | 0.56 | 7.29 |
| ethyl | 6.22 | 0.82 | 7.05 |
| propyl | 6.05 | 0.77 | 6.82 |
| butyl | 5.83 | 0.71 | 6.55 |
| pentyl | 5.58 | 0.76 | 6.34 |
| hexyl | 5.42 | 0.75 | 6.17 |
| R/R' = alkenes | |||
| phenyl | 5.10 | 0.71 | 5.81 |
| napthyl | 4.09 | 0.75 | 4.84 |
| R/R' = ethers | |||
| diphenyl ether | 4.30 | 0.62 | 4.92 |
| dimethyl ether | 4.57 | 1.36 | 5.93 |
| methoxy 1,4-dioxane | 4.23 | 1.37 | 5.60 |
| methoxy benzene | 3.48 | 1.43 | 4.90 |
| methoxy ethene | 3.84 | 1.39 | 5.23 |
| methoxy terbutyl | 4.44 | 1.26 | 5.70 |
| R/R' = amines | |||
| NH2 | 5.90 | 0.36 | 6.27 |
| dimethylamine | 4.08 | 1.12 | 5.20 |
| N-methyl-phenylamine | 2.90 | 1.81 | 4.70 |
| N-methyl-paraamino-phenylamine | 1.78 | ||
| para amino-phenyl | 3.85 | 0.39 | 4.24 |
| ortho diamino-phenyl | 3.37 | 0.71 | 4.08 |
| meta diamino-phenyl | 3.40 | 0.53 | 3.93 |
| ortho,para triamino-phenyl | 3.64 | 0.32 | 3.96 |
Discussion
Reduction stability and EW calculations of the variants in the combinatorial library show that the linear alkane substituted phospholanium cations feature better reductive stability and wider EWs. R/R' = CH3 features wider EW (6.73 V) among all the variants in the group and the EWs decrease with an increase in the alkyl chain lengths due to the increase in the solvation energies of corresponding radicals of C2–C6 alkanes. We find EDG functionlized (alkyl, alkene) CylP+5 ions feature wider EWs and better reduction stabilities against Li+/Li whereas EWG functionalized (ethers) CylP+5 ions feature narrower EW compared to others, and also marginal reduction stability against Li+/Li. The electron donating character of amines varies depending on the position in the ring. Cations with NH2 attached to base phospholanium and para − and ortho, para − aminophenyl substituted cations exhibit better reduction stabilities compared to meta − aminophenyl substituted models. Replacing hydrogens with methyl or phenyl groups also decreases the EDG character of − NH2 group and hence the reduction stability of methyl, phenylamine substituted models is lesser compared to others in the group.
The calculated EWs for the variants in the combinatorial library with reference to the redox potentials of currently used electrodes are shown in Figure 6. All the variants feature excellent oxidation stability against currently preferred cathode materials and are marginally unstable at the anode interfaces. However, as seen from our calculations, reduction stability of ILs can be tuned by varying the functionalities. Linear alkyls, cyclic alkenes, and primary amines show good oxidative stability among the variants in the combinatorial library and hence can be potential alternatives to currently preferred electrolytes. Linear alkyl functionalized phospholanium cations and other variants that are marginally unstable at Li interface can become potential alternatives to organic electrolytes when used in combination with SEI forming additives, such as fluoroethylene carbonate (FEC), that result in a thicker and uniform SEI altering the chemical reactivity of the anode/SEI interface thereby creating a barrier for electron transfers to the electrolyte.52,68–71 Besides the electrochemical stability of base phospholanium cations, chemical stability of the functionalities calculated from free energies of lithiation prior to reduction show that the functional substituents are stable and do not breakdown at the Li interface.
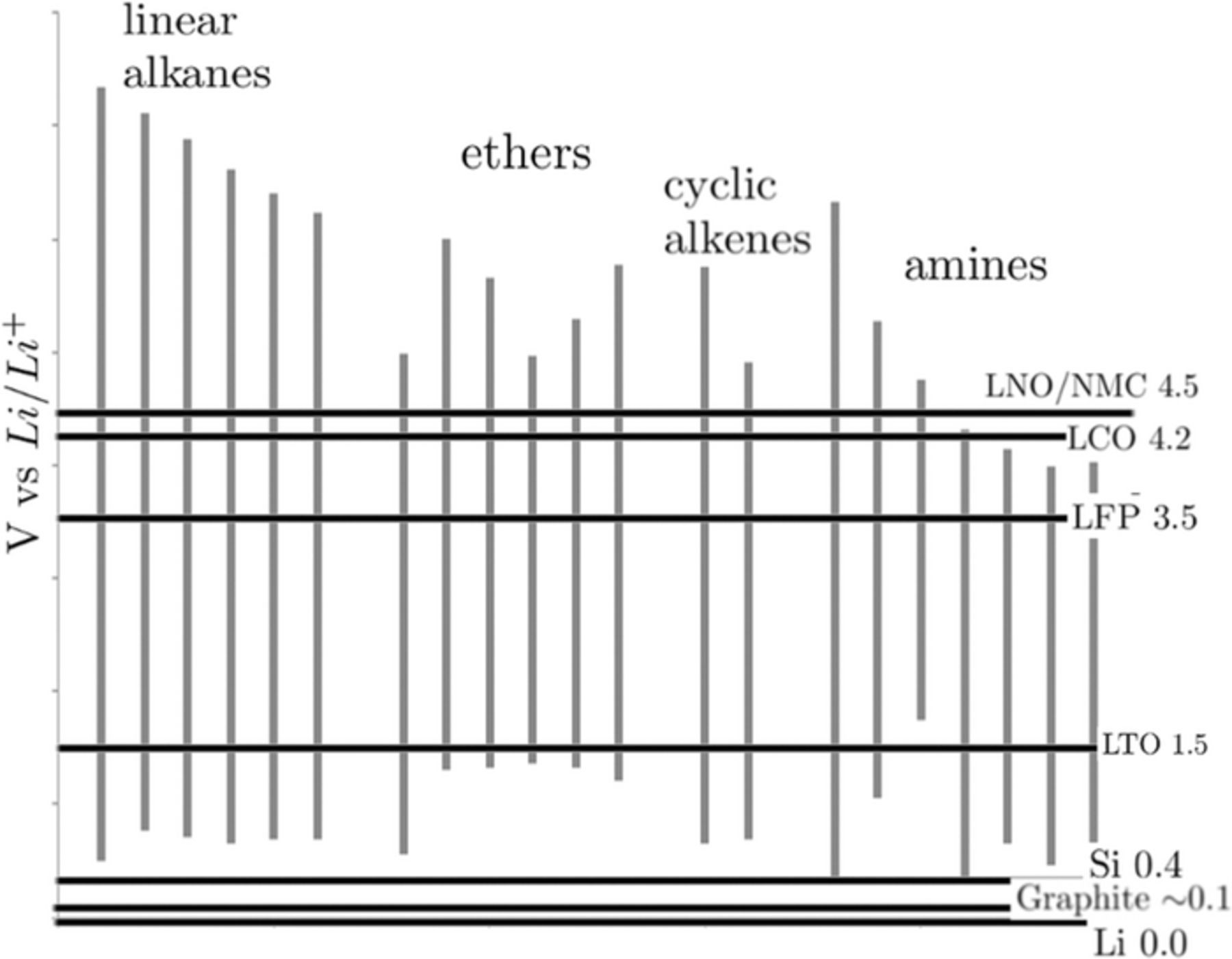
Figure 6. Electrochemical windows of linear alkane −, cyclic alkene −, ether − and amine − substituted CylP+5 cations. Redox potentials of commonly used electrodes are included for comparison.
Besides electrochemical stability at the electrode interfaces, viscosity and ionic conductivity of ionic liquids are also important when deployed as electrolytes in batteries. Smaller and symmetric cations (R = CH3 or NH2) based ILs are viscous compared to larger and asymmetric cations due to greater intermolecular interactions.72 Viscosity and density of nitrogen based and acyclic phosphonium based ILs can be altered by altering the flexibility and length of the alkyl/alkoxy chains.73–77 It is also reported that longer alkyl chains result in increased viscosity where as alkoxy functionalities result in decreased viscosity because of decreased intermolecular interactions in the latter case.74 Furthermore, the stable and bulkier substituents were also reported to increase the activation barrier for electron transfer resulting in increased kinetic stability of ILs. Combining the above arguements and the conclusions from this study that alkyls feature good electrochemical stabilities compared to ethers, it is further possible to design functionalized CylP+5 cations with longer alkyl chains terminated by alkoxy groups to achieve better electrochemical and thermo-physical properties.
Conclusions
CylP+n (n = 5 and 6) are phosphorous analogs of pyrrolidinium and piperidinium cations. CylP+5 cations feature better electrochemical stabilities compared to the nitrogen analogs. We generated a combinatorial library of functionalized CylP+5 ILs and demonstrated that the inherent redox stability of the CylP+5 ions can be tuned by the choice of the organic functional groups. We find that linear alkane functionalized CylP+5 ions feature good electrochemical stability among all the variants in the library and present opportunities to engineer thermophysical properties by facile chemical modifications. Hence, we conclude that the linear alkane functionalized CylP+n based ILs are potential candidate materials for use as electrolytes in advanced Li batteries and can be further pursued to engineer their electrochemical and thermo-physical properties.